
ORF-Interrupting Mutations in Monkeypox Virus Genomes from Washington and Ohio, 2022
 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Extraction, Library Preparation, and Whole Genome Sequencing
2.2. MPXV Bioinformatic Analysis
2.3. Confirmatory Deletion PCRs and ddPCR
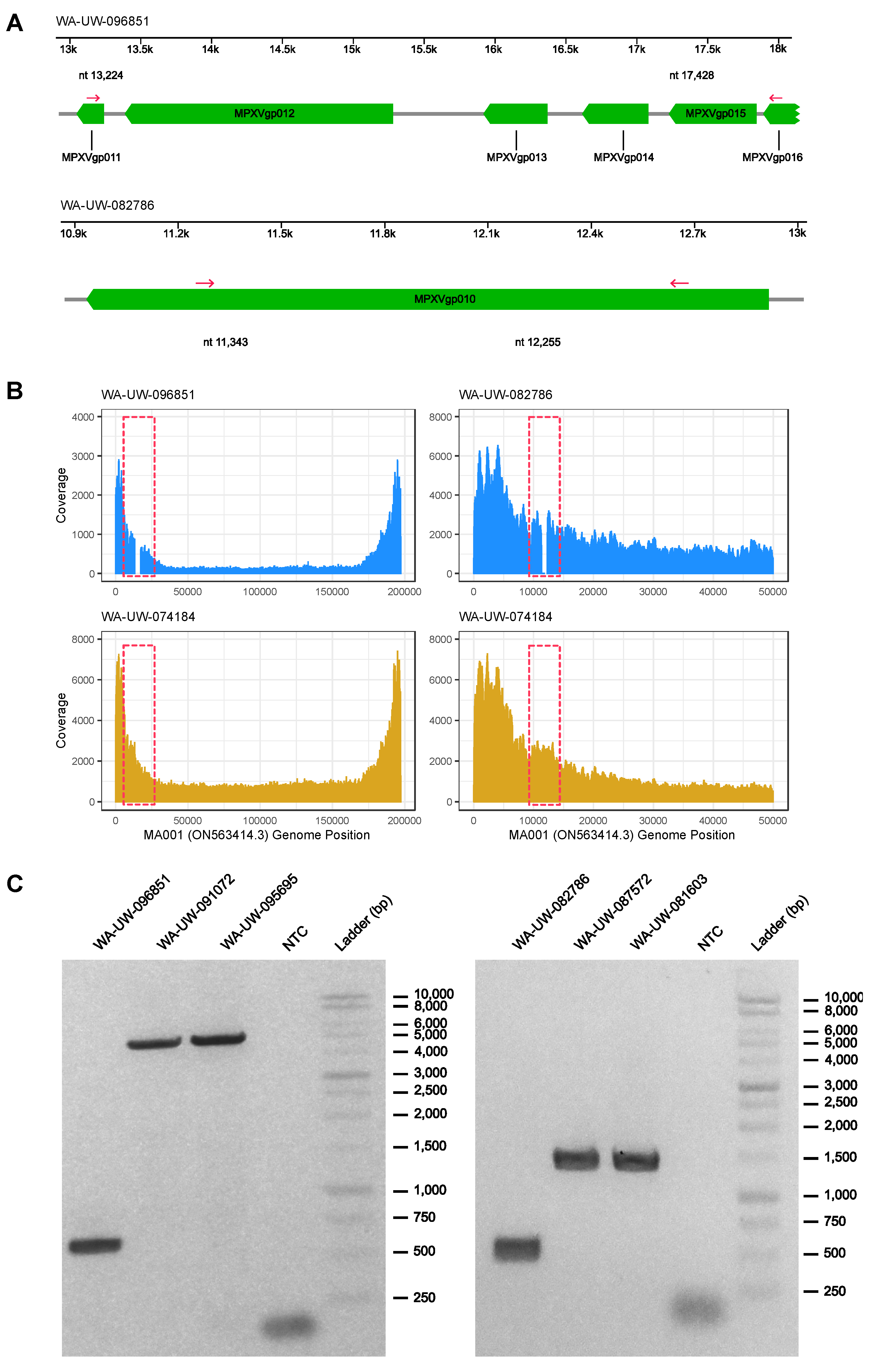
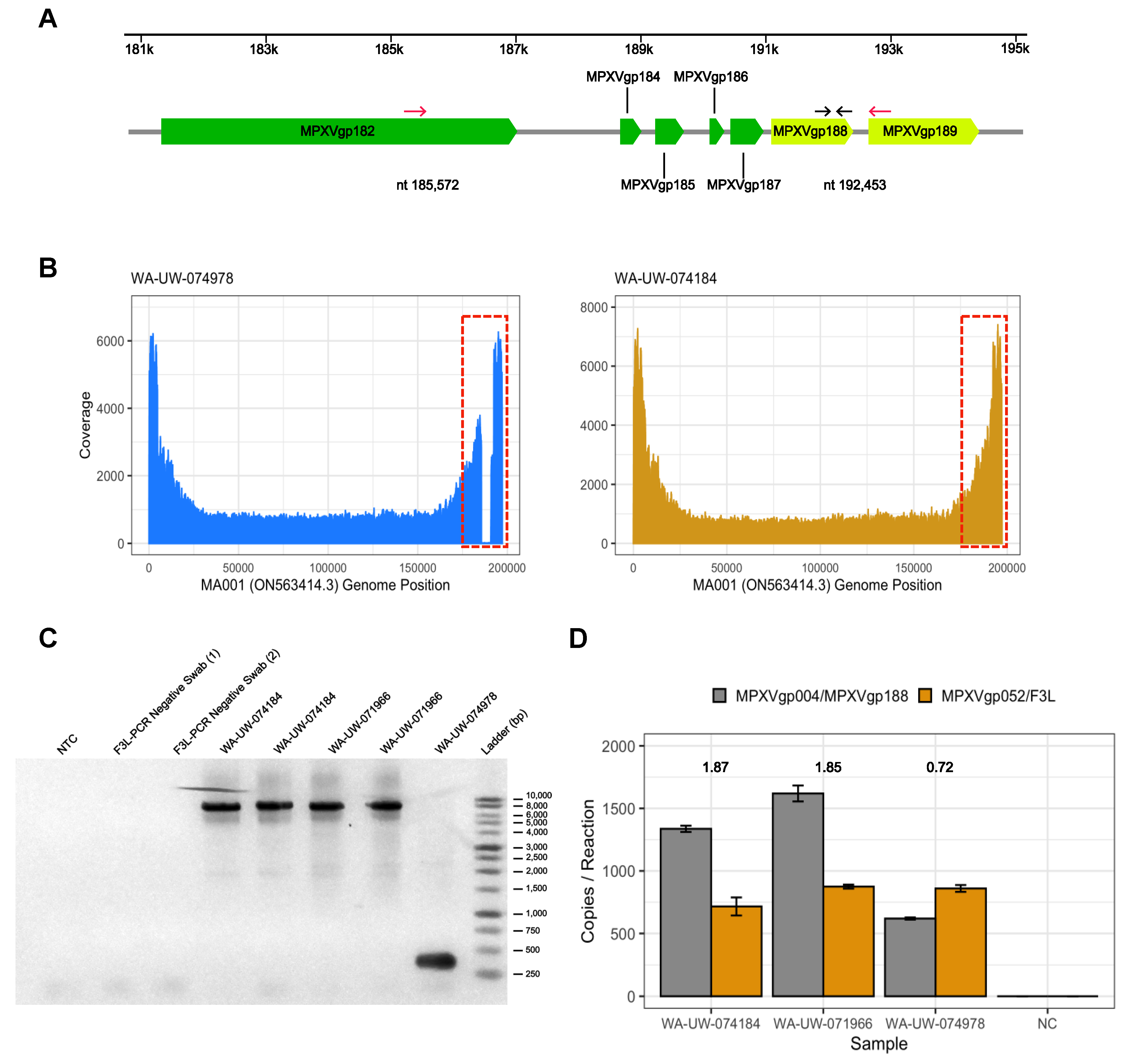
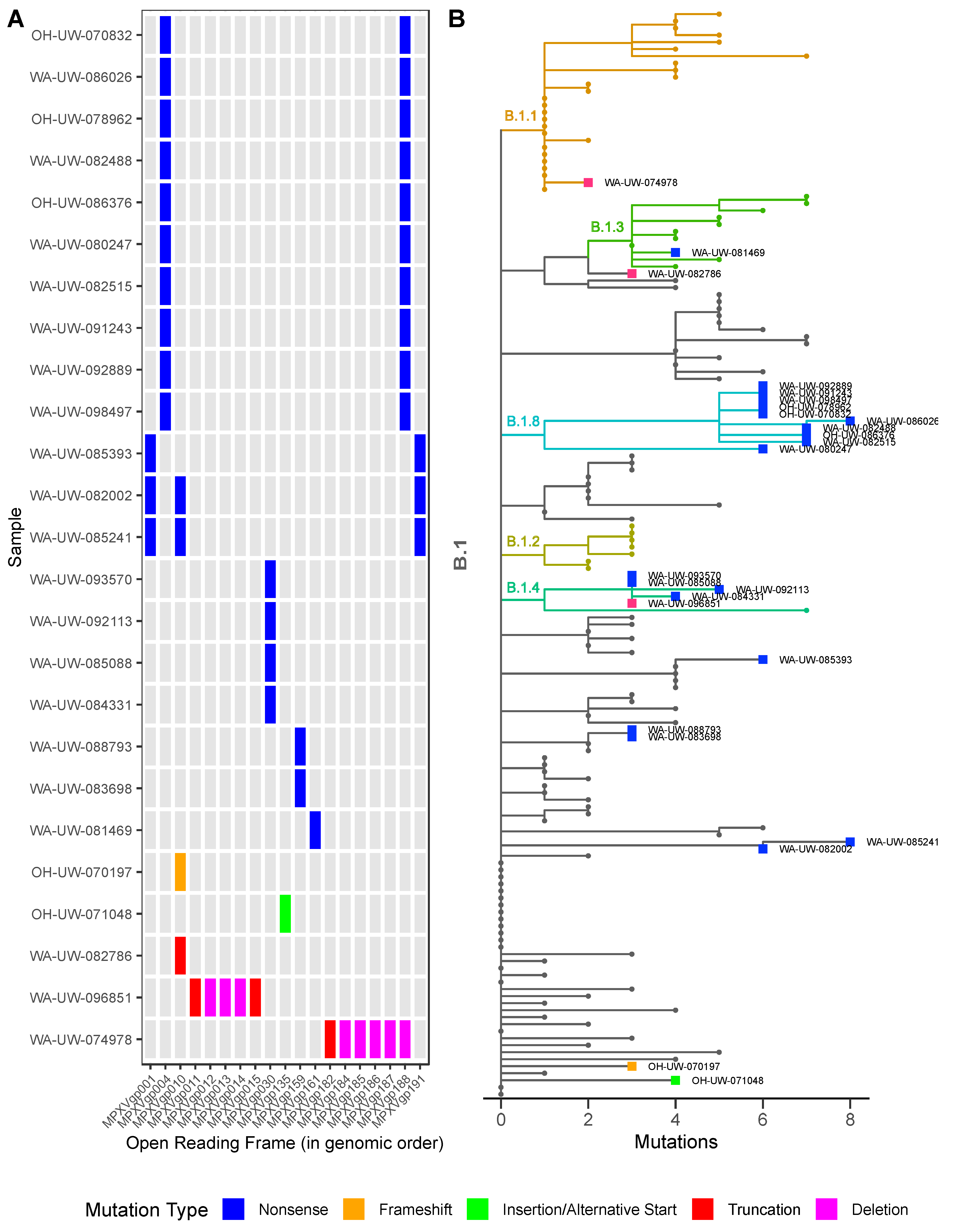
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alakunle, E.; Moens, U.; Nchinda, G.; Okeke, M.I. Monkeypox Virus in Nigeria: Infection Biology, Epidemiology, and Evolution. Viruses 2020, 12, 1257. [Google Scholar] [CrossRef] [PubMed]
- Kugelman, J.R.; Johnston, S.C.; Mulembakani, P.M.; Kisalu, N.; Lee, M.S.; Koroleva, G.; McCarthy, S.E.; Gestole, M.C.; Wolfe, N.D.; Fair, J.N.; et al. Genomic Variability of Monkeypox Virus among Humans, Democratic Republic of the Congo. Emerg. Infect. Dis. 2014, 20, 232–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteban, D.J.; Hutchinson, A.P. Genes in the Terminal Regions of Orthopoxvirus Genomes Experience Adaptive Molecular Evolution. BMC Genom. 2011, 12, 261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrickson, R.C.; Wang, C.; Hatcher, E.L.; Lefkowitz, E.J. Orthopoxvirus Genome Evolution: The Role of Gene Loss. Viruses 2010, 2, 1933–1967. [Google Scholar] [CrossRef] [PubMed]
- Firth, C.; Kitchen, A.; Shapiro, B.; Suchard, M.A.; Holmes, E.C.; Rambaut, A. Using Time-Structured Data to Estimate Evolutionary Rates of Double-Stranded DNA Viruses. Mol. Biol. Evol. 2010, 27, 2038–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidro, J.; Borges, V.; Pinto, M.; Sobral, D.; Santos, J.D.; Nunes, A.; Mixão, V.; Ferreira, R.; Santos, D.; Duarte, S.; et al. Phylogenomic Characterization and Signs of Microevolution in the 2022 Multi-Country Outbreak of Monkeypox Virus. Nat. Med. 2022, 28, 1569–1572. [Google Scholar] [CrossRef]
