
Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Compilation of Myotis Associated RABV Sequences
2.2. CST Quantification within the Myotis Genus in Comparison to Bats of Multiple Genera
2.3. Genetic and Ecological Host Predictors of CST within the Myotis Genus
2.3.1. Host Genetic Distance
2.3.2. Range Overlap
2.3.3. Foraging Niche Difference
2.3.4. Roosting Niche Overlap
2.3.5. Sample Size
2.4. Testing Predictors of RABV CST Frequency with the Myotis Genus
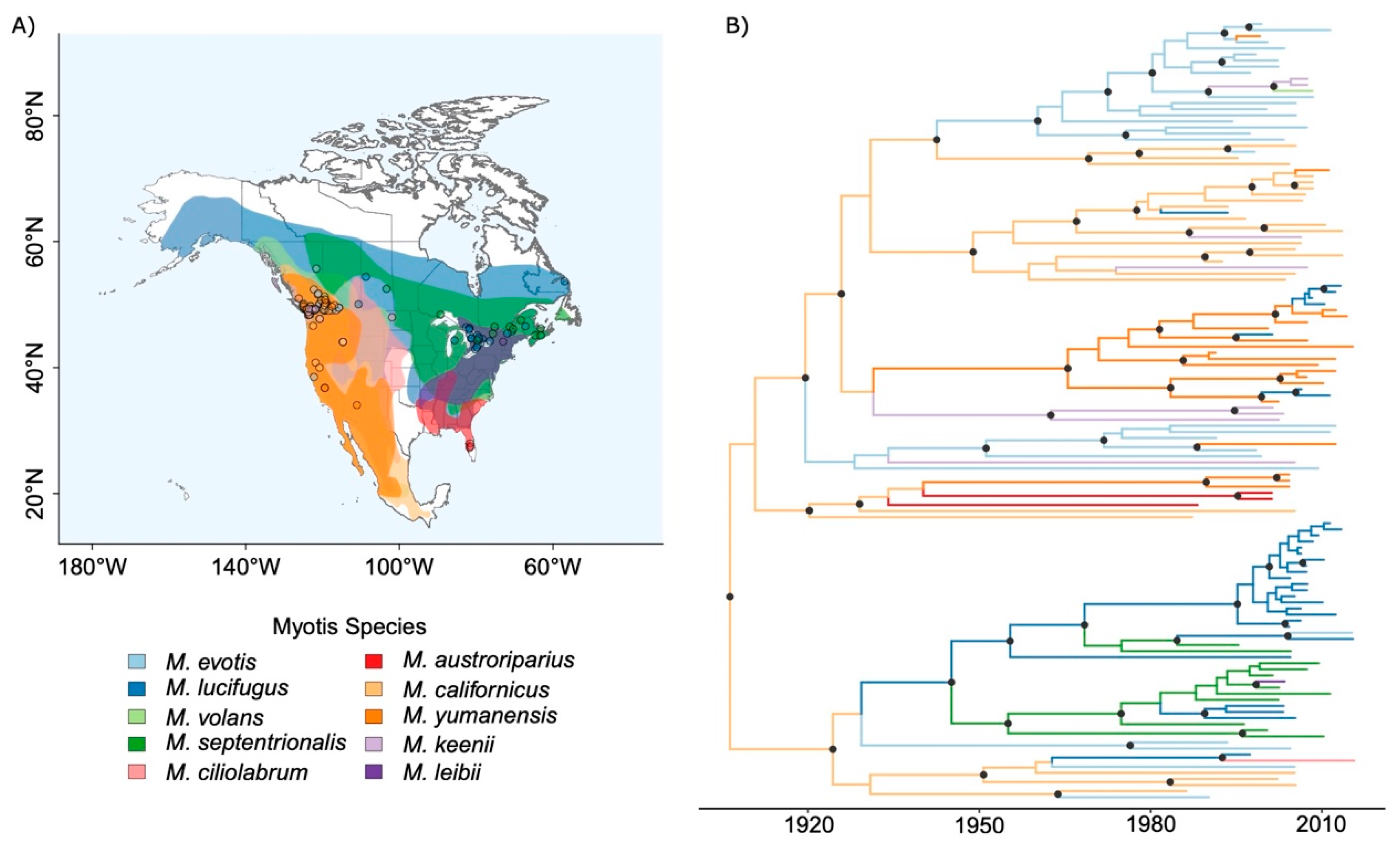
2.5. Clustering of Sequences Associated with Myotis Species
3. Results
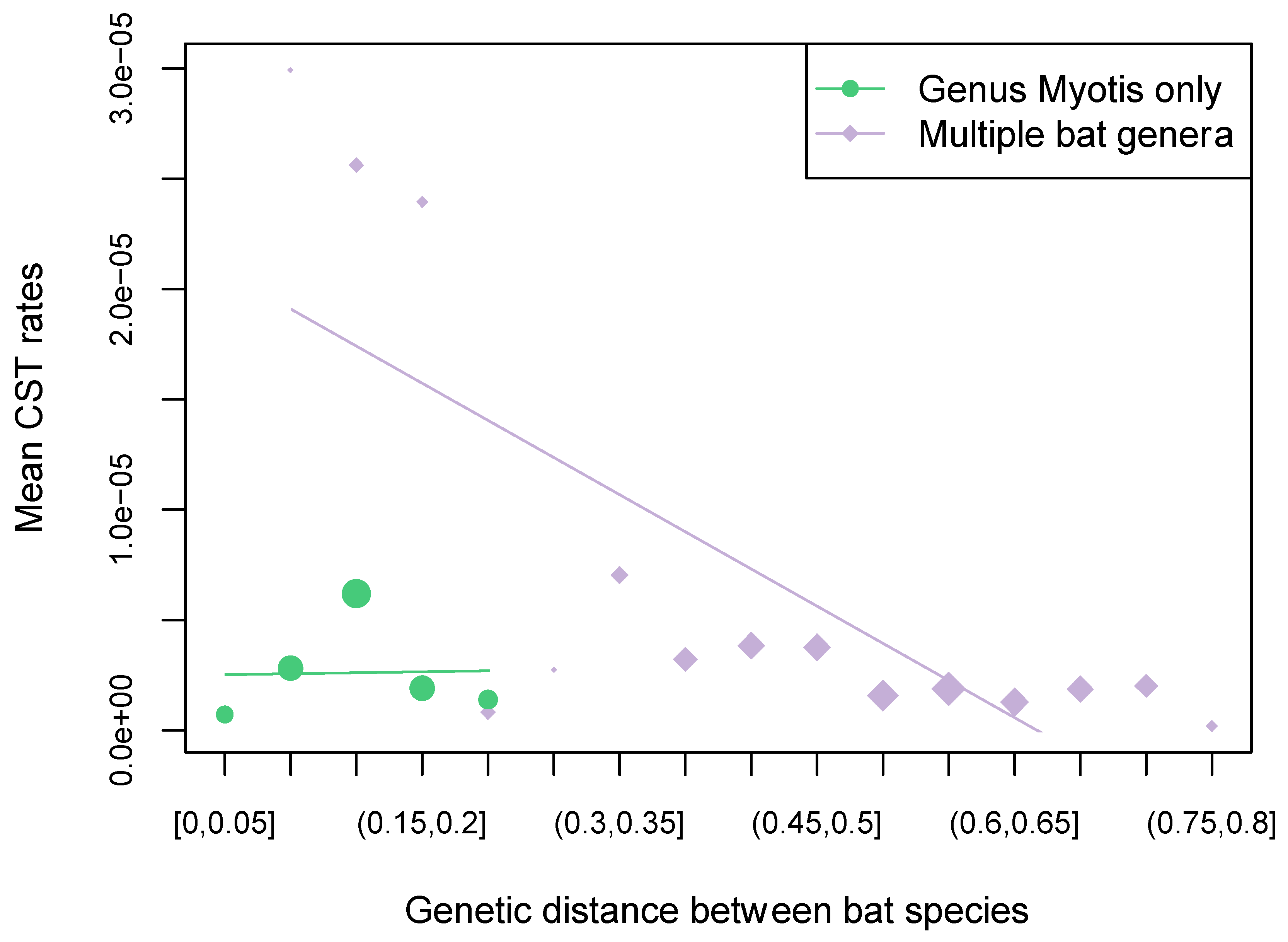
3.1. RABV Evolution and CST within North American Myotis Compared to a Multi-Genera Dataset
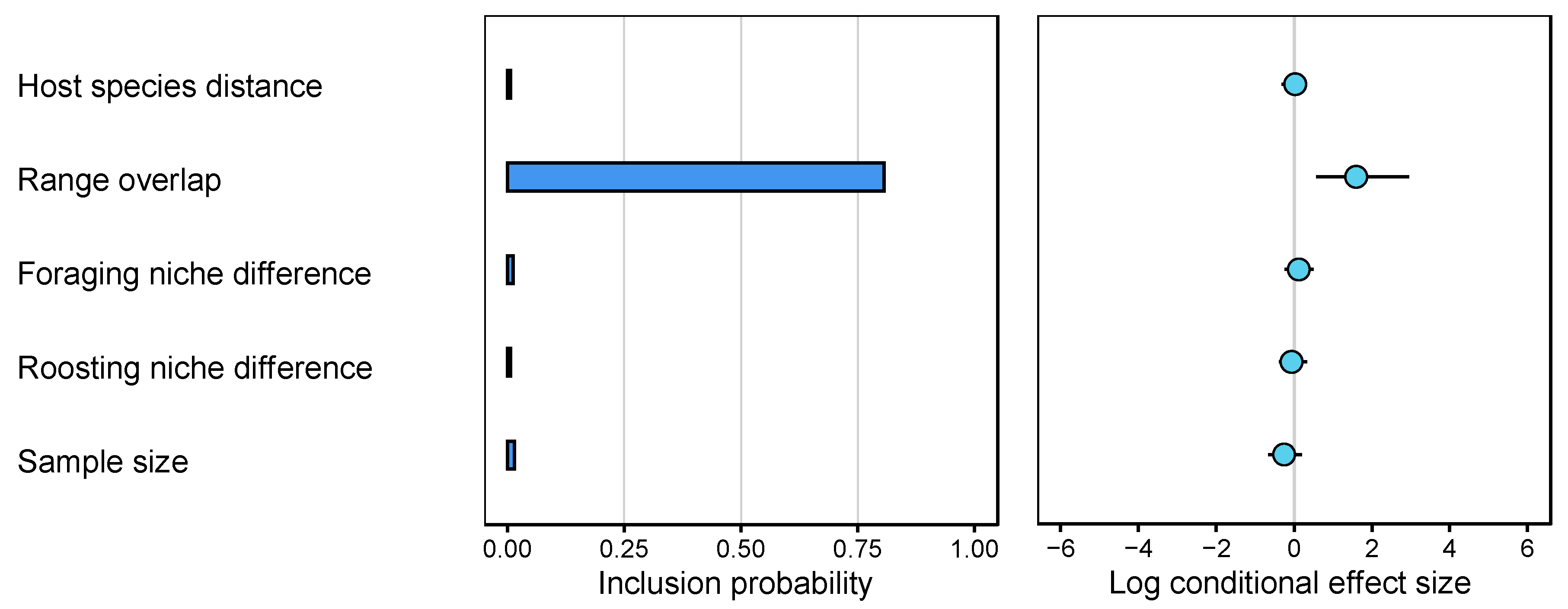
3.2. Testing Genetic and Ecological Host Predictors of CST in Myotis Bat Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holmes, E.C. The Comparative Genomics of Viral Emergence. Proc. Natl. Acad. Sci. USA 2010, 107, 1742–1746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longdon, B.; Brockhurst, M.A.; Russell, C.A.; Welch, J.J.; Jiggins, F.M. The Evolution and Genetics of Virus Host Shifts. PLoS Pathog. 2014, 10, e1004395. [Google Scholar] [CrossRef] [Green Version]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morens, D.M.; Folkers, G.K.; Fauci, A.S. Emerging Infections: A Perpetual Challenge. Lancet Infect. Dis. 2008, 8, 710–719. [Google Scholar] [CrossRef]
- Parrish, C.R.; Holmes, E.C.; Morens, D.M.; Park, E.