

Amino Acid Substitution within Seven-Octapeptide Repeat Insertions in the Prion Protein Gene Associated with Short-Term Course
Abstract
:1. Introduction
2. Methods
2.1. Study Design
2.2. Clinical and Laboratory Data
2.3. Laboratory Methods
2.3.1. Genetic Analyses
2.3.2. CSF 14-3-3 Protein Level Test and RT-QuIC Assay
2.3.3. CSF Tau Level Test
2.3.4. Electroencephalogram
2.3.5. Magnetic Resonance Imaging
2.3.6. Positron Emission Tomography
2.3.7. Statistical Analysis
3. Results
3.1. Clinical Presentation of Each Patient
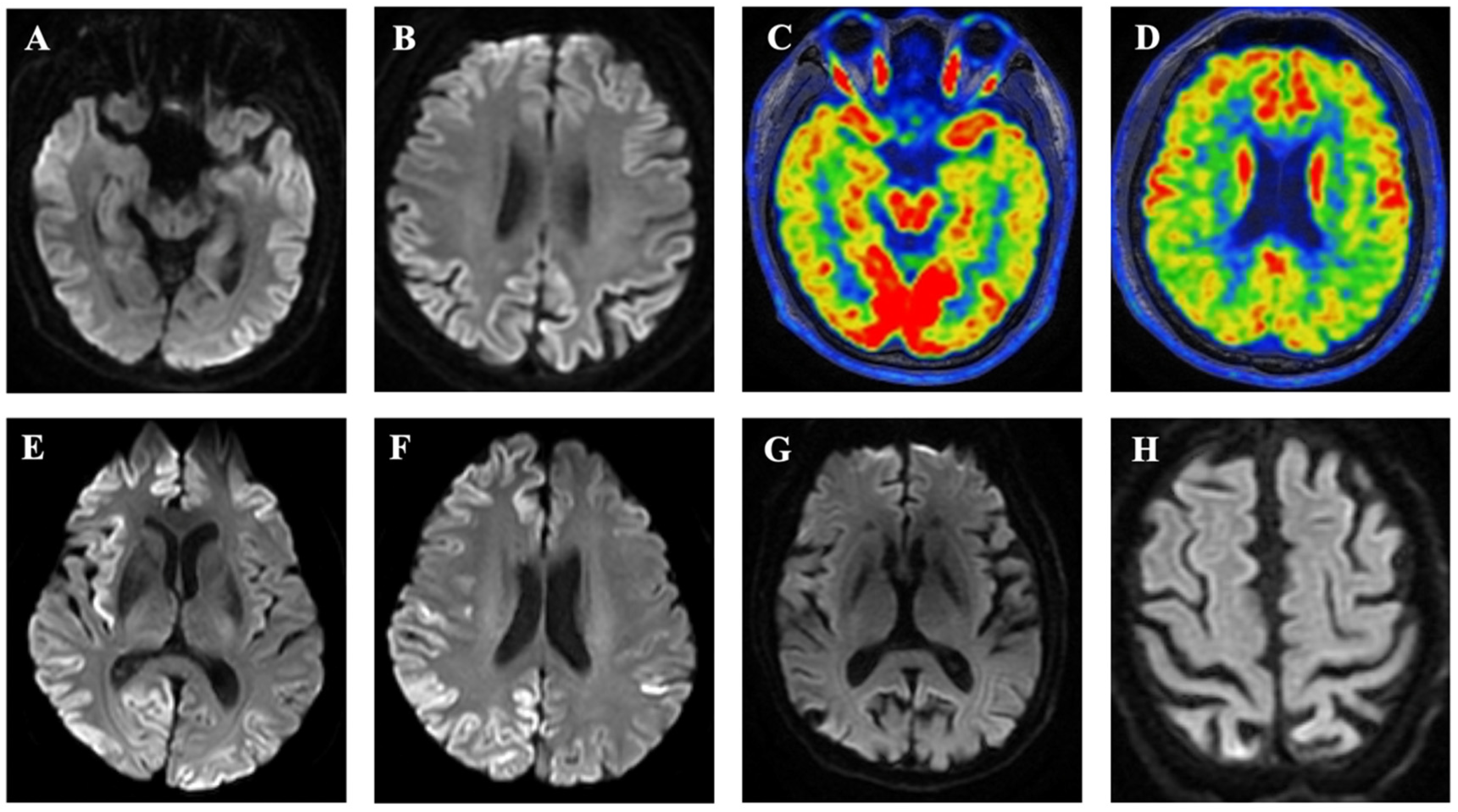
3.2. Subject IV-1
3.3. Subject III-4
3.4. Subject III-7
3.5. Subject III-2
3.6. Subjects I-1 and II-2
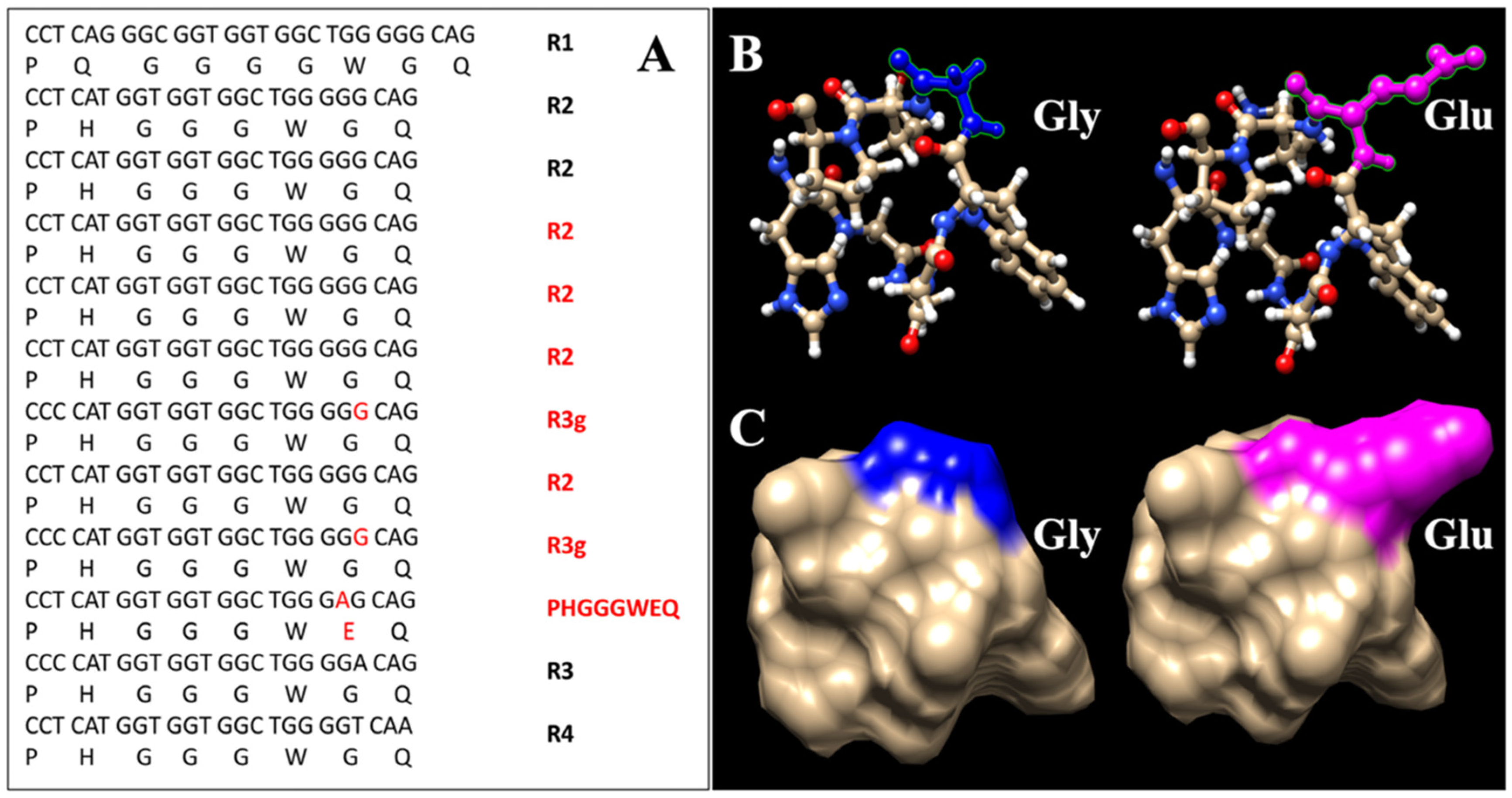
3.7. Genetic Analysis
3.8. Analysis of Combined Data
3.9. Comparison of Clinical and Ancillary Features between Cases with Short-Term and Long-Term Course
3.10. The Effect of Codon 129 Genotype and AAS within OPRIs on Disease Course
3.11. The Effect of the Neuropathological Changes on the Course of the Disease
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kim, M.-O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP Prion Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033134. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Chesebro, B. Abnormal properties of prion protein with insertional mutations in different cell types. J. Biol. Chem. 1998, 273, 11980–11985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, R.A.; Herzog, C.; Errett, J.; Kocisko, D.A.; Arnold, K.M.; Hayes, S.F.; Priola, S.A. Octapeptide repeat insertions increase the rate of protease-resistant prion protein formation. Protein Sci. 2006, 15, 609–619. [Google Scholar] [CrossRef]
- Hodak, M.; Chisnell, R.; Lu, W.; Bernholc, J. Functional implications of multistage copper binding to the prion protein. Proc. Natl. Acad. Sci. USA 2009, 106, 11576–11581. [Google Scholar] [CrossRef] [Green Version]
- Mead, S.; Poulter, M.; Beck, J.; Webb, T.E.F.; Campbell, T.A.; Linehan, J.M.; Desbruslais, M.; Joiner, S.; Wadsworth, J.D.F.; King, A.; et al. Inherited prion disease with six octapeptide repeat insertional mutation--molecular analysis of phenotypic heterogeneity. Brain 2006, 129 Pt 9, 2297–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takada, L.; Kim, M.