
Phylogenetic Characterization of HIV-1 Sub-Subtype A1 in Karachi, Pakistan
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. HIV-1 gag Gene Amplification and Sequencing
2.3. Subtyping Determination
2.4. Cluster Analysis
2.5. Time-Scaled Phylogenetic Reconstruction
2.6. Signature Variation(s) Analysis and Charge Distribution
2.7. Renaissance Counting
2.8. Epitope Mapping
2.9. Protein Structure Modeling
3. Results
3.1. Subtyping and Cluster Analysis of HIV-1 Sub-Subtype A1 in Pakistan
3.2. Date of Origin and Evolutionary Dynamics of HIV-1 Sub-Subtype A1 in Pakistan
3.3. Analysis of Molecular Properties between Pakistani and Kenyan HIV-1 Sub-Subtype A1 Sequences
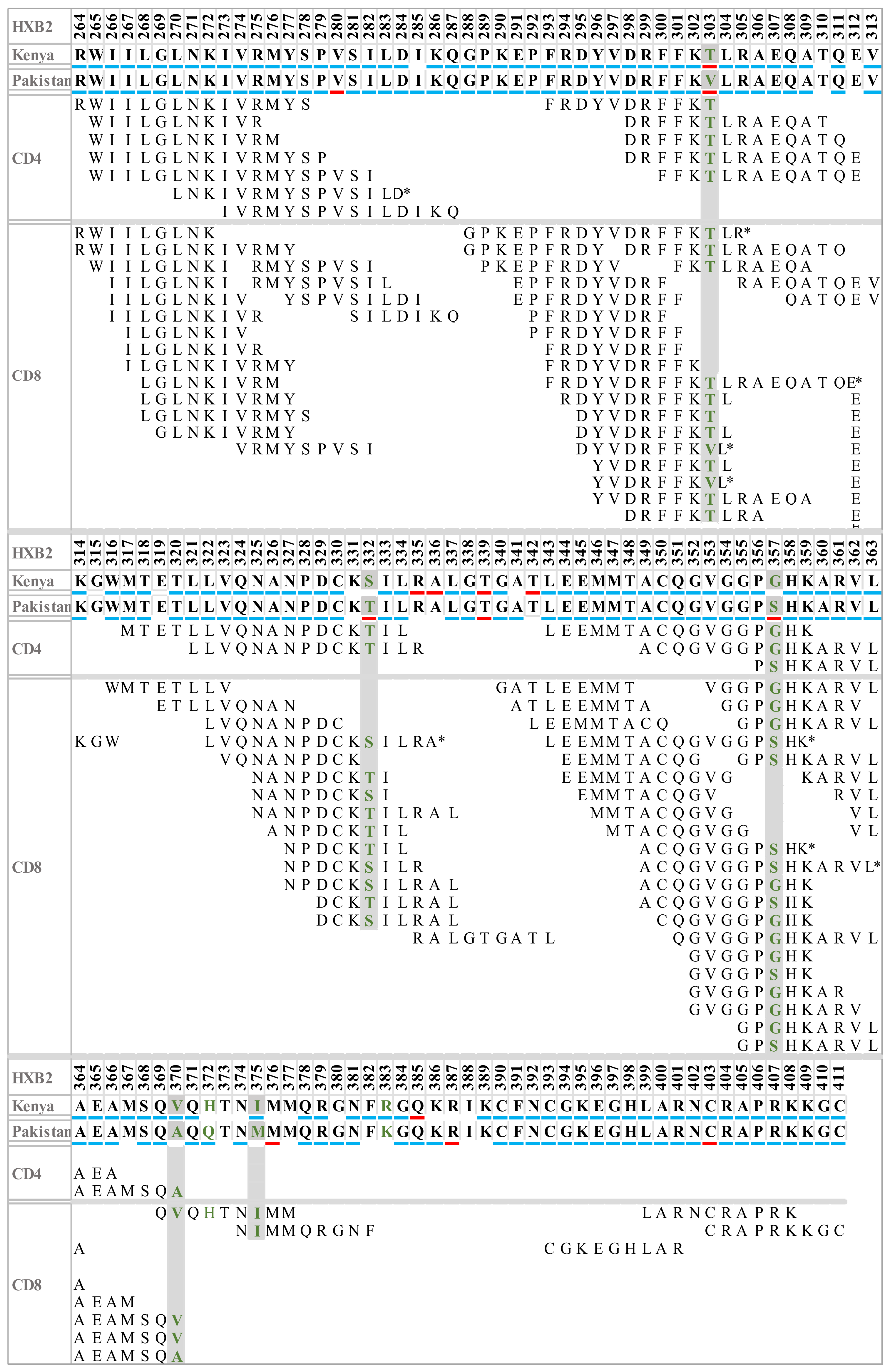
3.4. Gag Sites under Selection in Pakistani and Kenyan HIV-1 Sub-Subtype A1 Strains
3.5. Sequence Variation in HIV-1 Gag T-Cell Epitopes
3.6. Sequence Variation in HLA Binding Motifs and Epitopes
3.7. Structural Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- UNAIDS. UNAIDS Data 2020. 2020. Available online: https://www.unaids.org/en/resources/documents/2020/unaids-data (accessed on 1 January 2022).
