
Development and Characterization of Recombinase-Based Isothermal Amplification Assays (RPA/RAA) for the Rapid Detection of Monkeypox Virus
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of Monkeypox Virus and DNA Extraction
2.2. Preparation of Vaccinia Virus DNA and Varicella-Zoster Virus DNA
2.3. Real-Time PCR
2.4. Molecular DNA Standards, RPA Oligonucleotide, and crRNA Preparation
2.5. Real-Time RPA
2.6. RPA Combined with CRISPR-Cas Detection
2.7. RAA Combined with Lateral Flow Assays
3. Results
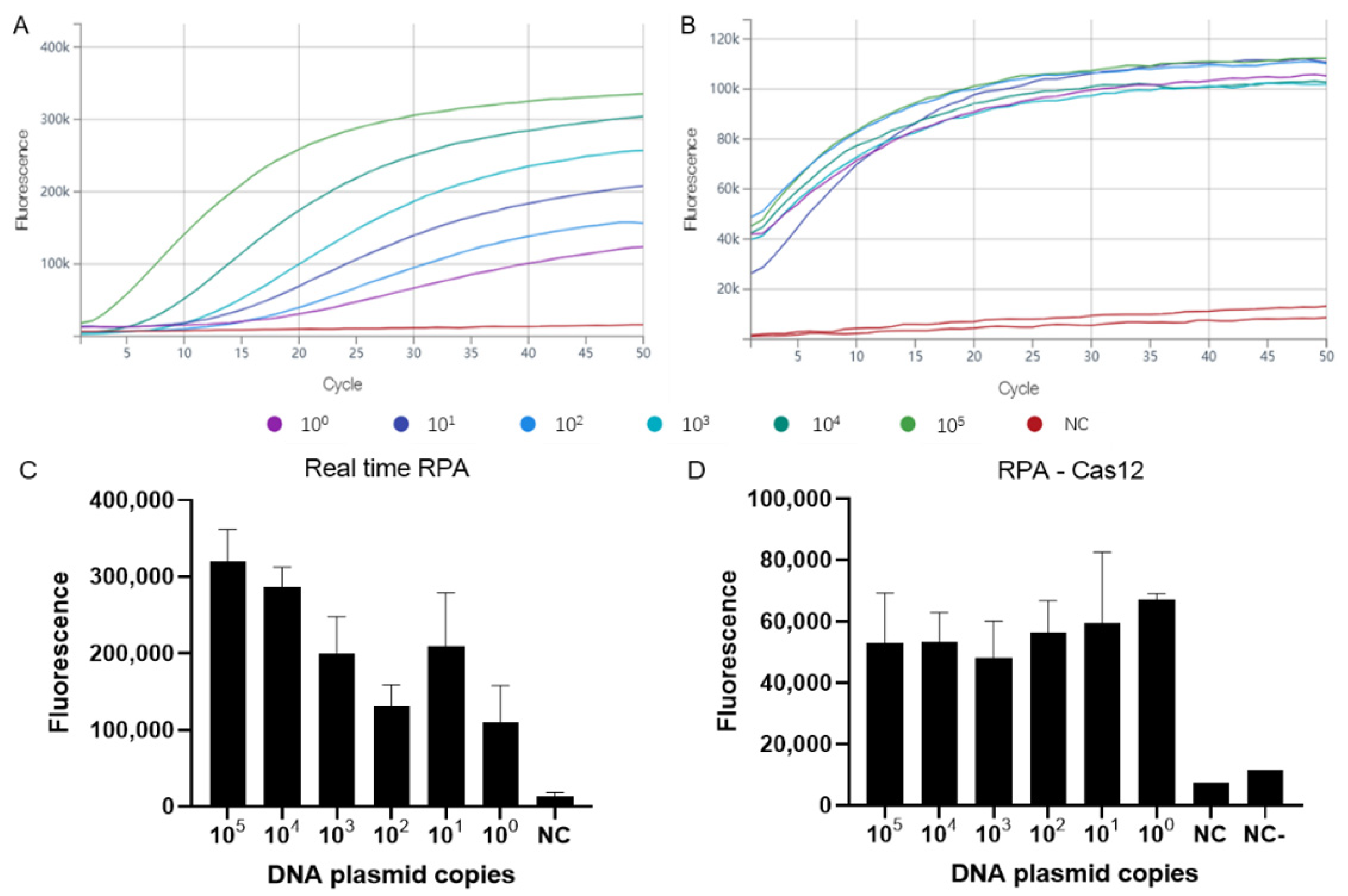
3.1. Assay Sensitivity and Specificity
3.2. Assay Performance on Clinical Samples as Compared to Traditional Real-Time PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Parker, S.; Nuara, A.; Buller, R.M.; Schultz, D.A. Human monkeypox: An emerging zoonotic disease. Future Microbiol. 2007, 2, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A tale of two clades: Monkeypox viruses. J. Gen. Virol. 2005, 86, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- von Magnus, P.; Andersen, E.K.; Petersen, K.B.; Birch-Andersen, A. A Pox-like Disease in Cynomolgus Monkeys. Acta Pathol. Microbiol. Scand. 1959, 46, 156–176. [Google Scholar] [CrossRef]
- Ladnyj, I.D.; Ziegler, P.; Kima, E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull. World Health Organ. 1972, 46, 593–597. [Google Scholar]
- Berthet, N.; Descorps-Declère, S.; Besombes, C.; Curaudeau, M.; Nkili Meyong, A.A.; Selekon, B.; Labouba, I.; Gonofio, E.C.; Ouilibona, R.S.; Simo Tchetgna, H.D.; et al. Genomic history of human monkey pox infections in the Central African Republic between 2001 and 2018. Sci. Rep. 2021, 11, 13085. [Google Scholar] [CrossRef] [PubMed]
- Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The detection of monkeypox in humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef]
- Nigeria Centre for Disease Control (NCDC). An Update of Monkeypox Outbreak in Nigeria for Week 43; Nigeria Centre for Disease Control (NCDC): Abuja, Nigeria, 2021.
- Hobson, G.; Adamson, J.; Adler, H.; Firth, R.; Gould, S.; Houlihan, C.; Johnson, C.; Porter, D.; Rampling, T.; Ratcliffe, L.; et al. Family cluster of three cases of monkeypox imported from Nigeria to the United Kingdom, May 2021. Eurosurveillance 2021, 26, 2100745. [Google Scholar] [CrossRef]
