
Implications of the Immune Polymorphisms of the Host and the Genetic Variability of SARS-CoV-2 in the Development of COVID-19

 , and
, and
Abstract
:1. Introduction
2. Genetic Variability of SARS-CoV-2: Implications on Antigenic Drift
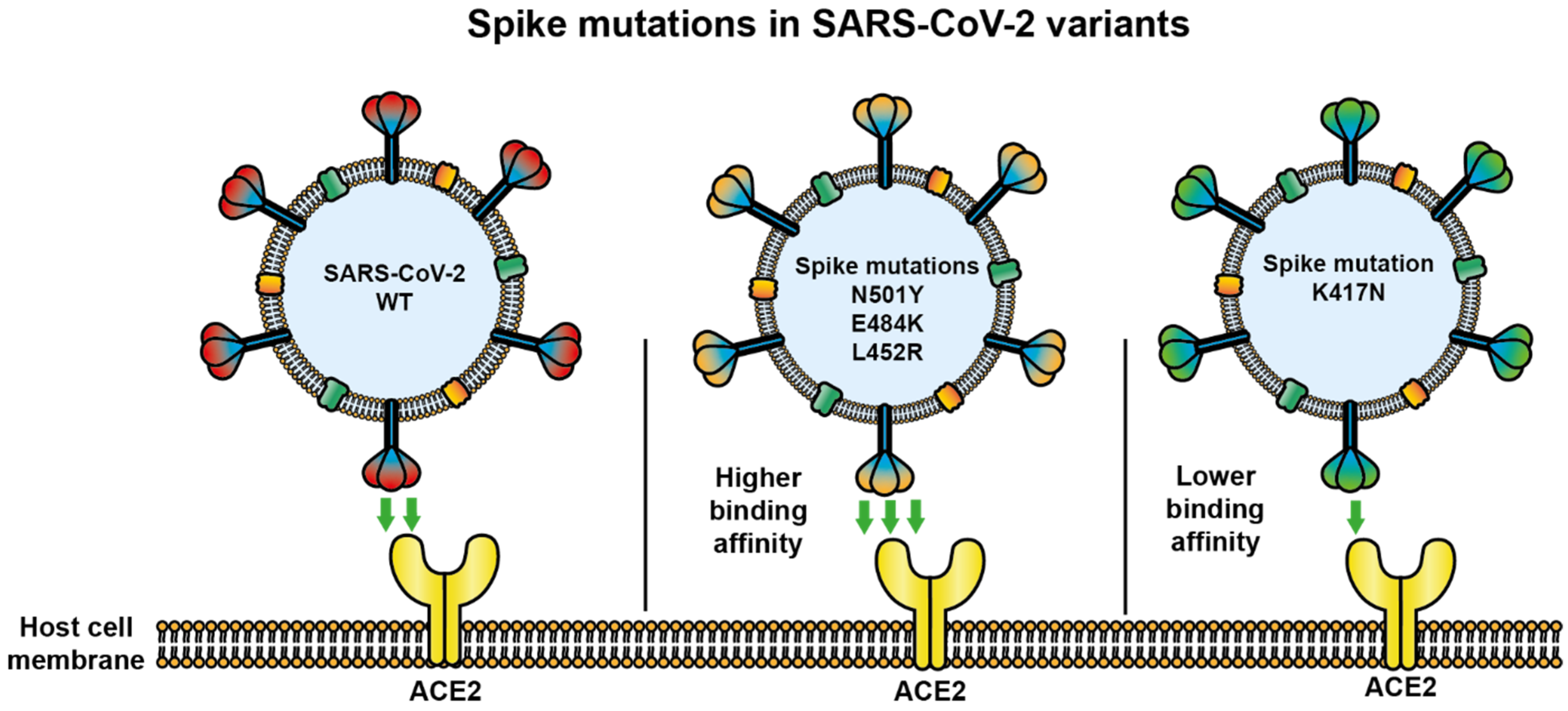
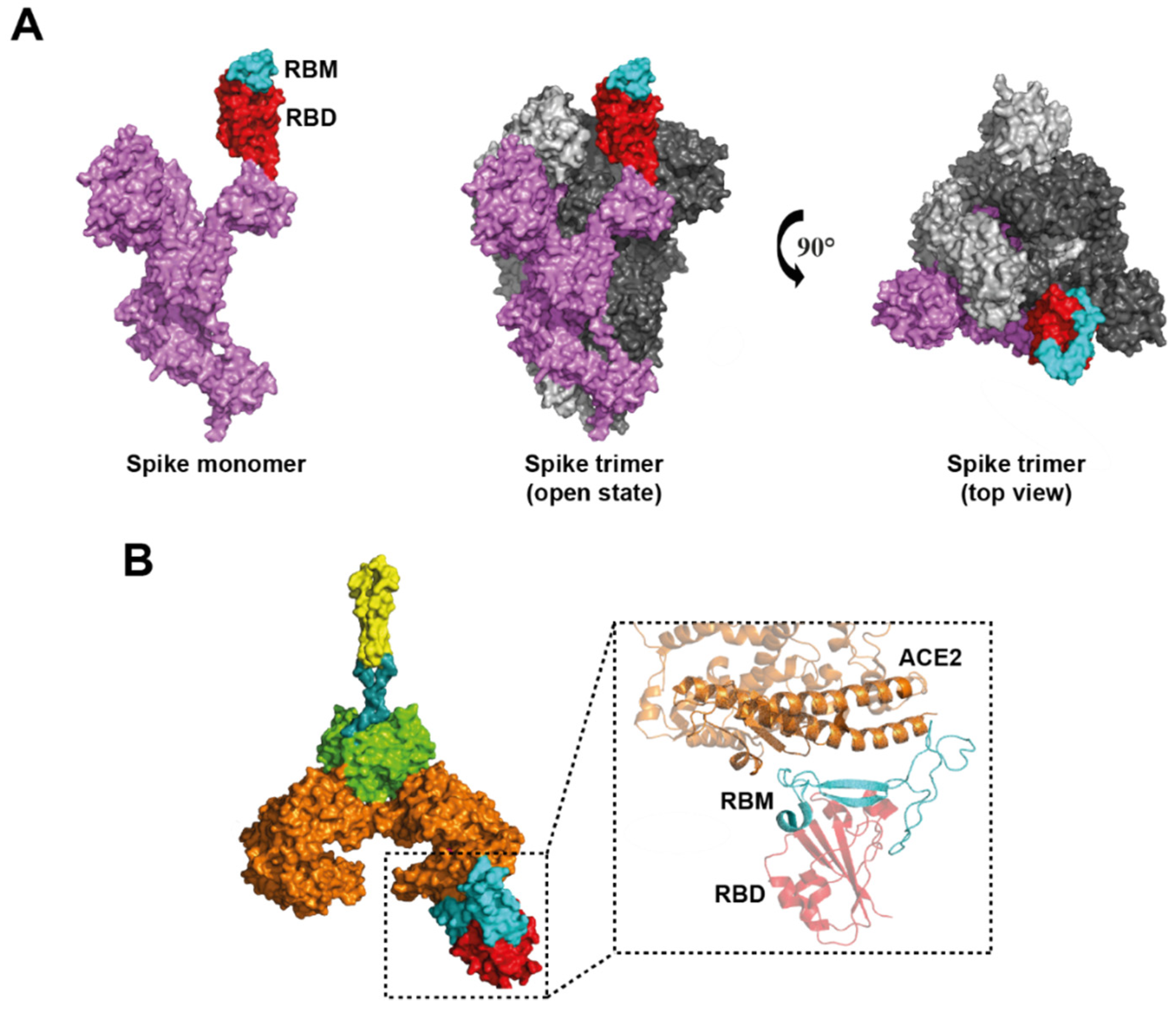
2.1. Mutations in S Glycoprotein
2.2. Mutations in Other SARS-CoV-2 Proteins
2.3. Antigenic Changes in SARS-CoV-2
3. SARS-CoV-2-Host Interaction
3.1. Transmission Mechanisms
3.2. Cells Permissive to SARS-CoV-2 Infection
3.3. SARS-CoV-2 Infection and Immunopathogenesis
4. ACE2 Polymorphisms in COVID-19
4.1. ACE-2: Global Structure
4.2. ACE2 of Animals and Cell Tropism
4.3. ACE2 Domains Involved in SARS-CoV-2 Infection
4.4. Is the ACE2 Variability Involved in COVID-19 Resistance?
4.5. ACE2 Regulation during Inflammation
5. Other Polymorphisms of Transmembrane Proteins, Cell Surface Molecules, or Enzymes Involved in COVID-19
5.1. TMPRSS2 and DC26 Polymorphisms
5.2. Tolloid Like-1 Polymorphism
5.3. Cathepsin B and L
5.4. ABO Polymorphism
6. Polymorphisms in Immune System Molecules
6.1. Cytokine Polymorphism
6.2. Polymorphisms in Tyrosine Kinases
6.3. Chemokine Polymorphisms
6.4. Polymorphisms in TLR and Their Signaling Molecules
6.5. HLA Polymorphism
6.6. Polymorphisms in Other Immune Proteins
7. Discussion
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Akkiz, H. Implications of the Novel Mutations in the SARS-CoV-2 Genome for Transmission, Disease Severity, and the Vaccine Development. Front. Med. 2021, 8, 636532. [Google Scholar] [CrossRef] [PubMed]