-O.; Cleveland, R.W.; Wong, K.; Forner, S.A.; Gala, I.I.; Fong, J.C.; Geschwind, M.D. Genetic prion disease: Experience of a rapidly progressive dementia center in the United States and a review of the literature. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 36–69. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Voet, W.; Head, M.W.; Parchi, P.; Yull, H.; Verrips, A.; Wesseling, P.; Meulstee, J.; Baas, F.; Van Gool, W.A.; et al. A novel seven-octapeptide repeat insertion in the prion protein gene (PRNP) in a Dutch pedigree with Gerstmann-Sträussler-Scheinker disease phenotype: Comparison with similar cases from the literature. Acta Neuropathol. 2011, 121, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, L.G.; Brown, P.; McCombie, W.R.; Goldgaber, D.; Swergold, G.D.; Wills, P.R.; Cervenakova, L.; Baron, H.; Gibbs, C.J.; Gajdusek, D.C. Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc. Natl. Acad. Sci. USA 1991, 88, 10926–10930. [Google Scholar] [CrossRef] [Green Version]
- Tateishi, J. Recent advances in the research of Creutzfeldt-Jakob disease (CJD) and Gerstmann-Strüssler syndrome (GSS). Rinsho Shinkeigaku 1991, 31, 1306–1308. [Google Scholar]
- Brown, P.; Goldfarb, L.G.; McCombie, W.R.; Nieto, A.; Squillacote, D.; Sheremata, W.; Little, B.W.; Godec, M.S.; Gibbs, C.J.; Gajdusek, D.C. Atypical Creutzfeldt-Jakob disease in an American family with an insert mutation in the PRNP amyloid precursor gene. Neurology 1992, 42, 422–427. [Google Scholar] [CrossRef]
- Dermaut, B.; Cruts, M.; Backhovens, H.; Lübcke, U.; Van Everbroeck, B.; Sciot, R.; Dom, R.; Martin, J.-J.; Van Broeckhoven, C.; Cras, P. Familial Creutzfeldt-Jakob disease in a patient carrying both a presenilin 1 missense substitution and a prion protein gene insertion. J. Neurol. 2000, 247, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Lewis, V.; Collins, S.; Hill, A.; Boyd, A.; McLean, C.A.; Smith, M.; Masters, C.L. Novel prion protein insert mutation associated with prolonged neurodegenerative illness. Neurology 2003, 60, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Cannella, M.; Martino, T.; Simonelli, M.; Ciammola, A.; Gradini, R.; Ciarmiello, A.; Gianfrancesco, F.; Squitieri, F. De novo seven extra repeat expanded mutation in the PRNP gene in an Italian patient with early onset dementia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1411–1413. [Google Scholar] [CrossRef] [PubMed]