-C.; Burke, D.S.; Calisher, C.H.; Laughlin, C.A.; Saif, L.J.; Daszak, P. Cross-Species Virus Transmission and the Emergence of New Epidemic Diseases. Microbiol. Mol. Biol. Rev. 2008, 72, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Wasik, B.R.; De Wit, E.; Munster, V.; Lloyd-Smith, J.O.; Martinez-Sobrido, L.; Parrish, C.R. Onward Transmission of Viruses: How Do Viruses Emerge to Cause Epidemics after Spillover? Philos. Trans. R. Soc. B Biol. Sci. 2019, 374, 20190017. [Google Scholar] [CrossRef] [Green Version]
- Longdon, B.; Hadfield, J.D.; Webster, C.L.; Obbard, D.J.; Jiggins, F.M. Host Phylogeny Determines Viral Persistence and Replication in Novel Hosts. PLoS Pathog. 2011, 7, e1002260. [Google Scholar] [CrossRef] [Green Version]
- Streicker, D.G.G.; Turmelle, A.S.S.; Vonhof, M.J.J.; Kuzmin, I.V.V.; McCracken, G.F.F.; Rupprecht, C.E.E. Host Phylogeny Constrains Cross-Species Emergence and Establishment of Rabies Virus in Bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef] [Green Version]
- Albery, G.F.; Eskew, E.A.; Ross, N.; Olival, K.J. Predicting the Global Mammalian Viral Sharing Network Using Phylogeography. Nat. Commun. 2020, 11, 2260. [Google Scholar] [CrossRef]
- Fisher, C.R.; Streicker, D.G.; Schnell, M.J. The Spread and Evolution of Rabies Virus: Conquering New Frontiers. Nat. Rev. Microbiol. 2018, 16, 241–255. [Google Scholar] [CrossRef]
- Singh, R.; Singh, K.P.; Cherian, S.; Saminathan, M.; Kapoor, S.; Manjunatha Reddy, G.B.; Panda, S.; Dhama, K. Rabies—Epidemiology, Pathogenesis, Public Health Concerns and Advances in Diagnosis and Control: A Comprehensive Review. Vet. Q. 2017, 37, 212–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzmin, I.V.; Shi, M.; Orciari, L.A.; Yager, P.A.; Velasco-Villa, A.; Kuzmina, N.A.; Streicker, D.G.; Bergman, D.L.; Rupprecht, C.E. Molecular Inferences Suggest Multiple Host Shifts of Rabies Viruses from Bats to Mesocarnivores in Arizona during 2001–2009. PLoS Pathog. 