- Vaughan, A.; Aarons, E.; Astbury, J.; Balasegaram, S.; Beadsworth, M.; Beck, C.R.; Chand, M.; O’Connor, C.; Dunning, J.; Ghebrehewet, S.; et al. Two cases of monkeypox imported to the United Kingdom, September 2018. Eurosurveillance 2018, 23, 1800509. [Google Scholar] [CrossRef]
- Erez, N.; Achdout, H.; Milrot, E.; Schwartz, Y.; Wiener-Well, Y.; Paran, N.; Politi, B.; Tamir, H.; Israely, T.; Weiss, S.; et al. Diagnosis of Imported Monkeypox, Israel, 2018. Emerg. Infect. Dis. 2019, 25, 980–983. [Google Scholar] [CrossRef]
- Ng, O.T.; Lee, V.; Marimuthu, K.; Vasoo, S.; Chan, G.; Lin, R.T.P.; Leo, Y.S. A case of imported Monkeypox in Singapore. Lancet Infect. Dis. 2019, 19, 1166. [Google Scholar] [CrossRef]
- Joana, I.; Vítor, B.; Miguel, P.; Rita, F.; Daniel, S.; Alexandra, N.; João, D.S.; Maria, J.B.; Sofia, N.; Ana, P.; et al. First draft genome sequence of Monkeypox virus associated with the suspected multi-country outbreak, May 2022 (confirmed case in Portugal). Eurosurveillance 2022, 27, 2200424. [Google Scholar]
- Petersen, E.; Kantele, A.; Koopmans, M.; Asogun, D.; Yinka-Ogunleye, A.; Ihekweazu, C.; Zumla, A. Human Monkeypox: Epidemiologic and Clinical Characteristics, Diagnosis, and Prevention. Infect. Dis. Clin. N. Am. 2019, 33, 1027–1043. [Google Scholar] [CrossRef]
- Khodakevich, L.; Jezek, Z.; Kinzanzka, K. Isolation of monkeypox virus from wild squirrel infected in nature. Lancet 1986, 1, 98–99. [Google Scholar] [CrossRef]
- Radonic, A.; Metzger, S.; Dabrowski, P.W.; Couacy-Hymann, E.; Schuenadel, L.; Kurth, A.; Matz-Rensing, K.; Boesch, C.; Leendertz, F.H.; Nitsche, A. Fatal monkeypox in wild-living sooty mangabey, Cote d’Ivoire, 2012. Emerg. Infect. Dis. 2014, 20, 1009–1011. [Google Scholar] [CrossRef]
- Patrono, L.V.; Pléh, K.; Samuni, L.; Ulrich, M.; Röthemeier, C.; Sachse, A.; Muschter, S.; Nitsche, A.; Couacy-Hymann, E.; Boesch, C.; et al. Monkeypox virus emergence in wild chimpanzees reveals distinct clinical outcomes and viral diversity. Nat. Microbiol. 2020, 5, 955–965. [Google Scholar] [CrossRef]
- Mariën, J.; Laudisoit, A.; Patrono, L.; Baelo, P.; Vredendaal, R.V.; Musaba, P.; Gembu, G.; Mande, C.; Ngoy, S.; Mussaw, M.; et al. Monkeypox viruses circulate in distantly-related small mammal species in the Democratic Republic of the Congo. Res. Sq. 2021; preprint. [Google Scholar] [CrossRef]