- Mauro, C.; Giaccone, G.; Piscosquito, G.; Lavorgna, A.; Nigro, M.; Di Fede, G.; Leonardi, A.; Coppola, C.; Formisano, S.; Tagliavini, F.; et al. A novel insertional mutation in the prion protein gene: Clinical and bio-molecular findings. J. Neurol. Neurosurg. Psychiatry 2008, 79, 1395–1398. [Google Scholar] [CrossRef]
- Wang, X.F.; Guo, Y.J.; Zhang, B.Y.; Zhao, W.Q.; Gao, J.M.; Wan, Y.Z.; Li, F.; Han, J.; Wang, D.X.; Dong, X.P. Creutzfeldt-Jakob disease in a Chinese patient with a novel seven extra-repeat insertion in PRNP. J. Neurol. Neurosurg. Psychiatry 2007, 78, 201–203. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.-J.; Wang, X.-F.; Han, J.; Zhang, B.-Y.; Shi, Q.; Wan, Y.-Z.; Gao, C.; Dong, X.-P.; Zhao, W.-Q.; Li, J.-M.; et al. A patient with Creutzfeldt-Jakob disease with an insertion of 7 octa-repeats in the PRNP gene: Molecular characteristics and clinical features. Am. J. Med. Sci. 2008, 336, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Cali, I.; Cracco, L.; Saracino, D.; Occhipinti, R.; Coppola, C.; Appleby, B.S.; Puoti, G. Case Report: Histopathology and Prion Protein Molecular Properties in Inherited Prion Disease With a De Novo Seven-Octapeptide Repeat Insertion. Front. Cell Neurosci. 2020, 14, 150. [Google Scholar] [CrossRef]
- Capellari, S.; Strammiello, R.; Saverioni, D.; Kretzschmar, H.; Parchi, P. Genetic Creutzfeldt-Jakob disease and fatal familial insomnia: Insights into phenotypic variability and disease pathogenesis. Acta Neuropathol. 2011, 121, 21–37. [Google Scholar] [CrossRef]
- Liu, L.; Cui, B.; Chu, M.; Cui, Y.; Jing, D.; Li, D.; Xie, K.; Kong, Y.; Xia, T.; Wang, C.; et al. The Frequency of Genetic Mutations Associated With Behavioral Variant Frontotemporal Dementia in Chinese Han Patients. Front. Aging Neurosci. 2021, 13, 699836. [Google Scholar] [CrossRef]
- Gao, C.; Shi, Q.; Tian, C.; Chen, C.; Han, J.; Zhou, W.; Zhang, B.-Y.; Jiang, H.-Y.; Zhang, J.; Dong, X.-P. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: Surveillance data from 2006 to 2010. PLoS ONE 2011, 6, e24231. [Google Scholar] [CrossRef] [Green Version]