- Van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for 2019-nCoV based on SARS-CoV immunological studies. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shah, V.K.; Firmal, P.; Alam, A.; Ganguly, D.; Chattopadhyay, S. Overview of Immune Response During SARS-CoV-2 Infection: Lessons From the Past. Front. Immunol. 2020, 11, 1949. [Google Scholar] [CrossRef]
- Molaei, S.; Dadkhah, M.; Asghariazar, V.; Karami, C.; Safarzadeh, E. The immune response and immune evasion characteristics in SARS-CoV, MERS-CoV, and SARS-CoV-2: Vaccine design strategies. Int. Immunopharmacol. 2021, 92, 107051. [Google Scholar] [CrossRef] [PubMed]
- Kowalik, M.M.; Trzonkowski, P.; Łasińska-Kowara, M.; Mital, A.; Smiatacz, T.; Jaguszewski, M. COVID-19—Toward a comprehensive understanding of the disease. Cardiol. J. 2020, 27, 99–114. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Costagliola, G.; Spada, E.; Consolini, R. Age-related differences in the immune response could contribute to determine the spectrum of severity of COVID-19. Immun. Inflamm. Dis. 2021, 9, 331–339. [Google Scholar] [CrossRef]
- Sang, E.; Tian, Y.; Miller, L.; Sang, Y. Epigenetic Evolution of ACE2 and IL-6 Genes: Non-Canonical Interferon-Stimulated Genes Correlate to COVID-19 Susceptibility in Vertebrates. Genes 2021, 12, 154. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.W.; Xu, D.; Zhang, H.; Zhou, W.; Wang, L.