- Andrews, S.M.; Zhang, Y.; Dong, T.; Rowland-Jones, S.L.; Gupta, S.; Esbjörnsson, J. Analysis of HIV-1 envelope evolution suggests antibody-mediated selection of common epitopes among Chinese former plasma donors from a narrow-source outbreak. Sci. Rep. 2018, 8, 5743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tariq, U.; Iftikhar, A.; Zahid, D.; Sultan, F.; Mahmood, S.F.; Naeem, S.; Ali, S.; Abidi, S.H. The emergence of an unassigned complex recombinant form in a Pakistani HIV-infected individual. Arch. Virol. 2020, 165, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Ciccozzi, M.; Parolin, C.; Borsetti, A. Molecular Epidemiology of HIV-1 in African Countries: A Comprehensive Overview. Pathogens 2020, 9, 1072. [Google Scholar] [CrossRef]
- Faria, N.R.; Vidal, N.; Lourenco, J.; Raghwani, J.; Sigaloff, K.C.E.; Tatem, A.J.; van de Vijver, D.A.M.; Pineda-Pena, A.C.; Rose, R.; Wallis, C.L.; et al. Distinct rates and patterns of spread of the major HIV-1 subtypes in Central and East Africa. PLoS Pathog. 2019, 15, e1007976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araujo, P.M.M.; Carvalho, A.; Pingarilho, M.; BEST-HOPE study group; Abecasis, A.B.; Osorio, N.S. Characterization of a large cluster of HIV-1 A1 infections detected in Portugal and connected to several Western European countries. Sci. Rep. 2019, 9, 7223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; Bozzi, G.; Franzetti, M.; Binda, F.; Simonetti, F.R.; De Luca, A.; Micheli, V.; Meraviglia, P.; Bagnarelli, P.; Di Biagio, A.; et al. HIV-1 A1 Subtype Epidemic in Italy Originated from Africa and Eastern Europe and Shows a High Frequency of Transmission Chains Involving Intravenous Drug Users. PLoS ONE 2016, 11, e0146097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloni, S.T.; Sankale, J.L.; Hamel, D.J.; Eisen, G.; Gueye-Ndiaye, A.; Mboup, S.; Kanki, P.J. Molecular epidemiology of human immunodeficiency virus type 1 sub-subtype A3 in Senegal from 1988 to 2001. J. Virol. 2004, 78, 12455–12461. [Google Scholar] [CrossRef] [Green Version]
- Palm, A.A.; Esbjornsson, J.; Mansson, F.; Kvist, A.; Isberg, P.E.; Biague, A.; da Silva, Z.J.; Jansson, M.; Norrgren, H.; Medstrand, P. Faster progression to AIDS and AIDS-related death among seroincident individuals infected with recombinant HIV-1 A3/CRF02_AG compared with sub-subtype A3. J. Infect. Dis. 2014, 209, 721–728. [Google Scholar] [CrossRef]
- Esbjornsson, J.; Mild, M.; Mansson, F.; Norrgren, H.; Medstrand, P. HIV-1 molecular epidemiology in Guinea-Bissau, West Africa: Origin, demography and migrations. PLoS ONE 2011, 6, e17025. [Google Scholar] [CrossRef] [Green Version]
- Junqueira, D.M.; Almeida, S.E. HIV-1 subtype B: Traces of a pandemic. Virology 2016, 495, 173–184. [Google Scholar] [CrossRef]
