

Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors

Abstract
:1. Introduction
2. Ankyrin Repeat Containing Protein
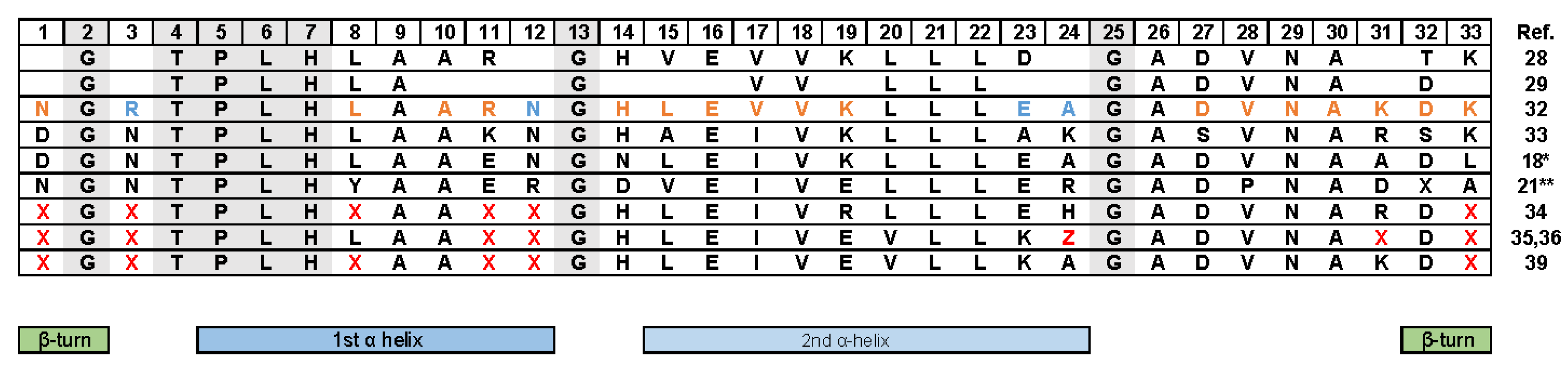
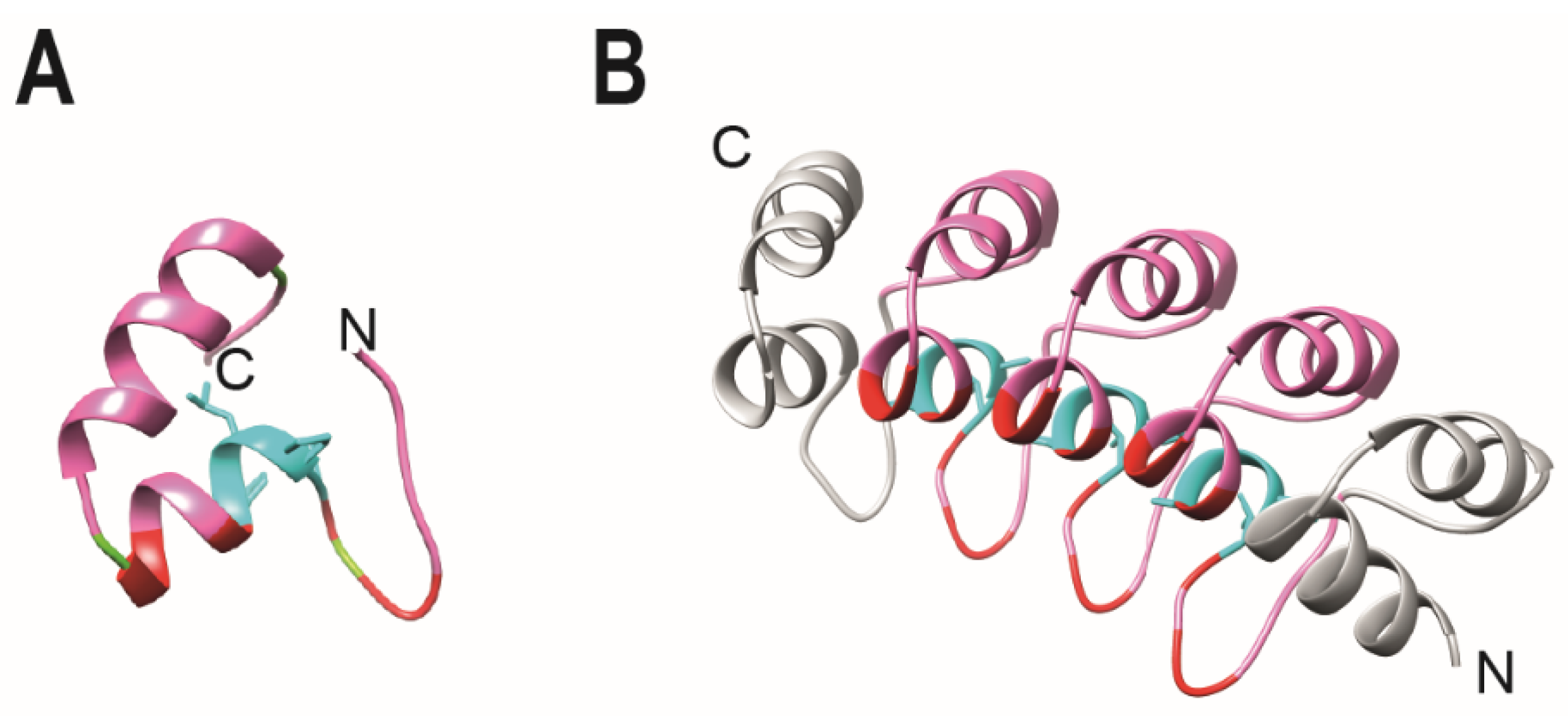
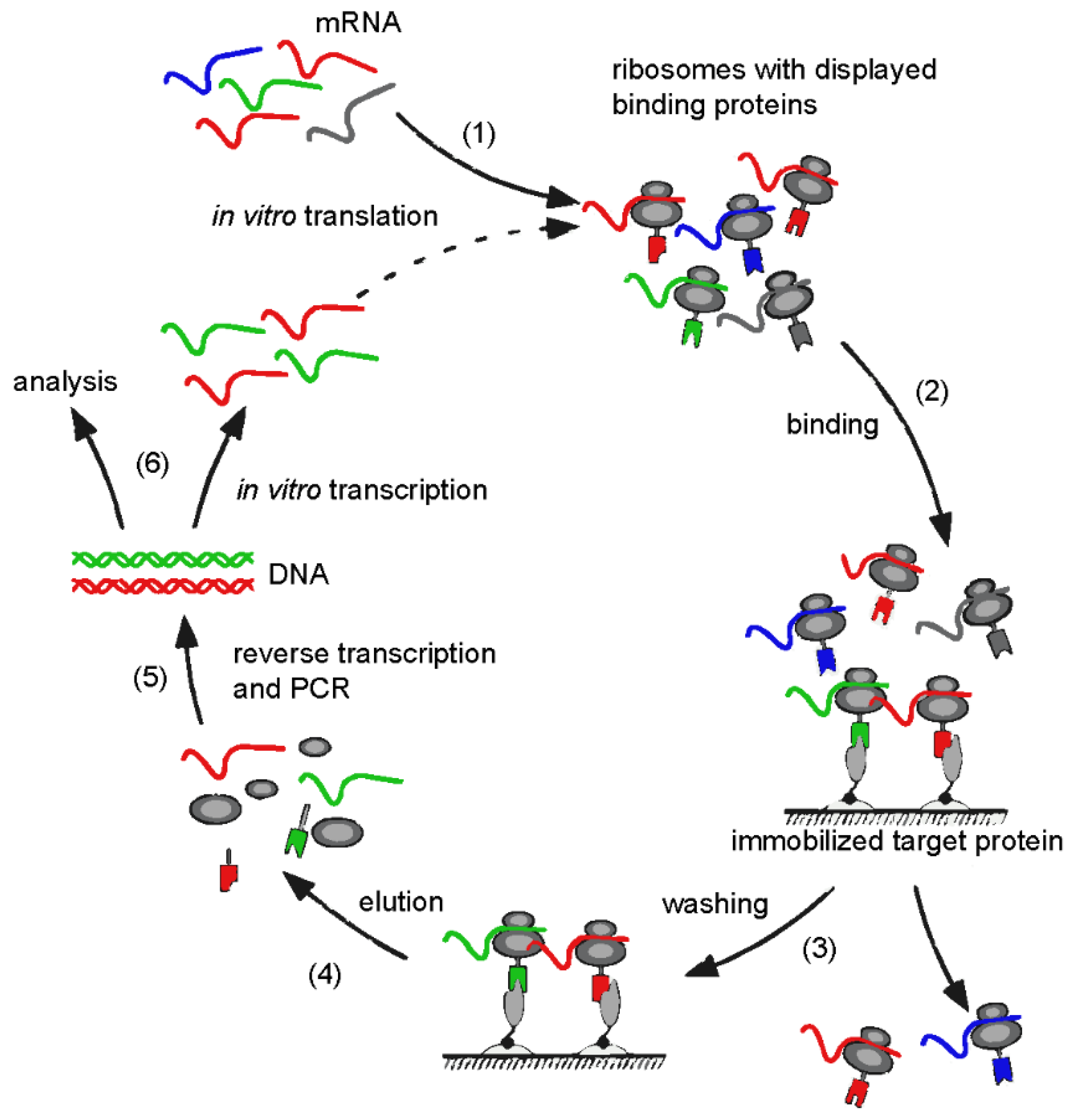
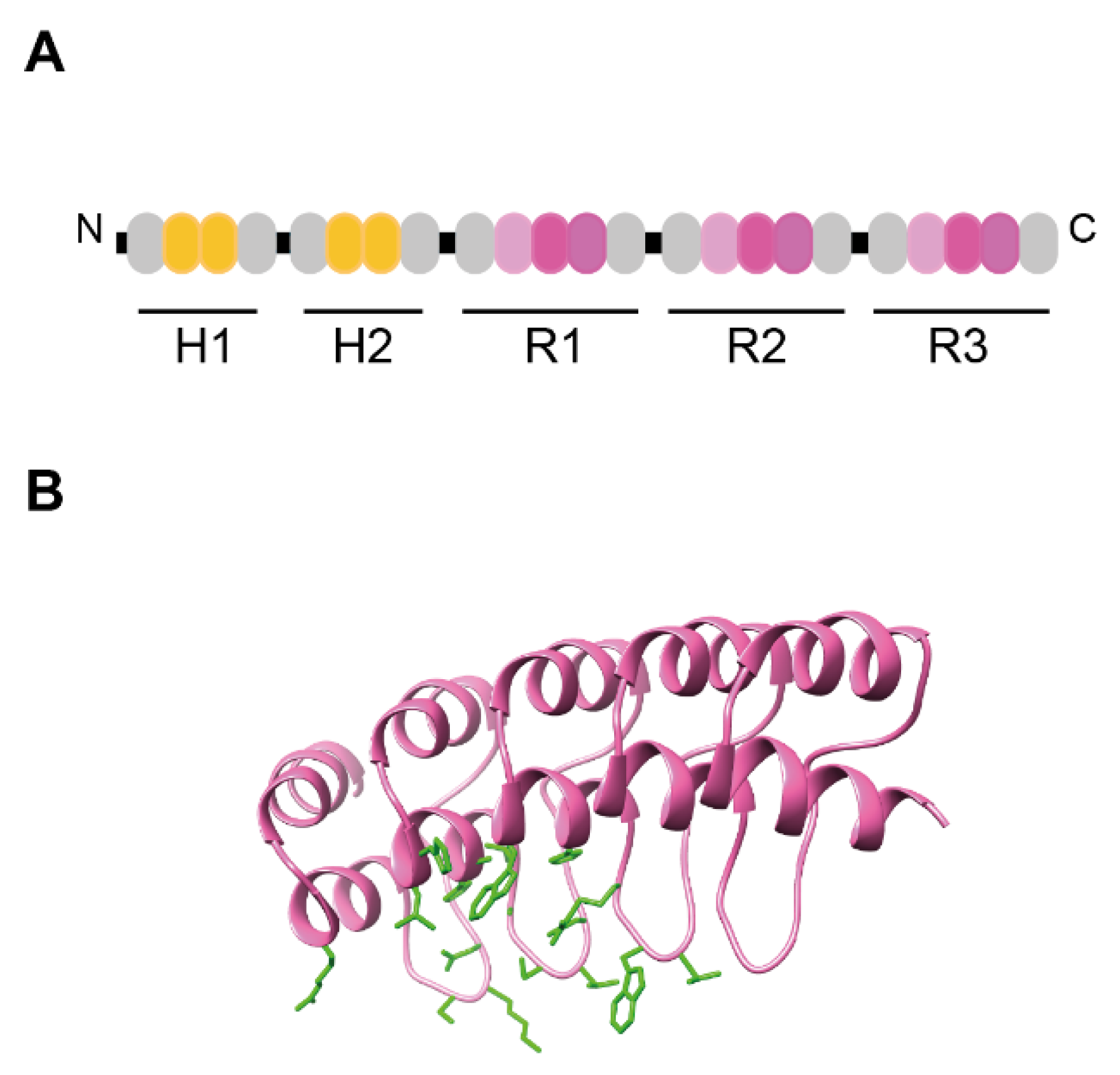
3. Design Ankyrin Repeat Proteins (DARPins)
4. Viral Applications

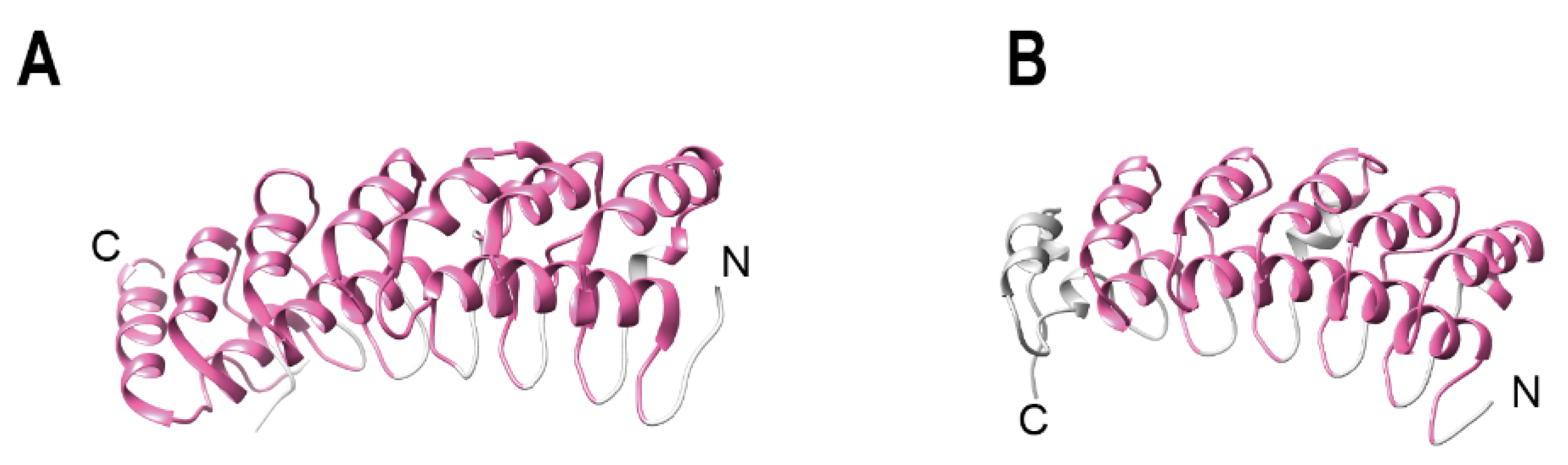{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DARPin Name | Target/Epitope | Mechanism of Action | Application | Ref. |
|---|---|---|---|---|
| 1D3 2E6 | Ad5 knob Her2 | Bispecific adapter to retarget Ad5 to tumor cells | Tumor retargeting of Ad5 | [51,58] |
| 9.26 9.01 9.16 G3 9.29 H14R | Her2 | Engineered MeV attachment protein Hemagglutinin unable to bind to its natural receptor genetically fused to a Her2-specific DARPin binding domain | Tumor retargeting of pseudotyped lentiviral vector | [61] |
| D29.2 | CD4 | Engineered MeV attachment protein Hemagglutinin unable to bind to its natural receptor genetically fused to a CD4-specific DARPin binding domain | Retargeting pseudotyped lentiviral vector to CD4+ cells | [62] |
| 9.16 9.29 9.26 G3 | Her2 | Engineered MeV attachment protein Hemagglutinin unable to bind to its natural receptor genetically fused to either Her2, EpCAM or EGFR-specific DARPin bindingdomain (Monospecific or bispecific G3 +EC4) | Tumor retargeting of recombinant MeV | [59] |
| C9 EC4 | EpCAM | |||
| E.01 E.68 E.69 | EGFR | |||
| E.01 | EGFR | Engineered MeV fusion protein (F) that can be activated by tumor-associated matrix metalloprotease, genetically fused to EGFR-specific DARPin binding domain | Tumor retargeting of recombinant MeV | [60] |
| 9.29 | Her2 | Engineered capsid protein VP2 fused to Her2-specific DARPin binding domain and ablated natural receptor binding site on VP3 | Tumor retargeting of AAV | [63,64] |
| EC1 | EpCAM | Engineered capsid protein VP2 fused to EpCAM-specific DARPin binding domain and ablated natural receptor binding site on VP3 | Tumor retargeting of AAV | [64] |
| D55.2 | CD4 | Engineered capsid protein VP2 fused to CD4-specific DARPin binding domain and ablated natural receptor binding site on VP3 | Retargeting AAV to CD4+ cells | [64] |
| E.01 | EGFR | Engineered capsid protein VP2 fused to human FK-binding protein (FKBP) and ablated natural receptor binding site on VP3. Adapter protein that consist of a modified FKBP rapamycin binding domain of mTOR fused to mCherry and EGFP-specific DARPin binding domain | Adaptor for the tumor retargeting of AAV | [74] |