- Xiao, K.; Shi, Q.; Zhou, W.; Zhang, B.Y.; Wang, Y.; Chen, C.; Ma, Y.; Gao, C.; Dong, X.P. T188K-Familial Creutzfeldt-Jacob Disease, Predominant Among Chinese, has a Reactive Pattern in CSF RT-QuIC Different from D178N-Fatal Familial Insomnia and E200K-Familial CJD. Neurosci. Bull. 2019, 35, 519–521. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, B.J.; Racker, S.; Herrendorf, G.; Poser, S.; Grosche, S.; Zerr, I.; Kretzschmar, H.; Weber, T. Accuracy and reliability of periodic sharp wave complexes in Creutzfeldt-Jakob disease. Arch. Neurol. 1996, 53, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Rahayel, S.; Tremblay, C.; Vo, A.; Zheng, Y.-Q.; Lehéricy, S.; Arnulf, I.; Vidailhet, M.; Corvol, J.-C.; Gagnon, J.-F.; Postuma, R.B.; et al. Brain atrophy in prodromal synucleinopathy is shaped by structural connectivity and gene expression. Brain 2022, 145, 3162–3178. [Google Scholar] [CrossRef] [PubMed]
- Kosami, K.; Ae, R.; Hamaguchi, T.; Sanjo, N.; Tsukamoto, T.; Kitamoto, T.; Yamada, M.; Mizusawa, H.; Nakamura, Y. Methionine homozygosity for PRNP polymorphism and susceptibility to human prion diseases. J. Neurol. Neurosurg. Psychiatry 2022, 93, 779–784. [Google Scholar] [CrossRef] [PubMed]
- Mead, S.; Burnell, M.; Lowe, J.; Thompson, A.; Lukic, A.; Porter, M.-C.; Carswell, C.; Kaski, D.; Kenny, J.; Mok, T.H.; et al. Clinical Trial Simulations Based on Genetic Stratification and the Natural History of a Functional Outcome Measure in Creutzfeldt-Jakob Disease. JAMA Neurol. 2016, 73, 447–455. [Google Scholar] [CrossRef]
- Hauw, J.-J.; Sazdovitch, V.; Laplanche, J.-L.; Peoc’H, K.; Kopp, N.; Kemeny, J.; Privat, N.; Delasnerie-Laupretre, N.; Brandel, J.P.; Deslys, J.P.; et al. Neuropathologic variants of sporadic Creutzfeldt-Jakob disease and codon 129 of PrP gene. Neurology 2000, 54, 1641–1646. [Google Scholar] [CrossRef]
- Montagna, P.; Cortelli, P.; Avoni, P.; Tinuper, P.; Plazzi, G.; Gallassi, R.; Portaluppi, F.; Julien, J.; Vital, C.; Delisle, M.B.; et al. Clinical features of fatal familial insomnia: Phenotypic variability in relation to a polymorphism at codon 129 of the prion protein gene. Brain Pathol. 1998, 8, 515–520. [Google Scholar] [CrossRef]
- Zhang, J.; Chu, M.; Tian, Z.; Xie, K.; Cui, Y.; Liu, L.; Meng, J.; Yan, H.; Ji, Y.-M.; Jiang, Z.; et al. Clinical profile of fatal familial insomnia: Phenotypic variation in 129 polymorphisms and geographical regions. J. Neurol. Neurosurg. Psychiatry 2022, 93, 291–297. [Google Scholar] [CrossRef]