- Soares, M.A.; De Oliveira, T.; Brindeiro, R.M.; Diaz, R.S.; Sabino, E.C.; Brigido, L.; Pires, I.L.; Morgado, M.G.; Dantas, M.C.; Barreira, D.; et al. A specific subtype C of human immunodeficiency virus type 1 circulates in Brazil. AIDS 2003, 17, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Cotton, L.A.; Kuang, X.T.; Le, A.Q.; Carlson, J.M.; Chan, B.; Chopera, D.R.; Brumme, C.J.; Markle, T.J.; Martin, E.; Shahid, A.; et al. Genotypic and functional impact of HIV-1 adaptation to its host population during the North American epidemic. PLoS Genet. 2014, 10, e1004295. [Google Scholar] [CrossRef] [PubMed]
- Boquett, J.A.; Bisso-Machado, R.; Zagonel-Oliveira, M.; Schüler-Faccini, L.; Fagundes, N.J. HLA diversity in Brazil. HLA 2020, 95, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Markov, P.V.; Pybus, O.G. Evolution and Diversity of the Human Leukocyte Antigen (HLA). Evol. Med. Public Health 2015, 2015, 1. [Google Scholar] [CrossRef] [Green Version]
- Abidi, S.H.; Shahid, A.; Lakhani, L.S.; Khanani, M.R.; Ojwang, P.; Okinda, N.; Shah, R.; Abbas, F.; Rowland-Jones, S.; Ali, S. Population-specific evolution of HIV Gag epitopes in genetically diverged patients. Infect. Genet. Evol. 2013, 16, 78–86. [Google Scholar] [CrossRef]
- UNAIDS. UNAIDS Data (Global and regional data). 2019. Available online: https://www.unaids.org/en/resources/documents/2019/2019-UNAIDS-data (accessed on 1 January 2022).
- Mubarak, N.; Hussain, I.; Raja, S.A.; Khan, T.M.; Zin, C.S. HIV outbreak of Ratodero, Pakistan requires urgent concrete measures to avoid future outbreaks. J. Pak. Med. Assoc. 2020, 70, 1475–1476. [Google Scholar] [CrossRef]
- Tariq, U.; Parveen, A.; Akhtar, F.; Mahmood, F.; Ali, S.; Abidi, S.H. Emergence of Circulating Recombinant Form 56_cpx in Pakistan. AIDS Res. Hum. Retrovir. 2018, 34, 1002–1004. [Google Scholar] [CrossRef]
- Tariq, U.; Mahmood, F.; Naeem, S.; Ali, S.; Abidi, S.H. Emergence of HIV-1 Unique DG Recombinant Form in Pakistan. AIDS Res. Hum. Retrovir. 2020, 36, 248–250. [Google Scholar] [CrossRef]
- Chen, Y.; Hora, B.; DeMarco, T.; Shah, S.A.; Ahmed, M.; Sanchez, A.M.; Su, C.; Carter, M.; Stone, M.; Hasan, R.; et al. Fast Dissemination of New HIV-1 CRF02/A1 Recombinants in Pakistan. PLoS ONE 2016, 11, e0167839. [Google Scholar] [CrossRef] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2006, 34, D16–D20. [Google Scholar] [CrossRef]
- Esbjörnsson, J.; Mild, M.; Audelin, A.; Fonager, J.; Skar, H.; Bruun Jørgensen, L.; Liitsola, K.; Björkman, P.; Bratt, G.; Gisslén, M.; et al. HIV-1 transmission between MSM and heterosexuals, and increasing proportions of circulating recombinant forms in the Nordic Countries. Virus Evol. 2016, 2, vew010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, V.; Baele, G. Bayesian estimation of past population dynamics in BEAST 1.10 using the Skygrid coalescent model. Mol. Biol. Evol. 2019, 36, 2620–2628. [Google Scholar] [CrossRef] [Green Version]
- Dellicour, S.; Gill, M.S.; Faria, N.R.; Rambaut, A.; Pybus, O.G.; Suchard, M.A.; Lemey, P. Relax, keep walking-a practical guide to continuous phylogeographic inference with BEAST. Mol. Biol. Evol. 2021, 38, 3486–3493. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4; Institute of Evolutionary Biology, University of Edinburgh: Edinburgh, UK, 2018; Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 January 2022).
