
In Silico Characterization of African Swine Fever Virus Nucleoprotein p10 Interaction with DNA
 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model Generation of wt:p10
2.2. Molecular Dynamics Simulations
2.3. MM/PBSA Calculations
2.4. Design of p10 Mutations
2.5. p10 and Mutants’ Production and Purification
2.6. Protein Quantification
2.7. ELISA Assays
2.8. Statistical Analysis
3. Results
3.1. wt:p10 Model Building and Its Characterization
3.2. Prediction of the Interaction of the Truncated wt:p10 with dsDNA
3.3. Design of p10 Mutants
3.4. In Silico Evaluation of the Truncated p10 Mutants and Their Interaction with DNA
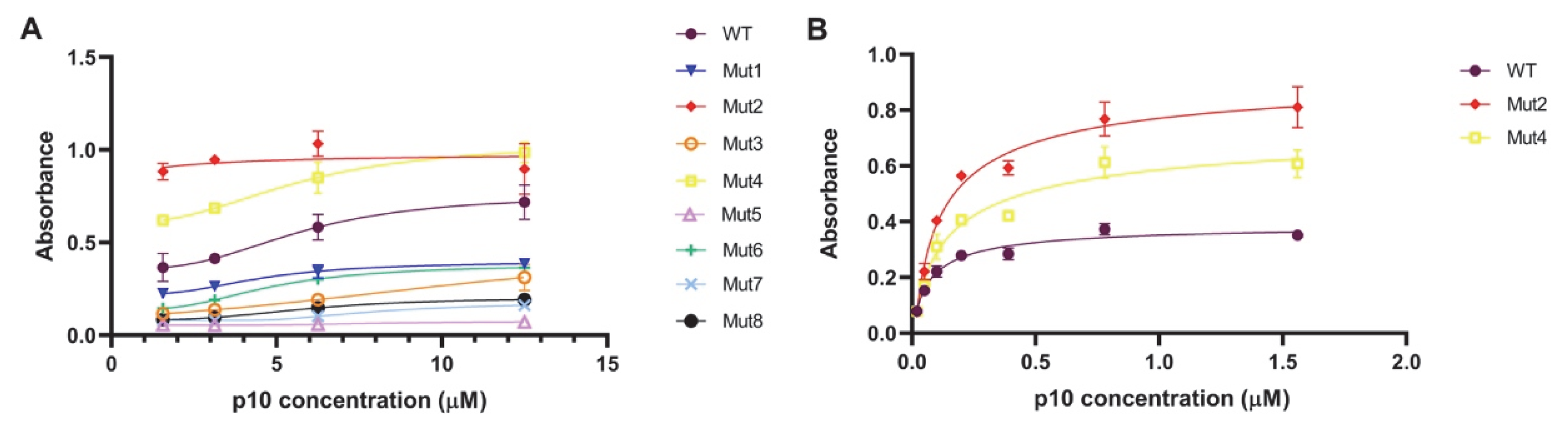
3.5. Experimental Evaluation of the Binding to DNA of the Mutated and Wild-Type p10
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Montgomery, R.E. On A Form of Swine Fever Occurring in British East Africa (Kenya Colony). J. Comp. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Penrith, M.-L.; Vosloo, W.; Jori, F.; Bastos, A.D.S. African swine fever virus eradication in Africa. Virus Res. 2013, 173, 228–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, L.K.; Sun, H.; Roberts, H. African Swine Fever. Antivir. Res. 2019, 165, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Barcos, L. International Agencies Provide Support to the Dominican Republic and Haiti to Prevent and Fight African Swine Fever (ASF). Available online: https://rr-americas.oie.int/en/news/int-ags-support-dominican-haiti-to-asf/ (accessed on 9 April 2022).
- Dixon, L.K.; Chapman, D.A.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Matamoros, T.; Guerra, M.; Andrés, G. A Proteomic Atlas of the African Swine Fever Virus Particle. J. Virol. 2018, 92, e01293-18. [Google Scholar] [CrossRef] [Green Version]
- Kollnberger, S.D.; Gutierrez-Castañeda, B.; Foster-Cuevas, M.; Corteyn, A.; Parkhouse, R.M.E. Identification of the principal serological immunodeterminants of African swine fever virus by screening a virus cDNA library with antibody. J. Gen. Virol. 2002, 83, 1331–1342. [Google Scholar] [CrossRef]
- Freije, J.M.P.; Salas, M.L.; Muñoz, M.; Viñuela, E.; López-Otín, C. Structure and expression inE. coli of the gene coding for protein p10 of African swine fever virus. Arch. Virol. 1993, 130, 93–107. [Google Scholar] [CrossRef]
- Nunes-Correia, I.; Rodríguez, J.M.; Eulálio, A.; Carvalho, A.L.; Citovsky, V.; Simões, S.; Faro, C.; Salas, M.L.; Pedroso de Lima, M.C. African Swine Fever Virus p10 Protein Exhibits Nuclear Import Capacity and Accumulates in the Nucleus during Viral Infection. Vet. Microbiol. 2008, 130, 47–59. [Google Scholar] [CrossRef] [Green Version]
- Yao, K.; Wu, Y.; Chen, Q.; Zhang, Z.; Chen, X.; Zhang, Y. The Arginine/Lysine-Rich Element within the DNA-Binding Domain Is Essential for Nuclear Localization and Function of the Intracellular Pathogen Resistance 1. PLoS ONE 2016, 11, e0162832. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. Nucleic Acids Force Field Based on Reference Quantum Chemical Calculations of Glycosidic Torsion Profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef] [PubMed]
- Zgarbová, M.; Šponer, J.; Otyepka, M.; Cheatham, T.E., III; Galindo-Murillo, R.; Jurečka, P. Refinement of the Sugar–Phosphate Backbone Torsion Beta for AMBER Force Fields Improves the Description of Z- and B-DNA. J. Chem. Theory Comput. 2015, 11, 5723–5736. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Grant, R.A.; Rould, M.A.; Klemm, J.D.; Pabo, C.O. Exploring the Role of Glutamine 50 in the Homeodomain−DNA Interface: Crystal Structure of Engrailed (Gln50 → Ala) Complex at 2.0 Å. Biochemistry 2000, 39, 8187–8192. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Neria, E.; Fischer, S.; Karplus, M. Simulation of activation free energies in molecular systems. J. Chem. Phys. 1996, 105, 1902–1921. [Google Scholar] [CrossRef]
- Luenberger, D.G.; Ye, Y. Linear and Nonlinear Programming; Springer International Publishing: Cham, Switzerland, 2015; ISBN 9783319188416. [Google Scholar]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Páll, S.; Hess, B. A flexible algorithm for calculating pair interactions on SIMD architectures. Comput. Phys. Commun. 2013, 184, 2641–2650. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. P-Lincs: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2007, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Kollman, P.A. Settle: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nosé, S.; Klein, M. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Williams, T.; Kelley, C. Gnuplot: An Interactive Plotting Program, Version 5.4 2013. Available online: http://www.gnuplot.info (accessed on 1 January 2021).