- Heinemann, U.; Krasnianski, A.; Meissner, B.; Varges, D.; Kallenberg, K.; Schulz-Schaeffer, W.J.; Steinhoff, B.J.; Grasbon-Frodl, E.M.; Kretzschmar, H.A.; Zerr, I. Creutzfeldt-Jakob disease in Germany: A prospective 12-year surveillance. Brain 2007, 130 Pt 5, 1350–1359. [Google Scholar] [CrossRef] [Green Version]
- Mead, S.; Webb, T.E.F.; Campbell, T.A.; Beck, J.; Linehan, J.M.; Rutherfoord, S.; Joiner, S.; Wadsworth, J.D.F.; Heckmann, J.; Wroe, S.; et al. Inherited prion disease with 5-OPRI: Phenotype modification by repeat length and codon 129. Neurology 2007, 69, 730–738. [Google Scholar] [CrossRef]
- Geschwind, M.D.; Potter, C.A.; Sattavat, M.; Garcia, P.A.; Rosen, H.J.; Miller, B.L.; DeArmond, S.J. Correlating DWI MRI with pathologic and other features of Jakob-Creutzfeldt disease. Alzheimer Dis. Assoc. Disord. 2009, 23, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Manners, D.N.; Parchi, P.; Tonon, C.; Capellari, S.; Strammiello, R.; Testa, C.; Tani, G.; Malucelli, E.; Spagnolo, C.; Cortelli, P.; et al. Pathologic correlates of diffusion MRI changes in Creutzfeldt-Jakob disease. Neurology 2009, 72, 1425–1431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, O.S.; Chapman, J.; Korczyn, A.D.; Nitsan, Z.; Appel, S.; Kahana, E.; Rosenmann, H.; Hoffmann, C. Disease duration in E200K familial Creutzfeldt-Jakob disease is correlated with clinical, radiological, and laboratory variables. J. Neural Transm. 2019, 126, 607–611. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total Patients (n = 17) | Short Clinical Course (n = 4) | Long Clinical Course (n = 13) | p | |
|---|---|---|---|---|
| Baseline characteristics | ||||
| Female, % | 7/15 (46.7) | 2/4 (50.0) | 5/11 (45.5) | 1.000 |
| Age at onset, year, mean ± SD | 40.6 ± 14.4 | 53.0 ± 7.3 | 35.0 ± 12.9 | 0.021 |
| Symptom’s duration, year, mean ± SD | 7.3 ± 5.2 | 0.9 ± 0.4 | 9.2 ± 4.3 | <0.001 |
| Family history, % | 16/17 (94.1) | 4/4 (100.0) | 12/13 (92.3) | 1.000 |
| Initial symptoms | ||||
| Cognitive defects, % | 11 (64.7) | 3 (75.0) | 8 (61.5) | 1.000 |
| Psychiatric disturbances, % | 6 (35.3) | 1 (25.0) | 5 (38.5) | 1.000 |
| Ataxia, % | 4 (25.3) | 1 (25.0) | 3 (23.1) | 1.000 |
| Speech disturbances, % | 3 (17.6) | 2 (50.0) | 1 (7.7) | 0.121 |
| Clinical features of prion diseases | ||||
| Cognitive dysfunction, % | 17 (100.0) | 4 (100.0) | 13 (100.0) | 1.000 |
| Psychiatric disturbances, % | 13 (76.5) | 3 (75.0) | 10 (76.9) | 1.000 |