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [Green Version]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; De Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E. ExPASy: SIB bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Korber, B.; Myers, G. Signature pattern analysis: A method for assessing viral sequence relatedness. AIDS Res. Hum. Retrovir. 1992, 8, 1549–1560. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Minin, V.N.; Bielejec Filip, K.P.; Sergei, L.; Suchard, M.A. A counting renaissance: Combining stochastic mapping and empirical Bayes to quickly detect amino acid sites under positive selection. Bioinformatics 2012, 28, 3248–3256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammensee, H.G.; Bachmann, J.; Stevanovic, S. MHC Ligands and Peptide Motifs; Springer: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.N.; Tan, K.P.; Madhusudhan, M.S. CLICK--topology-independent comparison of biomolecular 3D structures. Nucleic Acids Res. 2011, 39, W24–W28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biovia, D.S. Discovery Studio Visualizer. V12.2.0.16349; S.-Diego Dassault Systèmes: San Diego, CA, USA, 2016. [Google Scholar]
- Abidi, S.H.; Kalish, M.L.; Abbas, F.; Rowland-Jones, S.; Ali, S. HIV-1 subtype A gag variability and epitope evolution. PLoS ONE 2014, 9, e93415. [Google Scholar] [CrossRef]
- Khanani, M.R.; Somani, M.; Rehmani, S.S.; Veras, N.M.; Salemi, M.; Ali, S.H. The spread of HIV in Pakistan: Bridging of the epidemic between populations. PLoS ONE 2011, 6, e22449. [Google Scholar] [CrossRef] [Green Version]
- Nduva, G.M.; Hassan, A.S.; Nazziwa, J.; Graham, S.M.; Esbjornsson, J.; Sanders, E.J. HIV-1 Transmission Patterns Within and Between Risk Groups in Coastal Kenya. Sci. Rep. 2020, 10, 6775. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.S.; Esbjornsson, J.; Wahome, E.; Thiong’o, A.; Makau, G.N.; Price, M.A.; Sanders, E.J. HIV-1 subtype diversity, transmission networks and transmitted drug resistance amongst acute and early infected MSM populations from Coastal Kenya. PLoS ONE 2018, 13, e0206177. [Google Scholar] [CrossRef] [Green Version]
- Patino-Galindo, J.A.; Gonzalez-Candelas, F. The substitution rate of HIV-1 subtypes: A genomic approach. Virus Evol. 2017, 3, vex029. [Google Scholar] [CrossRef]
- Arimide, D.A.; Abebe, A.; Kebede, Y.; Adugna, F.; Tilahun, T.; Kassa, D.; Assefa, Y.; Balcha, T.T.; Bjorkman, P.; Medstrand, P. HIV-genetic diversity and drug resistance transmission clusters in Gondar, Northern Ethiopia, 2003-2013. PLoS ONE 2018, 13, e0205446. [Google Scholar] [CrossRef]
- Arif, F. HIV crisis in Sindh, Pakistan: The tip of the iceberg. Lancet Infect. Dis. 2019, 19, 695–696. [Google Scholar] [CrossRef] [Green Version]
- Usami, Y.; Popov, S.; Gottlinger, H.G. The Nef-like effect of murine leukemia virus glycosylated gag on HIV-1 infectivity is mediated by its cytoplasmic domain and depends on the AP-2 adaptor complex. J. Virol. 2014, 88, 3443–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sokalingam, S.; Raghunathan, G.; Soundrarajan, N.; Lee, S.G. A study on the effect of surface lysine to arginine mutagenesis on protein stability and structure using green fluorescent protein. PLoS ONE 2012, 7, e40410. [Google Scholar] [CrossRef] [Green Version]