- Wang, L.; Shang, J.; Weng, S.; Aliyari, S.R.; Ji, C.; Cheng, G.; Wu, A. Genomic Annotation and Molecular Evolution of Monkeypox Virus Outbreak in 2022. J. Med. Virol. 2022. [Google Scholar] [CrossRef]
- Jones, T.C.; Schneider, J.; Mühlemann, B.; Veith, T.; Beheim-Schwarzbach, J.; Tesch, J.; Schmidt, M.L.; Walper, F.; Bleicker, T.; Isner, C.; et al. Genetic Variability, Including Gene Duplication and Deletion, in Early Sequences from the 2022 European Monkeypox Outbreak. bioRxiv 2022. [Google Scholar] [CrossRef]
- Desingu, P.A.; Nagarajan, K. Genomic Regions Insertion and Deletion in Monkeypox Virus Causing Multi-Country Outbreak-2022. bioRxiv 2022. [Google Scholar] [CrossRef]
- Gigante, C.M.; Plumb, M.; Ruprecht, A.; Zhao, H.; Wicker, V.; Wilkins, K.; Matheny, A.; Khan, T.; Davidson, W.; Sheth, M.; et al. Genomic Deletions and Rearrangements in Monkeypox Virus from the 2022 Outbreak, USA. bioRxiv 2022. [Google Scholar] [CrossRef]
- Estep, R.D.; Messaoudi, I.; O’Connor, M.A.; Li, H.; Sprague, J.; Barron, A.; Engelmann, F.; Yen, B.; Powers, M.F.; Jones, J.M.; et al. Deletion of the Monkeypox Virus Inhibitor of Complement Enzymes Locus Impacts the Adaptive Immune Response to Monkeypox Virus in a Nonhuman Primate Model of Infection. J. Virol. 2011, 85, 9527–9542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.; Li, G.; Liszewski, M.K.; Atkinson, J.P.; Jahrling, P.B.; Feng, Z.; Schriewer, J.; Buck, C.; Wang, C.; Lefkowitz, E.J.; et al. Virulence Differences between Monkeypox Virus Isolates from West Africa and the Congo Basin. Virology 2005, 340, 46–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammarlund, E.; Lewis, M.W.; Carter, S.V.; Amanna, I.; Hansen, S.G.; Strelow, L.I.; Wong, S.W.; Yoshihara, P.; Hanifin, J.M.; Slifka, M.K. Multiple