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 2.4 2020; Schrödinger, LLC: New York, NY, USA.
- Campos, S.R.R.; Baptista, A.M. Conformational Analysis in a Multidimensional Energy Landscape: Study of an Arginylglutamate Repeat. J. Phys. Chem. B 2009, 113, 15989–16001. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Rizzo, R.C.; Aynechi, T.; Case, D.A.; Kuntz, I.D. Estimation of Absolute Free Energies of Hydration Using Continuum Methods: Accuracy of Partial Charge Models and Optimization of Nonpolar Contributions. J. Chem. Theory Comput. 2006, 2, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Sitkoff, D.; Sharp, K.A.; Honig, B. Accurate Calculation of Hydration Free Energies Using Macroscopic Solvent Models. J. Phys. Chem. 1994, 98, 1978–1988. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Šali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Fiser, A.; Do, R.K.; SSali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T.J.B. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.; Iyer, L. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef]
- Reis, A.L.; Parkhouse, M.; Penedos, A.; Martins, C.; Leitão, A. Systematic analysis of longitudinal serological responses of pigs infected experimentally with African swine fever virus. J. Gen. Virol. 2007, 88, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Medina, E.; Vuono, E.A.; Pruitt, S.; Rai, A.; Espinoza, N.; Valladares, A.; Silva, E.; Velazquez-Salinas, L.; Borca, M.V.; Gladue, D.P. Deletion of African Swine Fever Virus Histone-like Protein, A104R from the Georgia Isolate Drastically Reduces Virus Virulence in Domestic Pigs. Viruses 2022, 14, 1112. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference System | Amino Acid Sequence |
|---|---|
| WT | SSMHSGMLYKDMVNIARSRGIPIYQNGSRLTKSELEKKIKRSK |
| M1—Y59R | SSMHSGMLYKDMVNIARSRGIPIRQNGSRLTKSELEKKIKRSK |
| M2—N60R | SSMHSGMLYKDMVNIARSRGIPIYRNGSRLTKSELEKKIKRSK |
| M3—R76D | SSMHSGMLYKDMVNIARSRGIPIYQNGSRLTKSELEKKIKDSK |
| M4—S77R | SSMHSGMLYKDMVNIARSRGIPIYQNGSRLTKSELEKKIKRRK |
| M5—V48D,L70D,I74D | SSMHSGMLYKDMDNIARSRGIPIYQNGSRLTKSEDEKKDKRSK |
| M6—K72A,R76A,K78A | SSMHSGMLYKDMVNIARSRGIPIYQNGSRLTKSELEAKIKASA |
| M7—T66A,K67A,S68A | SSMHSGMLYKDMVNIARSRGIPIYQNGSRLAAAELEKKIKRSK |
| M8—S36D,S37D,S40D | DDMHDGMLYKDMVNIARSRGIPIYQNGSRLTKSELEKKIKRSK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Istrate, C.; Marques, J.; Bule, P.; Correia, S.; Aires-da-Silva, F.; Duarte, M.; Reis, A.L.; Machuqueiro, M.; Leitão, A.; Victor, B.L. In Silico Characterization of African Swine Fever Virus Nucleoprotein p10 Interaction with DNA. Viruses 2022, 14, 2348. https://doi.org/10.3390/v14112348
Istrate C, Marques J, Bule P, Correia S, Aires-da-Silva F, Duarte M, Reis AL, Machuqueiro M, Leitão A, Victor BL. In Silico Characterization of African Swine Fever Virus Nucleoprotein p10 Interaction with DNA. Viruses. 2022; 14(11):2348. https://doi.org/10.3390/v14112348
Chicago/Turabian StyleIstrate, Claudia, Jéssica Marques, Pedro Bule, Sílvia Correia, Frederico Aires-da-Silva, Marlene Duarte, Ana Luísa Reis, Miguel Machuqueiro, Alexandre Leitão, and Bruno L. Victor. 2022. "In Silico Characterization of African Swine Fever Virus Nucleoprotein p10 Interaction with DNA" Viruses 14, no. 11: 2348. https://doi.org/10.3390/v14112348