- Henderson, L.E.; Sowder, R.C.; Copeland, T.D.; Oroszlan, S.; Benveniste, R.E. Gag precursors of HIV and SIV are cleaved into six proteins found in the mature virions. J. Med. Primatol. 1990, 19, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Forshey, B.M.; von Schwedler, U.; Sundquist, W.I.; Aiken, C. Formation of a human immunodeficiency virus type 1 core of optimal stability is crucial for viral replication. J. Virol. 2002, 76, 5667–5677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teto, G.; Tagny, C.T.; Mbanya, D.; Fonsah, J.Y.; Fokam, J.; Nchindap, E.; Kenmogne, L.; Njamnshi, A.K.; Kanmogne, G.D. Gag P2/NC and pol genetic diversity, polymorphism, and drug resistance mutations in HIV-1 CRF02_AG- and non-CRF02_AG-infected patients in Yaounde, Cameroon. Sci. Rep. 2017, 7, 14136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, S.M. HIV CTL escape: At what cost? Retrovirology 2004, 1, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honeyborne, I.; Codoner, F.M.; Leslie, A.; Tudor-Williams, G.; Luzzi, G.; Ndung’u, T.; Walker, B.D.; Goulder, P.J.; Prado, J.G. HLA-Cw*03-restricted CD8+ T-cell responses targeting the HIV-1 gag major homology region drive virus immune escape and fitness constraints compensated for by intracodon variation. J. Virol. 2010, 84, 11279–11288. [Google Scholar] [CrossRef] [Green Version]
- Sakai, K.; Chikata, T.; Brumme, Z.L.; Brumme, C.J.; Gatanaga, H.; Oka, S.; Takiguchi, M. Lack of a significant impact of Gag-Protease-mediated HIV-1 replication capacity on clinical parameters in treatment-naive Japanese individuals. Retrovirology 2015, 12, 98. [Google Scholar] [CrossRef] [Green Version]
- Nowicka-Sans, B.; Protack, T.; Lin, Z.; Li, Z.; Zhang, S.; Sun, Y.; Samanta, H.; Terry, B.; Liu, Z.; Chen, Y.; et al. Identification and Characterization of BMS-955176, a Second-Generation HIV-1 Maturation Inhibitor with Improved Potency, Antiviral Spectrum, and Gag Polymorphic Coverage. Antimicrob. Agents Chemother. 2016, 60, 3956–3969. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category/Variable | Total No. (%) |
|---|---|
| Age (years) | |
| 15–24 | 11(18) |
| 25–64 | 50 (82) |
| Sex | |
| Male | 56 (92) |
| Female | 5 (8.12%) |
| Marital Status | |
| Married | 31 (51) |
| Single | 25 (41) |
| Not declared | 5 (8) |
| ART History | |
| Experienced | 39 (64) |
| Naïve | 19 (31) |
| Not declared | 3 (5) |
| Risk group | |
| PWID | 26 (43) |
| MSM | 8 (13) |
| HET | 3 (5) |
| SP | 5 (2) |
| NA | 2 (3) |
| Year of Diagnosis | |
| 2000–2005 | 8 (13) |
| 2006–2010 | 5 (8) |
| 2011–2015 | 37 (61) |
| Unknown | 11 (18) |
| Date of sampling | 2015 |
| Pakistan | V | T | S | A | Q | M | K |
|---|---|---|---|---|---|---|---|
| Frequency in Pakistan | 0.854 | 0.911 | 0.797 | 0.78 | 0.862 | 0.732 | 0.992 |
| Frequency in Kenya | 0.102 | 0.102 | 0.122 | 0.061 | 0.184 | 0 | 0.163 |
| Kenya | T | S | G | V | H | I | R |
| Frequency in Pakistan | 0.049 | 0.008 | 0.195 | 0.211 | 0.122 | 0.244 | 0.008 |