| E.01 | EGFR | Engineered capsid protein VP2 fused to EGFR-specific DARPin binding domain and ablated natural receptor binding site on VP3 | Tumor retargeting of AAV | [75] |
| F10 | Human HDAC6 zinc finger domain | Impairs interaction with ubiquitin and infection by influenza and Zika virus | antiviral | [65] |
| AnkGAG1D4 | HIV-1 Gag precursor | Interferes with late stages of HIV-1 capsid assembly | antiviral | [34,66,67,68,69] |
| D1.1-D6.1 D23.2 D25.2 D27.2 D29.2 D55.2 D57.2 | CD4 | Inhibit HIV-1 cell entry by blocking the binding to CD4, the main receptor of HIV-1 | antiviral | [70] |
| D_18 D_19 D_20 | Lactococcal phage TP901-1 BppU BppL complex | Neutralization of phage TP901-1 by blocking receptor binding | antiviral | [71] |
| bnD_1 bnD_2 bnD_3 | V3 crown of the HIV-1 envelope protein (gp120) | Neutralization of HIV-1 by blocking receptor binding | antiviral | [73] |
| 5m3_D12 | V3 loop of the HIV-1 envelope protein (gp120) | Neutralization of HIV-1 by blocking receptor binding | antiviral | [72] |
| R1 R2 R3 | RBD domain of SARS-CoV-2 spike | Neutralization of SARS-CoV-2 by blocking receptor binding | antiviral Therapeutic treatment of COVID-19 | [39] |
5. Ensovibep
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int. J. Biol. Macromol. 2021, 172, 524–541. [Google Scholar] [CrossRef] [PubMed]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Bonilla, H.; Jagannathan, P.; Shafer, R.W. SARS-CoV-2 Antiviral Therapy. Clin. Microbiol. Rev. 2021, 34, e0010921. [Google Scholar] [CrossRef] [PubMed]
- Pantaleo, G.; Correia, B.; Fenwick, C.; Joo, V.S.; Perez, L. Antibodies to combat viral infections: Development strategies and progress. Nat. Rev. Drug Discov. 2022, 21, 676–696. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 2021, 184, 3086–3108. [Google Scholar] [CrossRef] [PubMed]
- Iketani, S.; Liu, L.; Guo, Y.; Liu, L.; Chan, J.F.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; Consortium, C.-G.U.; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Takashita, E.; Yamayoshi, S.; Simon, V.; van Bakel, H.; Sordillo, E.M.; Pekosz, A.; Fukushi, S.; Suzuki, T.; Maeda, K.; Halfmann, P.; et al. Efficacy of Antibodies and Antiviral Drugs against Omicron BA.2.12.1, BA.4, and BA.5 Subvariants. N. Engl. J. Med. 2022, 387, 468–470. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kruger, N.; Schulz, S.; Cossmann, A.; Rocha, C.; Kempf, A.; Nehlmeier, I.; Graichen, L.; Moldenhauer, A.S.; Winkler, M.S.; et al. The Omicron variant is highly resistant against antibody-mediated neutralization: Implications for control of the COVID-19 pandemic. Cell 2022, 185, 447–456.e11. [Google Scholar] [CrossRef]