- Doty, J.B.; Malekani, J.M.; Kalemba, L.N.; Stanley, W.T.; Monroe, B.P.; Nakazawa, Y.U.; Mauldin, M.R.; Bakambana, T.L.; Liyandja Dja Liyandja, T.; Braden, Z.H.; et al. Assessing Monkeypox Virus Prevalence in Small Mammals at the Human-Animal Interface in the Democratic Republic of the Congo. Viruses 2017, 9, 283. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.; Leggat, P.A. Human Monkeypox: Current State of Knowledge and Implications for the Future. Trop. Med. Infect. Dis. 2016, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Olson, V.A.; Laue, T.; Laker, M.T.; Damon, I.K. Detection of monkeypox virus with real-time PCR assays. J. Clin. Virol. 2006, 36, 194–203. [Google Scholar] [CrossRef]
- Mojica, F.J.M.; Díez-Villaseñor, C.; García-Martínez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a target binding unleashes indiscriminate singlestranded DNase activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Xie, S.; Tian, R.; Wang, S.; Lu, Q.; Gao, D.; Lei, C.; Zhu, H.; Nie, Z. A CRISPR-Cas autocatalysis-driven feedback amplification network for supersensitive DNA diagnostics. Sci. Adv. 2021, 7, eabc7802. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Li, Z.; Wang, W.; Liu, J.; Liu, L.; Zhu, G.; Karthik, L.; Wang, M.; Wang, K.F.; Wang, Z.; et al. A CRISPR-Cas12a-derived biosensing platform for the highly sensitive detection of diverse small molecules. Nat. Commun. 2019, 10, 3672. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Yin, L.; Li, X.; Chen, S.; Peng, L.; Liu, G.; Ye, S.; Zhang, W.; Man, S. A smartphone-based visual biosensor for CRISPR-Cas powered SARS-CoV-2 diagnostics. Biosens. Bioelectron. 2022, 195, 113646. [Google Scholar] [CrossRef]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef]
- Yan, W.X.; Hunnewell, P.; Alfonse, L.E.; Carte, J.M.; Keston-Smith, E.; Sothiselvam, S.; Garrity, A.J.; Chong, S.; Makarova, K.S.; Koonin, E.V.; et al. Functionally diverse type V CRISPR-Cas systems. Science 2019, 363, 88–91. [Google Scholar] [CrossRef]
- Piepenburg, O.; Williams, C.H.; Stemple, D.L.; Armes, N.A. DNA detection using recombination proteins. PLoS Biol. 2006, 4, e204. [Google Scholar] [CrossRef]
- Cossio, A.; Jojoa, J.; Castro, M.D.M.; Castillo, R.M.; Osorio, L.; Shelite, T.R.; Gore Saravia, N.; Melby, P.C.; Travi, B.L. Diagnostic performance of a Recombinant Polymerase Amplification Test-Lateral Flow (RPA-LF) for cutaneous leishmaniasis in an endemic setting of Colombia. PLoS Negl. Trop. Dis. 2021, 15, e0009291. [Google Scholar] [CrossRef]