-H.; Cui, X.G. Identification of a potential mechanism of acute kidney injury during the COVID-19 outbreak: A study based on single-cell transcriptome analysis. Intensive Care Med. 2020, 46, 1114–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashemi, S.M.A.; Thijssen, M.; Hosseini, S.Y.; Tabarraei, A.; Pourkarim, M.R.; Sarvari, J. Human gene polymorphisms and their possible impact on the clinical outcome of SARS-CoV-2 infection. Arch. Virol. 2021, 166, 2089–2108. [Google Scholar] [CrossRef] [PubMed]
- Dhangadamajhi, G.; Rout, R. Association of TLR3 functional variant (rs3775291) with COVID-19 susceptibility and death: A population-scale study. Hum. Cell 2021, 34, 1025–1027. [Google Scholar] [CrossRef]
- Grolmusz, V.K.; Bozsik, A.; Papp, J.; Patócs, A. Germline Genetic Variants of Viral Entry and Innate Immunity May Influence Susceptibility to SARS-CoV-2 Infection: Toward a Polygenic Risk Score for Risk Stratification. Front. Immunol. 2021, 12, 653489. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Povysil, G.; Butler-Laporte, G.; Shang, N.; Wang, C.; Khan, A.; Alaamery, M.; Nakanishi, T.; Zhou, S.; Forgetta, V.; Eveleigh, R.J.M.; et al. Rare loss-of-function variants in type I IFN immunity genes are not associated with severe COVID-19. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Agwa, S.; Kamel, M.; Elghazaly, H.; Elsamee, A.A.; Hafez, H.; Girgis, S.; Elarab, H.E.; Ebeid, F.; Sayed, S.; Sherif, L.; et al. Association between Interferon-Lambda-3 rs12979860, TLL1 rs17047200 and DDR1 rs4618569 Variant Polymorphisms with the Course and Outcome of SARS-CoV-2 Patients. Genes 2021, 12, 830. [Google Scholar] [CrossRef]