- Chen, X.; Gentili, M.; Hacohen, N.; Regev, A. A cell-free nanobody engineering platform rapidly generates SARS-CoV-2 neutralizing nanobodies. Nat. Commun. 2021, 12, 5506. [Google Scholar] [CrossRef]
- Koenig, P.A.; Das, H.; Liu, H.; Kummerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Pluckthun, A. Designed ankyrin repeat proteins (DARPins): Binding proteins for research, diagnostics, and therapy. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Forrer, P.; Stumpp, M.T.; Binz, H.K.; Pluckthun, A. A novel strategy to design binding molecules harnessing the modular nature of repeat proteins. FEBS Lett. 2003, 539, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Stumpp, M.T.; Dawson, K.M.; Binz, H.K. Beyond Antibodies: The DARPin((R)) Drug Platform. BioDrugs 2020, 34, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Stumpp, M.T.; Binz, H.K.; Amstutz, P. DARPins: A new generation of protein therapeutics. Drug Discov. Today 2008, 13, 695–701. [Google Scholar] [CrossRef]
- Breeden, L.; Nasmyth, K. Similarity between cell-cycle genes of budding yeast and fission yeast and the Notch gene of Drosophila. Nature 1987, 329, 651–654. [Google Scholar] [CrossRef]
- Lux, S.E.; John, K.M.; Bennett, V. Analysis of cDNA for human erythrocyte ankyrin indicates a repeated structure with homology to tissue-differentiation and cell-cycle control proteins. Nature 1990, 344, 36–42. [Google Scholar] [CrossRef]
- Galpern, E.A.; Freiberger, M.I.; Ferreiro, D.U. Large Ankyrin repeat proteins are formed with similar and energetically favorable units. PLoS ONE 2020, 15, e0233865. [Google Scholar] [CrossRef]
- Al-Khodor, S.; Price, C.T.; Kalia, A.; Abu Kwaik, Y. Functional diversity of ankyrin repeats in microbial proteins. Trends Microbiol. 2010, 18, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Barrick, D.; Ferreiro, D.U.; Komives, E.A. Folding landscapes of ankyrin repeat proteins: Experiments meet theory. Curr. Opin. Struct. Biol. 2008, 18, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Mosavi, L.K.; Cammett, T.J.; Desrosiers, D.C.; Peng, Z.Y. The ankyrin repeat as molecular architecture for protein recognition. Protein Sci. 2004, 13, 1435–1448. [Google Scholar] [CrossRef] [Green Version]
- Jernigan, K.K.; Bordenstein, S.R. Ankyrin domains across the Tree of Life. PeerJ 2014, 2, e264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, E.I.; Spratt, D.E. Structural Insights into Ankyrin Repeat-Containing Proteins and Their Influence in Ubiquitylation. Int. J. Mol. Sci. 2021, 22, 609. [Google Scholar] [CrossRef] [PubMed]