- Crannell, Z.; Castellanos-Gonzalez, A.; Nair, G.; Mejia, R.; White, A.C.; Richards-Kortum, R. Multiplexed Recombinase Polymerase Amplification Assay to Detect Intestinal Protozoa. Anal. Chem. 2016, 88, 1610–1616. [Google Scholar] [CrossRef]
- Shelite, T.R.; Uscanga-Palomeque, A.C.; Castellanos-Gonzalez, A.; Melby, P.C.; Travi, B.L. Isothermal recombinase polymerase amplification-lateral flow detection of SARS-CoV-2, the etiological agent of COVID-19. J. Virol. Methods 2021, 296, 114227. [Google Scholar] [CrossRef]
- Saluzzo, J.F.; Gonzalez, J.P.; Hervé, J.P.; Georges, A.J. Epidemiological study of arboviruses in the Central African Republic: Demonstration of Chikungunya virus during 1978 and 1979. Bull. Soc. Pathol. Exot. Fil. 1980, 73, 390–399. [Google Scholar]
- Li, Y.; Zhao, H.; Wilkins, K.; Hughes, C.; Damon, I.K. Real-time PCR assays for the specific detection of monkeypox virus West African and Congo Basin strain DNA. J. Virol. Methods 2010, 169, 223–227. [Google Scholar] [CrossRef] [PubMed]
- McCollum, A.M.; Damon, I.K. Human monkeypox. Clin. Infect. Dis. 2014, 58, 260–267. [Google Scholar] [CrossRef]
- Vaughan, A.; Aarons, E.; Astbury, J.; Brooks, T.; Chand, M.; Flegg, P.; Hardman, A.; Harper, N.; Jarvis, R.; Mawdsley, S.; et al. Human-to-Human Transmission of Monkeypox Virus, United Kingdom, October 2018. Emerg. Infect. Dis. 2020, 26, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Fleischauer, A.T.; Kile, J.C.; Davidson, M.; Fischer, M.; Karem, K.L.; Teclaw, R.; Messersmith, H.; Pontones, P.; Beard, B.A.; Braden, Z.H.; et al. Evaluation of human-to-human transmission of monkeypox from infected patients to health care workers. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2005, 40, 689–694. [Google Scholar] [CrossRef] [PubMed]
- Yinka-Ogunleye, A.; Aruna, O.; Dalhat, M.; Ogoina, D.; McCollum, A.; Disu, Y.; Mamadu, I.; Akinpelu, A.; Ahmad, A.; Burga, J.; et al. Outbreak of human monkeypox in Nigeria in 2017-18: A clinical and epidemiological report. Lancet Infect. Dis. 2019, 19, 872–879. [Google Scholar] [CrossRef]
- WHO. WHO Director-General’s Statement at the Press Conference Following IHR Emergency Committee Regarding the Multi-Country Outbreak of Monkeypox. Available online: https://www.who.int/news-room/speeches/item/who-director-general-s-statement-on-the-press-conference-following-IHR-emergency-committee-regarding-the-multi--country-outbreak-of-monkeypox--23-july-2022 (accessed on 25 July 2022).