- Lam, S.D.; Bordin, N.; Waman, V.P.; Scholes, H.M.; Ashford, P.; Sen, N.; van Dorp, L.; Rauer, C.; Dawson, N.L.; Pang, C.S.M.; et al. SARS-CoV-2 spike protein predicted to form complexes with host receptor protein orthologues from a broad range of mammals. Sci. Rep. 2020, 10, 16471. [Google Scholar] [CrossRef]
- Sanjuán, R.; Domingo-Calap, P. Mechanisms of viral mutation. Cell. Mol. Life Sci. 2016, 73, 4433–4448. [Google Scholar] [CrossRef] [Green Version]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270–279. [Google Scholar] [CrossRef] [Green Version]
- Almubaid, Z.; Al-Mubaid, H. Analysis and comparison of genetic variants and mutations of the novel coronavirus SARS-CoV-2. Gene Rep. 2021, 23, 101064. [Google Scholar] [CrossRef]
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Kikkert, M. Innate Immune Evasion by Human Respiratory RNA Viruses. J. Innate Immun. 2020, 12, 4–20. [Google Scholar] [CrossRef] [PubMed]
- GISAID. Tracking of Variants. 2021. Available online: https://www.gisaid.org/hcov19-variants/ (accessed on 14 July 2021).
- CDC. SARS-CoV-2 Variant Classifications and Definitions. CDC. 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html%0Ahttps://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-info.html%0Ahttps://www.cdc.gov/coronavirus/2019-ncov/cases-updates/variant-surveillance/variant-in (accessed on 14 July 2021).