- Tao, Z.; Fusco, A.; Huang, D.B.; Gupta, K.; Young Kim, D.; Ware, C.F.; Van Duyne, G.D.; Ghosh, G. p100/IkappaBdelta sequesters and inhibits NF-kappaB through kappaBsome formation. Proc. Natl. Acad. Sci. USA 2014, 111, 15946–15951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskill, S.; Beg, A.A.; Tompkins, S.M.; Morris, J.S.; Yurochko, A.D.; Sampson-Johannes, A.; Mondal, K.; Ralph, P.; Baldwin, A.S., Jr. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell 1991, 65, 1281–1289. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Croy, C.H.; Bergqvist, S.; Huxford, T.; Ghosh, G.; Komives, E.A. Biophysical characterization of the free IkappaBalpha ankyrin repeat domain in solution. Protein Sci. 2004, 13, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Truhlar, S.M.; Torpey, J.W.; Komives, E.A. Regions of IkappaBalpha that are critical for its inhibition of NF-kappaB.DNA interaction fold upon binding to NF-kappaB. Proc. Natl. Acad. Sci. USA 2006, 103, 18951–18956. [Google Scholar] [CrossRef] [Green Version]
- Michaely, P.; Bennett, V. The ANK repeat: A ubiquitous motif involved in macromolecular recognition. Trends Cell Biol. 1992, 2, 127–129. [Google Scholar] [CrossRef]
- Sedgwick, S.G.; Smerdon, S.J. The ankyrin repeat: A diversity of interactions on a common structural framework. Trends Biochem. Sci. 1999, 24, 311–316. [Google Scholar] [CrossRef]
- Li, J.; Mahajan, A.; Tsai, M.D. Ankyrin repeat: A unique motif mediating protein-protein interactions. Biochemistry 2006, 45, 15168–15178. [Google Scholar] [CrossRef]
- Kumar, A.; Balbach, J. Folding and Stability of Ankyrin Repeats Control Biological Protein Function. Biomolecules 2021, 11, 840. [Google Scholar] [CrossRef] [PubMed]
- Mosavi, L.K.; Minor, D.L., Jr.; Peng, Z.Y. Consensus-derived structural determinants of the ankyrin repeat motif. Proc. Natl. Acad. Sci. USA 2002, 99, 16029–16034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripp, K.W.; Barrick, D. Enhancing the stability and folding rate of a repeat protein through the addition of consensus repeats. J. Mol. Biol. 2007, 365, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Nangola, S.; Urvoas, A.; Valerio-Lepiniec, M.; Khamaikawin, W.; Sakkhachornphop, S.; Hong, S.S.; Boulanger, P.; Minard, P.; Tayapiwatana, C. Antiviral activity of recombinant ankyrin targeted to the capsid domain of HIV-1 Gag polyprotein. Retrovirology 2012, 9, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, A.; Binz, H.K.; Forrer, P.; Stumpp, M.T.; Pluckthun, A.; Grutter, M.G. Designed to be stable: Crystal structure of a consensus ankyrin repeat protein. Proc. Natl. Acad. Sci. USA 2003, 100, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, J.; Munch, R.C.; Freiling, R.T.; Schneider, I.C.; Dreier, B.; Samukange, W.; Koch, J.; Seeger, M.A.; Pluckthun, A.; Buchholz, C.J. A Library-Based Screening Strategy for the Identification of DARPins as Ligands for Receptor-Targeted AAV and Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2018, 10, 128–143. [Google Scholar] [CrossRef]
- Forrer, P.; Binz, H.K.; Stumpp, M.T.; Pluckthun, A. Consensus design of repeat proteins. Chembiochem 2004, 5, 183–189. [Google Scholar] [CrossRef]
- Schilling, J.; Schoppe, J.; Pluckthun, A. From DARPins to LoopDARPins: Novel LoopDARPin design allows the selection of low picomolar binders in a single round of ribosome display. J. Mol. Biol. 2014, 426, 691–721. [Google Scholar] [CrossRef]
- Rothenberger, S.; Hurdiss, D.L.; Walser, M.; Malvezzi, F.; Mayor, J.; Ryter, S.; Moreno, H.; Liechti, N.; Bosshart, A.; Iss, C.; et al. The trispecific DARPin ensovibep inhibits diverse SARS-CoV-2 variants. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Pluckthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Binz, H.K.; Amstutz, P.; Kohl, A.; Stumpp, M.T.; Briand, C.; Forrer, P.; Grutter, M.G.; Pluckthun, A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004, 22, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Schilling, J.; Jost, C.; Ilie, I.M.; Schnabl, J.; Buechi, O.; Eapen, R.S.; Truffer, R.; Caflisch, A.; Forrer, P. Thermostable designed ankyrin repeat proteins (DARPins) as building blocks for innovative drugs. J. Biol. Chem. 2021, 298, 101403. [Google Scholar] [CrossRef] [PubMed]
- Interlandi, G.; Wetzel, S.K.; Settanni, G.; Pluckthun, A.; Caflisch, A. Characterization and further stabilization of designed ankyrin repeat proteins by combining molecular dynamics simulations and experiments. J. Mol. Biol. 2008, 375, 837–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pluckthun, A. Ribosome display: A perspective. Methods Mol. Biol. 2012, 805, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.; Forrer, P.; Stumpp, M.T.; Pluckthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 2006, 24, 823–831. [Google Scholar] [CrossRef]
- Steiner, D.; Forrer, P.; Pluckthun, A. Efficient selection of DARPins with sub-nanomolar affinities using SRP phage display. J. Mol. Biol. 2008, 382, 1211–1227. [Google Scholar] [CrossRef] [Green Version]