- Antinori, A.; Mazzotta, V.; Vita, S.; Carletti, F.; Tacconi, D.; Lapini, L.E.; D’Abramo, A.; Cicalini, S.; Lapa, D.; Pittalis, S.; et al. Epidemiological, clinical and virological characteristics of four cases of monkeypox support transmission through sexual contact, Italy, May 2022. Eurosurveillance 2022, 27, 2200421. [Google Scholar] [CrossRef] [PubMed]
- Ajmera, K.M.; Goyal, L.; Pandit, T.; Pandit, R. Monkeypox- An Emerging Pandemic. IDCases 2022, 29, e01587. [Google Scholar] [CrossRef]
- Jezek, Z.; Szczeniowski, M.; Paluku, K.M.; Mutombo, M.; Grab, B. Human monkeypox: Confusion with chickenpox. Acta Trop. 1988, 45, 297–307. [Google Scholar]
- MacNeil, A.; Reynolds, M.G.; Carroll, D.S.; Karem, K.; Braden, Z.; Lash, R.; Moundeli, A.; Mombouli, J.-V.; Jumaan, A.O.; Schmid, D.S.; et al. Monkeypox or varicella? Lessons from a rash outbreak investigation in the Republic of the Congo. Am. J. Trop. Med. Hyg. 2009, 80, 503–507. [Google Scholar] [CrossRef]
- Mande, G.; Akonda, I.; De Weggheleire, A.; Brosius, I.; Liesenborghs, L.; Bottieau, E.; Ross, N.; Gembu, G.-C.; Colebunders, R.; Verheyen, E.; et al. Enhanced surveillance of monkeypox in Bas-Uélé, Democratic Republic of Congo: The limitations of symptom-based case definitions. Int. J. Infect. Dis. 2022, 122, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.M. Antiviral therapy for varicella and herpes zoster. Semin. Pediatr. Infect. Dis. 2002, 13, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.; McCollum, A.M.; Radford, K.; Hughes, C.; Lopez, A.S.; Guagliardo, S.A.J.; Nguete, B.; Likafi, T.; Kabamba, J.; Malekani, J.; et al. Varicella in Tshuapa Province, Democratic Republic of Congo, 2009-2014. Trop. Med. Int. Health 2019, 24, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Yu, L.; Liu, C.; Ye, S.; Chen, W.; Li, D.; Huang, W. One-tube SARS-CoV-2 detection platform based on RT-RPA and CRISPR/Cas12a. J. Transl. Med. 2021, 19, 74. [Google Scholar] [CrossRef] [PubMed]
- Lobato, I.M.; O’Sullivan, C.K. Recombinase polymerase amplification: Basics, applications and recent advances. Trends Analyt. Chem. 2018, 98, 19–35. [Google Scholar] [CrossRef]
- Iizuka, I.; Saijo, M.; Shiota, T.; Ami, Y.; Suzaki, Y.; Nagata, N.; Hasegawa, H.; Sakai, K.; Fukushi, S.; Mizutani, T.; et al. Loop-mediated isothermal amplification-based diagnostic assay for monkeypox virus infections. J. Med. Virol. 2009, 81, 1102–1108. [Google Scholar] [CrossRef]
- Davi, S.D.; Kissenkotter, J.; Faye, M.; Bohlken-Fascher, S.; Stahl-Hennig, C.; Faye, O.; Faye, O.; Sall, A.A.; Weidmann, M.; Ademowo, O.G.; et al. Recombinase polymerase amplification assay for rapid detection of Monkeypox virus. Diagn Microbiol. Infect. Dis. 2019, 95, 41–45. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Name | Sequence (5′ to 3′) | Assays |