| Frequency in Kenya | 0.673 | 0.898 | 0.878 | 0.898 | 0.653 | 0.959 | 0.837 |
| HXB2 Position | 303 | 332 | 357 | 370 | 372 | 375 | 383 |
| Pakistan | Restricted HLA | HXB2 Position | Epitope | Anchors | Kenya | Restricted HLA | HXB2 Position | Epitope | Anchors | |
| B*5103, Cw*0601, Cw*0602 | 295-303 | DYVDRFFKV | ........V | DRB1*0301 or DRB3*0201 | 300-303 | FFKT | F..T | |||
| DPA1*0102, DPB1*0201 | 296-303 | YVDRFFKV | Y...F..V | B*7801 | 325-333 | NANPDCKSI | .A.....S. | |||
| DRB1*1501 | 297-303 | VDRFFKV | V..F..V | B*7801 | 356-364 | PGHKARVLA | .G....... | |||
| DPA1*0201/DPB1*0401 | 297-306 | VDRFFKVLRA | V.....V..A | A*0201, A*0202, A*0214 | 362-370 | VLAEAMSQV | .L......V | |||
| DRB1*0901 or DRB4*0101 | 300-303 | FFKV | F..V | B*5103, Cw*0601, Cw*0602 | ........V | |||||
| DRB1*0401 or DRB4*0101 | 300-308 | FFKVLRAEQ | F..V.RA.Q | DRB1*1501 or DRB5*0101 | 363-372 | LAEAMSQVQH | L........H | |||
| A*0206, B*3501 | 302-310 | KVLRAEQAT | .V....... | B*1517 | 367-375 | MSQVQHTNI | .S......I | |||
| DRB1*0401 | 303-311 | VLRAEQATQ | V.......Q | B*3801, B*5101, B*5103, Cw*0601, Cw*0602 | ........I | |||||
| DRB1*0401 or DRB4*0101 | V..A.QA.Q | A*0206 | 369-377 | QVQHTNIMM | .V....... | |||||
| DQA1*0102, DQB1*0602 | 352-360 | GVGGPSHKA | .....S..A | A*3001 | .V......M | |||||
| DQA1*0301, DQB1*0302 | 357-365 | SHKARVLAE | S.......E | DRB1*0301 or DRB3*0201 | 370-373 | VQHT | V..T | |||
| B*7801 | 363-371 | LAEAMSQAQ | .A.....A. | DRB1*0401 | 370-378 | VQHTNIMMQ | V.......Q | |||
| DQA1*0301, DQB1*0301 | 365-370 | EAMSQA | E..S.A | DRB1*1501 or DRB5*0101 | 370-379 | VQHTNIMMQR | V........R | |||
| A*3004, B*1502 | 367-375 | MSQAQQTNM | ........M | DQA1*0301/DQB1*0301 | 371-375 | QHTNI | ..T.I | |||
| B*5101, Cw*0303, Cw*0305-0306, Cw*0308-0309, Cw*0801-Cw*0806, Cw*1502, Cw*1503, Cw*1506, Cw*1507 | 369-377 | QAQQTNMMM | .A......M | A*2601, A*2602, A*2603 | 374-382 | NIMMQRGNF | .I......F | |||
| B*7801 | .A....... | DRB1*0301 or DRB3*0201 | 375-378 | IMMQ | I..Q | |||||
| A*0206 | 371-379 | QQTNMMMQR | .Q....... | DRB1*0801 | 375-379 | IMMQR | I...R | |||
| B*1512 | 374-382 | NMMMQRGNF | .M......F | A*3101, A*3303 | 375-383 | IMMQRGNFR | ........R | |||
| B*2703 | .M....... | B*2703, DPB1*0301 | 382-390 | FRGQKRIKC | .R....... | |||||
| DRB5*0101 | 375-383 | MMMQRGNFK | M..Q....K | Signature Amino Amino Acid: Bold and underline | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tariq, U.; Nazziwa, J.; Sasinovich, S.; Shah, S.A.; Naeem, S.; Abidi, S.H.; Esbjörnsson, J. Phylogenetic Characterization of HIV-1 Sub-Subtype A1 in Karachi, Pakistan. Viruses 2022, 14, 2307. https://doi.org/10.3390/v14102307
Tariq U, Nazziwa J, Sasinovich S, Shah SA, Naeem S, Abidi SH, Esbjörnsson J. Phylogenetic Characterization of HIV-1 Sub-Subtype A1 in Karachi, Pakistan. Viruses. 2022; 14(10):2307. https://doi.org/10.3390/v14102307
Chicago/Turabian StyleTariq, Uroosa, Jamirah Nazziwa, Sviataslau Sasinovich, Sharaf Ali Shah, Sadaf Naeem, Syed Hani Abidi, and Joakim Esbjörnsson. 2022. "Phylogenetic Characterization of HIV-1 Sub-Subtype A1 in Karachi, Pakistan" Viruses 14, no. 10: 2307. https://doi.org/10.3390/v14102307