2012, 8, e1002786. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Villa, A.; Mauldin, M.R.; Shi, M.; Escobar, L.E.; Gallardo-Romero, N.F.; Damon, I.; Olson, V.A.; Streicker, D.G.; Emerson, G. The History of Rabies in the Western Hemisphere. Antivir. Res. 2017, 146, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Wallace, R.M.; Gilbert, A.; Slate, D.; Chipman, R.; Singh, A.; Wedd, C.; Blanton, J.D. Right Place, Wrong Species: A 20-Year Review of Rabies Virus Cross Species Transmission among Terrestrial Mammals in the United States. PLoS ONE 2014, 9, e107539. [Google Scholar] [CrossRef]
- Mollentze, N.; Biek, R.; Streicker, D.G. The Role of Viral Evolution in Rabies Host Shifts and Emergence. Curr. Opin. Virol. 2014, 8, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Mollentze, N.; Streicker, D.G.; Murcia, P.R.; Hampson, K.; Biek, R. Virulence Mismatches in Index Hosts Shape the Outcomes of Cross-Species Transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 28859–28866. [Google Scholar] [CrossRef]
- Borucki, M.K.; Chen-Harris, H.; Lao, V.; Vanier, G.; Wadford, D.A.; Messenger, S.; Allen, J.E. Ultra-Deep Sequencing of Intra-Host Rabies Virus Populations during Cross-Species Transmission. PLoS Negl. Trop. Dis. 2013, 7, e2555. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Bininda-Emonds, O.R.P.; Stephens, P.R.; Gittleman, J.L.; Altizer, S. Phylogenetically Related and Ecologically Similar Carnivores Harbour Similar Parasite Assemblages. J. Anim. Ecol. 2014, 83, 671–680. [Google Scholar] [CrossRef]