|---|---|---|
| MPXV-RPA-F | ATCCAATGGAAAATGTAAAGACAACGAATACAG | Real-time RPA, RPA-Cas12a, RAA-LFS |
| MPXV-RPA-R | TCGTGTTACACGATCGCGTCTCTACCTGATTA | Real-time RPA, RPA-Cas12a |
| MPXV-exo-Probe | GTGATAGCAAGACTAATACACAATGTACGCCGFHQGGTTCGGATACCTTT-BLOCK | Real-time RPA |
| MPXV-nfo-Probe | 6-FAM-GTGATAGCAAGACTAATACACAATGTACGCCGTHTGGTTCGGATACCTTT-BLOCK | RAA-LFS |
| MPXV-nfo-R | Biotin-TCGTGTTACACGATCGCGTCTCTACCTGATTA | RAA-LFS |
| MPXV-PCR-F | GGAAAATGTAAAGACAACGAATACAG | Real-time PCR |
| MPXV-PCR-R | GCTATCACATAATCTGGAAGCGTA | Real-time PCR |
| MPXV-PCR-Probe | AAGCCGTAATCTATGTTGTCTATCGTGTCC | Real-time PCR |
| Sample ID | Sample Type | Assay Results | |||
|---|---|---|---|---|---|
| Real-Time PCR (Average Ct for Positive Results) | Real-Time RPA | RPA-Cas12a | RAA-LFS | ||
| 1 | Pus | Negative | - | - | - |
| 2 | Crusts | 37.95451 (4/6) | + (4/6) | − (2/6) | + (2/3) |
| 3 | Mice brain biopsy (from crusts) | 20.68634 | + | + | + |
| 4 | Pus | 33.41898 | + | + | + |
| 5 | Crusts | 26.33024 | + | + | + |
| 6 | Mice brain biopsy (from crusts) | 25.58751 | + | + | + |
| 7 | Crusts | 31.04101 | + | + | + |
| 8 | Mice brain biopsy (from crusts) | 23.25146 | + | + | + |
| 9 | Pus | 22.62705 | + | + | + |
| 10 | Pus | 25.66506 | + | + | + |
| 11 | Pus | 30.93425 | + | + | + |
| 12 | Pus | 22.09036 | + | + | + |
| 13 | Pus | 24.65622 | + | + | + |
| 14 | Pus | 19.49808 | + | + | + |
| 15 | Pus | 23.24219 | + | + | + |
| 16 | Pus | 23.97732 | + | + | + |
| 17 | Serum | 31.4181 | + | + | + |
| 18 | Serum | 38.22707 (4/6) | + (5/6) | - | + (2/3) |
| 19 | Serum | 37.72934 (2/6) | + (5/6) | - | + (2/3) |
| NC | Negative | - | - | - | |
| VACV | Negative | - | - | - | |
| VZV | Crusts | Negative | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mao, L.; Ying, J.; Selekon, B.; Gonofio, E.; Wang, X.; Nakoune, E.; Wong, G.; Berthet, N. Development and Characterization of Recombinase-Based Isothermal Amplification Assays (RPA/RAA) for the Rapid Detection of Monkeypox Virus. Viruses 2022, 14, 2112. https://doi.org/10.3390/v14102112
Mao L, Ying J, Selekon B, Gonofio E, Wang X, Nakoune E, Wong G, Berthet N. Development and Characterization of Recombinase-Based Isothermal Amplification Assays (RPA/RAA) for the Rapid Detection of Monkeypox Virus. Viruses. 2022; 14(10):2112. https://doi.org/10.3390/v14102112
Chicago/Turabian StyleMao, Lingjing, Jiaxu Ying, Benjamin Selekon, Ella Gonofio, Xiaoxia Wang, Emmanuel Nakoune, Gary Wong, and Nicolas Berthet. 2022. "Development and Characterization of Recombinase-Based Isothermal Amplification Assays (RPA/RAA) for the Rapid Detection of Monkeypox Virus" Viruses 14, no. 10: 2112. https://doi.org/10.3390/v14102112