- Schutz, M.; Batyuk, A.; Klenk, C.; Kummer, L.; de Picciotto, S.; Gulbakan, B.; Wu, Y.; Newby, G.A.; Zosel, F.; Schoppe, J.; et al. Generation of Fluorogen-Activating Designed Ankyrin Repeat Proteins (FADAs) as Versatile Sensor Tools. J. Mol. Biol. 2016, 428, 1272–1289. [Google Scholar] [CrossRef]
- Zahnd, C.; Wyler, E.; Schwenk, J.M.; Steiner, D.; Lawrence, M.C.; McKern, N.M.; Pecorari, F.; Ward, C.W.; Joos, T.O.; Pluckthun, A. A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 2007, 369, 1015–1028. [Google Scholar] [CrossRef]
- Hanes, J.; Schaffitzel, C.; Knappik, A.; Pluckthun, A. Picomolar affinity antibodies from a fully synthetic naive library selected and evolved by ribosome display. Nat. Biotechnol. 2000, 18, 1287–1292. [Google Scholar] [CrossRef]
- Amstutz, P.; Binz, H.K.; Parizek, P.; Stumpp, M.T.; Kohl, A.; Grutter, M.G.; Forrer, P.; Pluckthun, A. Intracellular kinase inhibitors selected from combinatorial libraries of designed ankyrin repeat proteins. J. Biol. Chem. 2005, 280, 24715–24722. [Google Scholar] [CrossRef]
- Dreier, B.; Mikheeva, G.; Belousova, N.; Parizek, P.; Boczek, E.; Jelesarov, I.; Forrer, P.; Pluckthun, A.; Krasnykh, V. Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J. Mol. Biol. 2011, 405, 410–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, K.; March, K.; Alexaki, A.; Fabozzi, G.; Moysi, E.; Petrovas, C. Immunogenicity of Protein Therapeutics: A Lymph Node Perspective. Front. Immunol. 2020, 11, 791. [Google Scholar] [CrossRef] [PubMed]
- Safdari, Y.; Farajnia, S.; Asgharzadeh, M.; Khalili, M. Antibody humanization methods—A review and update. Biotechnol. Genet. Eng. Rev. 2013, 29, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Moussa, E.M.; Panchal, J.P.; Moorthy, B.S.; Blum, J.S.; Joubert, M.K.; Narhi, L.O.; Topp, E.M. Immunogenicity of Therapeutic Protein Aggregates. J. Pharm. Sci. 2016, 105, 417–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, R.D.; Linossi, C.; Middleton, M.; Lord, S.; Harris, A.; Rodon, J.; Zitt, C.; Fiedler, U.; Dawson, K.M.; Leupin, N.; et al. First-in-Human Phase I Study of MP0250, a First-in-Class DARPin Drug Candidate Targeting VEGF and HGF, in Patients With Advanced Solid Tumors. J. Clin. Oncol. 2021, 39, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D. Filamentous Phage Display of Designed Ankyrin Repeat Proteins: From Conception to Applications. 2007. Available online: https://www.zora.uzh.ch/id/eprint/163768/1/20080424_002127557.pdf (accessed on 22 September 2022).