- Laffeber, C.; de Koning, K.; Kanaar, R.; Lebbink, J.H. Experimental Evidence for Enhanced Receptor Binding by Rapidly Spreading SARS-CoV-2 Variants. J. Mol. Biol. 2021, 433, 167058. [Google Scholar] [CrossRef] [PubMed]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Ascoli, C.A. Could mutations of SARS-CoV-2 suppress diagnostic detection? Nat. Biotechnol. 2021, 39, 274–275. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Lam, E.C.; Denis, K.S.; Nitido, A.D.; Garcia, Z.H.; Hauser, B.M.; Feldman, J.; Pavlovic, M.N.; Gregory, D.J.; Poznansky, M.C.; et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell 2021, 184, 2372–2383.e9. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Daniloski, Z.; Jordan, T.X.; Ilmain, J.K.; Guo, X.; Bhabha, G.; Tenoever, B.R.; Sanjana, E.N. The Spike D614G mutation increases SARS-CoV-2 infection of multiple human cell types. eLife 2021, 10, 65365. [Google Scholar] [CrossRef]
- Hu, J.; He, C.-L.; Gao, Q.-Z.; Zhang, G.-J.; Cao, X.-X.; Long, Q.-X.; Deng, H.-J.; Huang, L.-Y.; Chen, J.; Wang, K.; et al. The D614G mutation of SARS-CoV-2 spike protein enhances viral infectivity and decreases neutralization sensitivity to individual convalescent sera. BioRxiv 2020. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Li, C.; Tian, X.; Jia, X.; Wan, J.; Lu, L.; Jiang, S.; Lan, F.; Lu, Y.; Wu, Y.; Ying, T. The impact of receptor-binding domain natural mutations on antibody recognition of SARS-CoV-2. Signal Transduct. Target. Ther. 2021, 6, 1–3. [Google Scholar] [CrossRef]
- Ali, F.; Kasry, A.; Amin, M. The new SARS-CoV-2 strain shows a stronger binding affinity to ACE2 due to N501Y mutant. Med. Drug Discov. 2021, 10, 100086. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Zhao, S.; Lou, J.; Chong, M.; Cao, L.; Zheng, H.; Chen, Z.; Chan, R.; Zee, B.; Chan, P.; Wang, M. Inferring the Association between the Risk of COVID-19 Case Fatality and N501Y Substitution in SARS-CoV-2. Viruses 2021, 13, 638. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.W.; Toovey, O.T.R.; Harvey, K.N.; Hui, D.D.S. Introduction of the South African SARS-CoV-2 variant 501Y.V2 into the UK. J. Infect. 2021, 82, e8–e10. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.-H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef] [PubMed]
- Baum, A.; Fulton, B.O.; Wloga, E.; Copin, R.; Pascal, K.E.; Russo, V.; Giordano, S.; Lanza, K.; Negron, N.; Ni, M.; et al. Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies. Science 2020, 369, 1014–1018. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.-M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef] [PubMed]
- Andreano, E.; Piccini, G.; Licastro, D.; Casalino, L.; Johnson, N.V.; Paciello, I.; Dal Monego, S.; Pantano, E.; Manganaro, N.; Manenti, A. SARS-CoV-2 escape in vitro from a highly neutralizing COVID-19 convalescent plasma. bioRxiv 2020. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Ikeda, T.; Saito, A.; Tan, T.S.; Ngare, I.; Nasser, H.; Kimura, I.; Uriu, K. An emerging SARS-CoV-2 mutant evading cellular immunity and increasing viral infectivity. BioRxiv 2021. [Google Scholar] [CrossRef]
- McCallum, M.; Bassi, J.; De Marco, A.; Chen, A.; Walls, A.C.; Di Iulio, J.; Tortorici, M.A.; Navarro, M.-J.; Silacci-Fregni, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Sarkar, R.; Mitra, S.; Lo, M.; Dutta, S.; Chawla-Sarkar, M. The Novel Coronavirus Enigma: Phylogeny and Analyses of Coevolving Mutations Among the SARS-CoV-2 Viruses Circulating in India. JMIR Bioinform. Biotechnol. 2020, 1, e20735. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Näsvall, S.J.; Chen, P.; Björk, G.R. The wobble hypothesis revisited: Uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 2007, 13, 2151–2164. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, F.K. The Proteins of Severe Acute Respiratory Syndrome Coronavirus-2 (SARS CoV-2 or n-COV19), the Cause of COVID-19. J. Protein Chem. 2020, 39, 198–216. [Google Scholar] [CrossRef]
- Saha, A.; Sharma, R.; Bhattacharya, M.; Sharma, G.; Lee, S. Since January 2020 Elsevier Has Created a COVID-19 Resource Centre with Free Information in English and Mandarin on the Novel Coronavirus COVID-19. The COVID-19 Resource Centre is Hosted on Elsevier Connect, the Company’s Public News and Information. January 2020. Available online: https://cdn.who.int/media/docs/default-source/whhd-2021/scientific-publications/2.jhi_5may2021.pdf?sfvrsn=6526a2a5_5 (accessed on 22 December 2021).