Diagnostic Techniques Identify Previously Vaccinated Individuals with Protective Immunity against Monkeypox. Nat. Med. 2005, 11, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, N.A.P.; Mathias, P.C.; Bradley, B.T.; Greninger, A.L. Clinical Performance and Trends During the First Two Months of Monkeypox Virus PCR Testing at Two United States Reference Labs. medRxiv 2022. [Google Scholar] [CrossRef]
- Maksyutov, R.A.; Gavrilova, E.V.; Shchelkunov, S.N. Species-Specific Differentiation of Variola, Monkeypox, and Varicella-Zoster Viruses by Multiplex Real-Time PCR Assay. J Virol Methods 2016, 236, 215–220. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner; Lawrence Berkeley National Lab.: Berkeley, CA, USA, 2014. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup the Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Gangavarapu, K.; Quick, J.; Matteson, N.L.; De Jesus, J.G.; Main, B.J.; Tan, A.L.; Paul, L.M.; Brackney, D.E.; Grewal, S.; et al. An Amplicon-Based Sequencing Framework for Accurately Measuring Intrahost Virus Diversity Using PrimalSeq and IVar. Genome Biol. 2019, 20, 8. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The Conserved Domain Database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teufel, F.; Almagro Armenteros, J.J.; Johansen, A.R.; Gíslason, M.H.; Pihl, S.I.; Tsirigos, K.D.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 6.0 Predicts All Five Types of Signal Peptides Using Protein Language Models. Nat. Biotechnol. 2022, 40, 1023–1025. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhury, P.; Sereewit, J.; Xie, H.; Nunley, E.; Lieberman, N.A.P.; Greninger, A.L. Genomic Analysis of Early Spread of Monkeypox Virus in Washington State. MedRxiv 2022. [Google Scholar] [CrossRef]