- Faria, N.R.; Suchard, M.A.; Rambaut, A.; Streicker, D.G.; Lemey, P. Simultaneously Reconstructing Viral Cross-Species Transmission History and Identifying the Underlying Constraints. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2013, 368, 20120196. [Google Scholar] [CrossRef] [Green Version]
- Bray, T.C.; Mohammed, O.B.; Alagaili, A.N. Phylogenetic and Demographic Insights into Kuhl’s Pipistrelle, Pipistrellus Kuhlii, in the Middle East. PLoS ONE 2013, 8, e57306. [Google Scholar] [CrossRef]
- Miller-Butterworth, C.M.; Murphy, W.J.; O’Brien, S.J.; Jacobs, D.S.; Springer, M.S.; Teeling, E.C. A Family Matter: Conclusive Resolution of the Taxonomic Position of the Long-Fingered Bats, Miniopterus. Mol. Biol. Evol. 2007, 24, 1553–1561. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Suleski, M.; Blair Hedges, S. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef]
- Nadin-Davis, S.; Alnabelseya, N.; Knowles, M.K.; Streicker, D.; Lemey, P. The Phylogeography of Myotis Bat-Associated Rabies Viruses across Canada. PLoS Negl. Trop. Dis. 2017, 11, e0005541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.; Suchard, M. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minin, V.N.; Suchard, M.A. Fast, Accurate and Simulation-Free Stochastic Mapping. Philos. Trans. R. Soc. B Biol. Sci. 2008, 363, 3985–3995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Gascuel, O. A Simple, Fast, and Accurate Algorithm to Estimate Large Phylogenies by Maximum Likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, 699–710. [Google Scholar] [CrossRef]
- Minin, V.N.; Bloomquist, E.W.; Suchard, M.A. Smooth Skyride through a Rough Skyline: Bayesian Coalescent-Based Inference of Population Dynamics. Mol. Biol. Evol. 2008, 25, 1459–1471. [Google Scholar] [CrossRef]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum Likelihood Phylogenetic Analysis Using Quartets and Parallel Computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R Language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef] [Green Version]