| Parkinsonism, % | 12 (70.6) | 3 (75.0) | 9 (69.2) | 1.000 |
| Cerebellar signs, % | 12 (70.6) | 2 (50.0) | 10 (76.9) | 0.538 |
| Myoclonus, % | 8 (47.1) | 3 (75.0) | 5(38.5) | 0.294 |
| Pyramidal signs, % | 8 (47.1) | 2 (50.0) | 6 (46.2) | 1.000 |
| Speech disorders, % | 6 (35.3) | 2 (50.0) | 4 (30.8) | 0.584 |
| Mutism, % | 4 (23.5) | 2 (50.0) | 2 (15.4) | 0.219 |
| Seizure, % | 3 (17.6) | 0 | 3 (23.1) | 0.541 |
| Visual signs, % | 2 (11.8) | 0 | 2 (15.4) | 1.000 |
| Laboratory features | ||||
| PSWCs on EEG, % | 2/11 (18.2) | 1/3 (33.3) | 1/8 (12.5) | 0.491 |
| Positive CSF 14-3-3 protein, % | 2/3 (66.7) | 2/2 (100.0) | 0/1 | 0.333 |
| Elevated CSF tau protein, % | 2/3 (66.7) | 2/2 (100.0) | 0/1 | 0.333 |
| Positive RT-QuIC, % | 2/2 (100.0) | 2/2 (100.0) | 0/0 | - |
| Hyperintensity on MRI, % | 3/7 (41.9) | 3/3 (100.0) | 0/4 | 0.029 |
| Condon 129 | 0.066 | |||
| 129 cis-M | 12/14 (85.7) | 2/4 (50.0) | 10/10 (100.0) | |
| 129 cis-V | 2/14 (14.3) | 2/4 (50.0) | 0/10 | |
| AAS within OPRIs | 0.516 | |||
| Yes | 4/15 (26.7) | 2/4 (50.0) | 2/11 (18.2) | |
| No | 11/15 (73.3) | 2/4 (50.0) | 9/11 (81.8) | |
| Neuropathology | ||||
| Spongiosis | 9/11 (81.8) | 1/1 (100.0) | 8/10 (80.0) | 1.000 |
| Multicentric plaques | 3/11 (27.3) | 1/1 (100.0) | 2/10 (20.0) | 0.273 |
| Elongated plaques | 3/11 (27.3) | 0/1 | 3/10 (30.0) | 1.000 |
| Kuru-like plaques | 1/11(9.1) | 0/1 | 1/10 (10.0) | 1.000 |
| Nonspecific change | 1/11 (9.1) | 0/1 | 1/10 (10.0) | 1.000 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Z.; Nan, H.; Kong, Y.; Chu, M.; Liu, L.; Zhang, J.; Wang, L.; Wu, L. Amino Acid Substitution within Seven-Octapeptide Repeat Insertions in the Prion Protein Gene Associated with Short-Term Course. Viruses 2022, 14, 2245. https://doi.org/10.3390/v14102245
Chen Z, Nan H, Kong Y, Chu M, Liu L, Zhang J, Wang L, Wu L. Amino Acid Substitution within Seven-Octapeptide Repeat Insertions in the Prion Protein Gene Associated with Short-Term Course. Viruses. 2022; 14(10):2245. https://doi.org/10.3390/v14102245
Chicago/Turabian StyleChen, Zhongyun, Haitian Nan, Yu Kong, Min Chu, Li Liu, Jing Zhang, Lin Wang, and Liyong Wu. 2022. "Amino Acid Substitution within Seven-Octapeptide Repeat Insertions in the Prion Protein Gene Associated with Short-Term Course" Viruses 14, no. 10: 2245. https://doi.org/10.3390/v14102245