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petropoulos, F.; Makridakis, S. Forecasting the novel coronavirus COVID-19. PLoS ONE 2020, 15, e0231236. [Google Scholar] [CrossRef] [PubMed]
- Emameh, R.Z.; Nosrati, H.; Taheri, R.A. Combination of Biodata Mining and Computational Modelling in Identification and Characterization of ORF1ab Polyprotein of SARS-CoV-2 Isolated from Oronasopharynx of an Iranian Patient. Biol. Proced. Online 2020, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Randhawa, P.K.; Scanlon, K.; Rappaport, J.; Gupta, M.K. Modulation of Autophagy by SARS-CoV-2: A Potential Threat for Cardiovascular System. Front. Physiol. 2020, 11, 611275. [Google Scholar] [CrossRef]
- Benvenuto, D.; Angeletti, S.; Giovanetti, M.; Bianchi, M.; Pascarella, S.; Cauda, R.; Ciccozzi, M.; Cassone, A. Evolutionary analysis of SARS-CoV-2: How mutation of Non-Structural Protein 6 (NSP6) could affect viral autophagy. J. Infect. 2020, 81, e24–e27. [Google Scholar] [CrossRef]
- Li, X.; Zai, J.; Zhao, Q.; Nie, Q.; Li, Y.; Foley, B.T.; Chaillon, A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J. Med. Virol. 2020, 92, 602–611. [Google Scholar] [CrossRef]
- Mirza, M.U.; Froeyen, M. Structural elucidation of SARS-CoV-2 vital proteins: Computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J. Pharm. Anal. 2020, 10, 320–328. [Google Scholar] [CrossRef]
- Yuen, C.-K.; Lam, J.-Y.; Wong, W.-M.; Mak, L.-F.; Wang, X.; Chu, H.; Cai, J.-P.; Jin, D.-Y.; To, K.K.-W.; Chan, J.F.-W.; et al. SARS-CoV-2 nsp13, nsp14, nsp15 and orf6 function as potent interferon antagonists. Emerg. Microbes Infect. 2020, 9, 1418–1428. [Google Scholar] [CrossRef]
- Ugurel, O.M.; Ata, O.; Turgut-Balik, D. An updated analysis of variations in SARS-CoV-2 genome. Turk. J. Biol. 2020, 44, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 418–423. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huang, Z.; Chen, X.; He, S.; Zhou, Z.; Zhou, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA binding domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Genet. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Dhama, K.; Patel, S.K.; Sharun, K.; Pathak, M.; Tiwari, R.; Yatoo, M.I.; Malik, Y.S.; Sah, R.; Rabaan, A.A.; Panwar, P.K.; et al. SARS-CoV-2 jumping the species barrier: Zoonotic lessons from SARS, MERS and recent advances to combat this pandemic virus. Travel Med. Infect. Dis. 2020, 37, 101830. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.L.; Parrish, C.R.; Cobey, S.; Glass, G.E.; Bush, R.M.; Leighton, T.J. Anticipating the Species Jump: Surveillance for Emerging Viral Threats. Zoonoses Public Health 2011, 59, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Baxi, P.; Saxena, S.K. Emergence and Reemergence of Severe Acute Respiratory Syndrome (SARS) Coronaviruses; Springer: Singapore, 2020; pp. 151–163. [Google Scholar] [CrossRef]
- Brun, J.; Vasiljevic, S.; Gangadharan, B.; Hensen, M.; Chandran, A.V.; Hill, M.L.; Kiappes, J.; Dwek, R.A.; Alonzi, D.S.; Struwe, W.B.; et al. Assessing Antigen Structural Integrity through Glycosylation Analysis of the SARS-CoV-2 Viral Spike. ACS Cent. Sci. 2021, 7, 586–593. [Google Scholar] [CrossRef]