- Minhaj, F.S.; Ogale, Y.P.; Whitehill, F.; Schultz, J.; Foote, M.; Davidson, W.; Hughes, C.M.; Wilkins, K.; Bachmann, L.; Chatelain, R.; et al. Monkeypox Outbreak—Nine States, May 2022. MMWR Morb. Mortal. Wkly. Rep. 2022, 71, 764–769. [Google Scholar] [CrossRef]
- Elde, N.C.; Child, S.J.; Eickbush, M.T.; Kitzman, J.O.; Rogers, K.S.; Shendure, J.; Geballe, A.P.; Malik, H.S. Poxviruses Deploy Genomic Accordions to Adapt Rapidly against Host Antiviral Defenses. Cell 2012, 150, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Assis, F.L.; Almeida, G.M.F.; Oliveira, D.B.; Franco-Luiz, A.P.M.; Campos, R.K.; Guedes, M.I.M.; Fonseca, F.G.; Trindade, G.S.; Drumond, B.P.; Kroon, E.G.; et al. Characterization of a New Vaccinia Virus Isolate Reveals the C23L Gene as a Putative Genetic Marker for Autochthonous Group 1 Brazilian Vaccinia Virus. PLoS ONE 2012, 7, e50413. [Google Scholar] [CrossRef] [Green Version]
- Caillat, C.; Topalis, D.; Agrofoglio, L.A.; Pochet, S.; Balzarini, J.; Deville-Bonne, D.; Meyer, P. Crystal Structure of Poxvirus Thymidylate Kinase: An Unexpected Dimerization Has Implications for Antiviral Therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 16900–16905. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Cao, S.; Martens, C.A.; Porcella, S.F.; Xie, Z.; Ma, M.; Shen, B.; Moss, B. Deciphering Poxvirus Gene Expression by RNA Sequencing and Ribosome Profiling. J. Virol. 2015, 89, 6874–6886. [Google Scholar] [CrossRef] [Green Version]
- Mauldin, M.R.; McCollum, A.M.; Nakazawa, Y.J.; Mandra, A.; Whitehouse, E.R.; Davidson, W.; Zhao, H.; Gao, J.; Li, Y.; Doty, J.; et al. Exportation of Monkeypox Virus from the African Continent. J. Infect. Dis. 2022, 225, 1367–1376. [Google Scholar] [CrossRef]
- Shchelkunov, S.N. Orthopoxvirus Genes That Mediate Disease Virulence and Host Tropism. Adv. Virol. 2012, 2012, 524743. [Google Scholar] [CrossRef]