- Buchholz, C.J.; Friedel, T.; Buning, H. Surface-Engineered Viral Vectors for Selective and Cell Type-Specific Gene Delivery. Trends Biotechnol. 2015, 33, 777–790. [Google Scholar] [CrossRef]
- Dreier, B.; Honegger, A.; Hess, C.; Nagy-Davidescu, G.; Mittl, P.R.; Grutter, M.G.; Belousova, N.; Mikheeva, G.; Krasnykh, V.; Pluckthun, A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc. Natl. Acad. Sci. USA 2013, 110, E869–E877. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, K.; Hanauer, J.R.; Prufer, S.; Munch, R.C.; Volker, I.; Filippis, C.; Jost, C.; Hanschmann, K.M.; Cattaneo, R.; Peng, K.W.; et al. DARPin-targeting of measles virus: Unique bispecificity, effective oncolysis, and enhanced safety. Mol. Ther. 2013, 21, 849–859. [Google Scholar] [CrossRef] [Green Version]
- Hanauer, J.R.H.; Koch, V.; Lauer, U.M.; Muhlebach, M.D. High-Affinity DARPin Allows Targeting of MeV to Glioblastoma Multiforme in Combination with Protease Targeting without Loss of Potency. Mol. Ther. Oncolytics 2019, 15, 186–200. [Google Scholar] [CrossRef] [Green Version]
- Munch, R.C.; Muhlebach, M.D.; Schaser, T.; Kneissl, S.; Jost, C.; Pluckthun, A.; Cichutek, K.; Buchholz, C.J. DARPins: An efficient targeting domain for lentiviral vectors. Mol. Ther. 2011, 19, 686–693. [Google Scholar] [CrossRef]
- Zhou, Q.; Uhlig, K.M.; Muth, A.; Kimpel, J.; Levy, C.; Munch, R.C.; Seifried, J.; Pfeiffer, A.; Trkola, A.; Coulibaly, C.; et al. Exclusive Transduction of Human CD4+ T Cells upon Systemic Delivery of CD4-Targeted Lentiviral Vectors. J. Immunol. 2015, 195, 2493–2501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, R.C.; Janicki, H.; Volker, I.; Rasbach, A.; Hallek, M.; Buning, H.; Buchholz, C.J. Displaying high-affinity ligands on adeno-associated viral vectors enables tumor cell-specific and safe gene transfer. Mol. Ther. 2013, 21, 109–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, R.C.; Muth, A.; Muik, A.; Friedel, T.; Schmatz, J.; Dreier, B.; Trkola, A.; Pluckthun, A.; Buning, H.; Buchholz, C.J. Off-target-free gene delivery by affinity-purified receptor-targeted viral vectors. Nat. Commun. 2015, 6, 6246. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Moreira, E.A.; Kempf, G.; Miyake, Y.; Oliveira Esteves, B.I.; Fahmi, A.; Schaefer, J.V.; Dreier, B.; Yamauchi, Y.; Alves, M.P.; et al. Disrupting the HDAC6-ubiquitin interaction impairs infection by influenza and Zika virus and cellular stress pathways. Cell Rep. 2022, 39, 110736. [Google Scholar] [CrossRef]
- Moonmuang, S.; Maniratanachote, R.; Chetprayoon, P.; Sornsuwan, K.; Thongkum, W.; Chupradit, K.; Tayapiwatana, C. Specific Interaction of DARPin with HIV-1 CANTD Disturbs the Distribution of Gag, RNA Packaging, and Tetraspanin Remodelling in the Membrane. Viruses 2022, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Khamaikawin, W.; Saoin, S.; Nangola, S.; Chupradit, K.; Sakkhachornphop, S.; Hadpech, S.; Onlamoon, N.; Ansari, A.A.; Byrareddy, S.N.; Boulanger, P.; et al. Combined Antiviral Therapy Using Designed Molecular Scaffolds Targeting Two Distinct Viral Functions, HIV-1 Genome Integration and Capsid Assembly. Mol. Ther. Nucleic Acids 2015, 4, e249. [Google Scholar] [CrossRef] [PubMed]
- Sakkhachornphop, S.; Hadpech, S.; Wisitponchai, T.; Panto, C.; Kantamala, D.; Utaipat, U.; Praparattanapan, J.; Kotarathitithum, W.; Taejaroenkul, S.; Yasamut, U.; et al. Broad-Spectrum Antiviral Activity of an Ankyrin Repeat Protein on Viral Assembly against Chimeric NL4-3 Viruses Carrying Gag/PR Derived from Circulating Strains among Northern Thai Patients. Viruses 2018, 10, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sornsuwan, K.; Thongkhum, W.; Pamonsupornwichit, T.; Carraway, T.S.; Soponpong, S.; Sakkhachornphop, S.; Tayapiwatana, C.; Yasamut, U. Performance of Affinity-Improved DARPin Targeting HIV Capsid Domain in Interference of Viral Progeny Production. Biomolecules 2021, 11, 1437. [Google Scholar] [CrossRef]
- Schweizer, A.; Rusert, P.; Berlinger, L.; Ruprecht, C.R.; Mann, A.; Corthesy, S.; Turville, S.G.; Aravantinou, M.; Fischer, M.; Robbiani, M.; et al. CD4-specific designed ankyrin repeat proteins are novel potent HIV entry inhibitors with unique characteristics. PLoS Pathog. 2008, 4, e1000109. [Google Scholar] [CrossRef] [Green Version]
- Veesler, D.; Dreier, B.; Blangy, S.; Lichiere, J.; Tremblay, D.; Moineau, S.; Spinelli, S.; Tegoni, M.; Pluckthun, A.; Campanacci, V.; et al. Crystal structure and function of a DARPin neutralizing inhibitor of lactococcal phage TP901-1: Comparison of DARPin and camelid VHH binding mode. J. Biol. Chem. 2009, 284, 30718–30726. [Google Scholar] [CrossRef]
- Mann, A.; Friedrich, N.; Krarup, A.; Weber, J.; Stiegeler, E.; Dreier, B.; Pugach, P.; Robbiani, M.; Riedel, T.; Moehle, K.; et al. Conformation-dependent recognition of HIV gp120 by designed ankyrin repeat proteins provides access to novel HIV entry inhibitors. J. Virol. 2013, 87, 5868–5881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, N.; Stiegeler, E.; Glogl, M.; Lemmin, T.; Hansen, S.; Kadelka, C.; Wu, Y.; Ernst, P.; Maliqi, L.; Foulkes, C.; et al. Distinct conformations of the HIV-1 V3 loop crown are targetable for broad neutralization. Nat. Commun. 2021, 12, 6705. [Google Scholar] [CrossRef] [PubMed]
- Horner, M.; Kaufmann, B.; Cotugno, G.; Wiedtke, E.; Buning, H.; Grimm, D.; Weber, W. A chemical switch for controlling viral infectivity. Chem. Commun. 2014, 50, 10319–10322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagen, S.; Baumann, T.; Wagner, H.J.; Morath, V.; Kaufmann, B.; Fischer, A.; Bergmann, S.; Schindler, P.; Arndt, K.M.; Muller, K.M. Modular adeno-associated virus (rAAV) vectors used for cellular virus-directed enzyme prodrug therapy. Sci. Rep. 2014, 4, 3759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clinicaltrial.gov. Clinical Trials Identifiers NCT04834856, NCT04828161, NCT04870164, NCT04501978.