- Bivand, R.; Lewin-Koh, N. Maptools: Tools for Reading and Handling Spatial Objects; R Package Version 0.8-39; 2011; Available online: https://cran.r-project.org/web/packages/maptools/index.html (accessed on 27 October 2022).
- Pebesma, E.J.; Bivand, R.S. Classes and Methods for Spatial Data in R. R News 2005, 5, 9–13. [Google Scholar]
- Baddeley, A.; Rubak, E.; Turner, R. Spatial Point Patterns: Methodology and Application with R; Chapman and Hall: London, UK; CRC Press: Boca Raton, FL, USA, 2015. [Google Scholar]
- Hijmans, R.J. Raster: Geographic Data Analysis and Modeling; R Package Version 2.5-8; 2016; Available online: https://rdrr.io/cran/raster/ (accessed on 27 October 2022).
- Farney, J.; Fleharty, E.D. Aspect Ratio, Loading, Wing Span, and Membrane Areas of Bats. J. Mammal. 1969, 50, 362–367. [Google Scholar] [CrossRef]
- Krutzsch, P.H. Notes of the Habits of the Bat, Myotis Californicus. J. Mammal. 1954, 35, 539–545. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. Unifying Viral Genetics and Human Transportation Data to Predict the Global Transmission Dynamics of Human Influenza H3N2. PLoS Pathog. 2014, 10, e1003932. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Gorjanc, G.; Grothendieck, G.; Korosec, A.; Lumley, T.; MacQueen, D.; Magnusson, A.; Rogers, J.; Al, E. Gdata: Various R Programming Tools for Data Manipulation; R Package Version 2.18.0; 2017; Available online: https://rdrr.io/cran/gdata/ (accessed on 27 October 2022).