- Graham, S.C.; Bahar, M.W.; Cooray, S.; Chen, R.A.-J.; Whalen, D.M.; Abrescia, N.G.A.; Alderton, D.; Owens, R.J.; Stuart, D.I.; Smith, G.L.; et al. Vaccinia Virus Proteins A52 and B14 Share a Bcl-2-like Fold but Have Evolved to Inhibit NF-KappaB Rather than Apoptosis. PLoS Pathog. 2008, 4, e1000128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.A.; Trossman, D.S.; Yokoyama, W.M.; Carayannopoulos, L.N. Zoonotic Orthopoxviruses Encode a High-Affinity Antagonist of NKG2D. J. Exp. Med. 2007, 204, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Alzhanova, D.; Hammarlund, E.; Reed, J.; Meermeier, E.; Rawlings, S.; Ray, C.A.; Edwards, D.M.; Bimber, B.; Legasse, A.; Planer, S.; et al. T Cell Inactivation by Poxviral B22 Family Proteins Increases Viral Virulence. PLoS Pathog. 2014, 10, e1004123. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sereewit, J.; Lieberman, N.A.P.; Xie, H.; Bakhash, S.A.K.M.; Nunley, B.E.; Chung, B.; Mills, M.G.; Roychoudhury, P.; Greninger, A.L. ORF-Interrupting Mutations in Monkeypox Virus Genomes from Washington and Ohio, 2022. Viruses 2022, 14, 2393. https://doi.org/10.3390/v14112393
Sereewit J, Lieberman NAP, Xie H, Bakhash SAKM, Nunley BE, Chung B, Mills MG, Roychoudhury P, Greninger AL. ORF-Interrupting Mutations in Monkeypox Virus Genomes from Washington and Ohio, 2022. Viruses. 2022; 14(11):2393. https://doi.org/10.3390/v14112393
Chicago/Turabian StyleSereewit, Jaydee, Nicole A. P. Lieberman, Hong Xie, Shah A. K. Mohamed Bakhash, B. Ethan Nunley, Benjamin Chung, Margaret G. Mills, Pavitra Roychoudhury, and Alexander L. Greninger. 2022. "ORF-Interrupting Mutations in Monkeypox Virus Genomes from Washington and Ohio, 2022" Viruses 14, no. 11: 2393. https://doi.org/10.3390/v14112393