- Clinicaltrial.gov. Clinical Trials Identifier NCT04049903.
- Clinicaltrial.gov. Clinical Trials Identifiers NCT03335852, NCT03539549, NCT02859766, NCT02462486, NCT02462928, NCT02181517, NCT02181504, NCT02186119.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 93–116. [Google Scholar] [CrossRef]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Bosch, B.J.; Frenz, B.; Rottier, P.J.M.; DiMaio, F.; Rey, F.A.; Veesler, D. Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 2016, 531, 114–117. [Google Scholar] [CrossRef]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlQahtani, A.D.; O’Connor, D.; Domling, A.; Goda, S.K. Strategies for the production of long-acting therapeutics and efficient drug delivery for cancer treatment. Biomed. Pharmacother. 2019, 113, 108750. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.; Merz, F.W.; Sonderegger, I.; Gulotti-Georgieva, M.; Villemagne, D.; Phillips, D.J.; Forrer, P.; Stumpp, M.T.; Zitt, C.; Binz, H.K. Half-life extension using serum albumin-binding DARPin(R) domains. Protein Eng. Des. Sel. 2017, 30, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Available online: https://investors.molecularpartners.com/news-releases/news-release-details/molecular-partners-and-novartis-report-positive-topline-data (accessed on 22 September 2022).
- Rosenberg, R. Detecting the emergence of novel, zoonotic viruses pathogenic to humans. Cell Mol. Life Sci. 2015, 72, 1115–1125. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and viral traits predict zoonotic spillover from mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef]
- Zhang, Y.Z.; Wu, W.C.; Shi, M.; Holmes, E.C. The diversity, evolution and origins of vertebrate RNA viruses. Curr. Opin. Virol. 2018, 31, 9–16. [Google Scholar] [CrossRef]
- Bohan, D.; Maury, W. Enveloped RNA virus utilization of phosphatidylserine receptors: Advantages of exploiting a conserved, widely available mechanism of entry. PLoS Pathog. 2021, 17, e1009899. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468–470, 565–580. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Helenius, A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 2008, 320, 531–535. [Google Scholar] [CrossRef]
- Mayor, J.; Torriani, G.; Rothenberger, S.; Engler, O. T-cell immunoglobulin and mucin (TIM) contributes to the infection of human airway epithelial cells by pseudotype viruses containing Hantaan virus glycoproteins. Virology 2020, 543, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.M.; et al. Axl Mediates ZIKA Virus Entry in Human Glial Cells and Modulates Innate Immune Responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B., Jr.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Shimojima, M.; Takada, A.; Ebihara, H.; Neumann, G.; Fujioka, K.; Irimura, T.; Jones, S.; Feldmann, H.; Kawaoka, Y. Tyro3 family-mediated cell entry of Ebola and Marburg viruses. J. Virol. 2006, 80, 10109–10116. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef]
- Liu, H.Y.; Yang, P.L. Small-Molecule Inhibition of Viral Fusion Glycoproteins. Annu. Rev. Virol. 2021, 8, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Mast, F.D.; Fridy, P.C.; Ketaren, N.E.; Wang, J.; Jacobs, E.Y.; Olivier, J.P.; Sanyal, T.; Molloy, K.R.; Schmidt, F.; Rutkowska, M.; et al. Highly synergistic combinations of nanobodies that target SARS-CoV-2 and are resistant to escape. Elife 2021, 10, e73027. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Lu, B.; McTamney, P.; Palaszynski, S.; Diallo, S.; Ren, K.; Ulbrandt, N.D.; Kallewaard, N.; Wang, W.; Fernandes, F.; et al. Prevalence and Significance of Substitutions in the Fusion Protein of Respiratory Syncytial Virus Resulting in Neutralization Escape From Antibody MEDI8897. J. Infect. Dis. 2018, 218, 572–580. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Walser, M.; Mayor, J.; Rothenberger, S. Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors. Viruses 2022, 14, 2242. https://doi.org/10.3390/v14102242
Walser M, Mayor J, Rothenberger S. Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors. Viruses. 2022; 14(10):2242. https://doi.org/10.3390/v14102242
Chicago/Turabian StyleWalser, Marcel, Jennifer Mayor, and Sylvia Rothenberger. 2022. "Designed Ankyrin Repeat Proteins: A New Class of Viral Entry Inhibitors" Viruses 14, no. 10: 2242. https://doi.org/10.3390/v14102242