- Goslee, S.; Urban, D. The Ecodist Package: Dissimilarity-Based Functions for Ecological Analysis. J. Stat. Softw. 2007, 22, 1–19. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Bedford, T.; Faria, N.R.; Bielejec, F.; Baele, G.; Russell, C.A.; Smith, D.J.; Pybus, O.G.; Brockmann, D.; et al. The Seasonal Flight of Influenza: A Unified Framework for Spatiotemporal Hypothesis Testing. arXiv 2012, arXiv:1210.5877. [Google Scholar] [CrossRef]
- Jeffreys, H. Theory of Probability, 3rd ed.; Clarendon Press: Oxford, UK, 1961; Volume 2. [Google Scholar]
- Morales, A.E.; Carstens, B.C. Evidence That Myotis Lucifugus “Subspecies” Are Five Nonsister Species, Despite Gene Flow. Syst. Biol. 2018, 67, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murtagh, F.; Legendre, P. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? J. Classif. 2014, 31, 274–295. [Google Scholar] [CrossRef] [Green Version]
- George, D.B.; Webb, C.T.; Farnsworth, M.L.; O’Shea, T.J.; Bowen, R.A.; Smith, D.L.; Stanley, T.R.; Ellison, L.E.; Rupprecht, C.E. Host and Viral Ecology Determine Bat Rabies Seasonality and Maintenance. Proc. Natl. Acad. Sci. USA 2011, 108, 10208–10213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streicker, D.G.; Lemey, P.; Velasco-Villa, A.; Rupprecht, C.E. Rates of Viral Evolution Are Linked to Host Geography in Bat Rabies. PLoS Pathog. 2012, 8, e1002720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadelmann, B.; Lin, L.K.; Kunz, T.H.; Ruedi, M. Molecular Phylogeny of New World Myotis (Chiroptera, Vespertilionidae) Inferred from Mitochondrial and Nuclear DNA Genes. Mol. Phylogenet. Evol. 2007, 43, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Streicker, D.G.; Altizer, S.M.; Velasco-Villa, A.; Rupprecht, C.E. Variable Evolutionary Routes to Host Establishment across Repeated Rabies Virus Host Shifts among Bats. Proc. Natl. Acad. Sci. USA 2012, 109, 19715–19720. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, A.B.; Davies, T.J. Cross-Species Pathogen Transmission and Disease Emergence in Primates. Ecohealth 2009, 6, 496–508. [Google Scholar] [CrossRef]
- Condori-Condori, R.E.; Streicker, D.G.; Cabezas-Sanchez, C.; Velasco-Villa, A. Enzootic and Epizootic Rabies Associated with Vampire Bats, Peru. Emerg. Infect. Dis. 2013, 19, 1463–1469. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Huang, W.; Armstrong, J.; Casey, G.A.; Bahloul, C.; Tordo, N.; Wandeler, A.I. Antigenic and Genetic Divergence of Rabies Viruses from Bat Species Indigenous to Canada. Virus Res. 2001, 74, 139–156. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jacquot, M.; Wallace, M.A.; Streicker, D.G.; Biek, R. Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species. Viruses 2022, 14, 2399. https://doi.org/10.3390/v14112399
Jacquot M, Wallace MA, Streicker DG, Biek R. Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species. Viruses. 2022; 14(11):2399. https://doi.org/10.3390/v14112399
Chicago/Turabian StyleJacquot, Maude, Megan A. Wallace, Daniel G. Streicker, and Roman Biek. 2022. "Geographic Range Overlap Rather than Phylogenetic Distance Explains Rabies Virus Transmission among Closely Related Bat Species" Viruses 14, no. 11: 2399. https://doi.org/10.3390/v14112399