- Zheng, L.; Ma, Y.; Chen, M.; Wu, G.; Yan, C.; Zhang, X.-E. SARS-CoV-2 spike protein receptor-binding domain N-glycans facilitate viral internalization in respiratory epithelial cells. Biochem. Biophys. Res. Commun. 2021, 579, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Choi, Y.K.; Frank, M.; Woo, H.; Park, S.-J.; Yeom, M.S.; Seok, C.; Im, W. Dynamic Interactions of Fully Glycosylated SARS-CoV-2 Spike Protein with Various Antibodies. J. Chem. Theory Comput. 2021, 17, 6559–6569. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, W.; Mao, Y.; Chen, Y.; Wang, S.; Zhong, Y.; Su, T.; Gong, M.; Du, D.; Lu, X.; et al. Site-specific N-glycosylation Characterization of Recombinant SARS-CoV-2 Spike Proteins. Mol. Cell. Proteom. 2021, 20, 100058. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, W.; Mao, Y.; Chen, Y.; Zheng, S.; Cao, W.; Zhu, J.; Hu, L.; Gong, M.; Cheng, J.; et al. O-Glycosylation Landscapes of SARS-CoV-2 Spike Proteins. Front. Chem. 2021, 9, 689521. [Google Scholar] [CrossRef] [PubMed]
- Rahnama, S.; Irani, M.A.; Amininasab, M.; Ejtehadi, M.R. S494 O-glycosylation site on the SARS-CoV-2 RBD affects the virus affinity to ACE2 and its infectivity; a molecular dynamics study. Sci. Rep. 2021, 11, 15162. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, M.; Su, J.; Yao, X.; Luo, H. FURIN correlated with immune infiltration serves as a potential biomarker in SARS-CoV-2 infection-related lung adenocarcinoma. Clin. Exp. Med. 2021, 1–14. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Zhang, L.; Mann, M.; Syed, Z.; Reynolds, H.M.; Tian, E.; Samara, N.L.; Zeldin, D.C.; Tabak, L.A.; Ten Hagen, K.G. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Guo, S.; Liu, K.; Zheng, J. The Genetic Variant of SARS-CoV-2: Would it matter for Controlling the Devastating Pandemic? Int. J. Biol. Sci. 2021, 17, 1476–1485. [Google Scholar] [CrossRef]
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Nechipurenko, Y.D.; Anashkina, A.A.; Matveeva, O.V. Change of Antigenic Determinants of SARS-CoV-2 Virus S-Protein as a Possible Cause of Antibody-Dependent Enhancement of Virus Infection and Cytokine Storm. Biophysics 2020, 65, 703–709. [Google Scholar] [CrossRef]
- Thomson, E.C.; Rosen, L.E.; Shepherd, J.G.; Spreafico, R.; Filipe, A.D.S.; Wojcechowskyj, J.A.; Davis, C.; Piccoli, L.; Pascall, D.J.; Dillen, J.; et al. Circulating SARS-CoV-2 spike N439K variants maintain fitness while evading antibody-mediated immunity. Cell 2021, 184, 1171–1187.e20. [Google Scholar] [CrossRef]
- Kemp, S.A.; Harvey, W.T.; Datir, R.P.; Collier, D.A.; Ferreira, I.A.; Carabelli, A.M.; Gupta, R.K.; Meng, B. Recurrent emergence and transmission of a SARS-CoV-2 spike deletion H69/V70. bioRxiv 2021. [Google Scholar] [CrossRef]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, K.R.; Rennick, L.J.; Nambulli, S.; Robinson-McCarthy, L.R.; Bain, W.G.; Haidar, G.; Duprex, W.P. Recurrent deletions in the SARS-CoV-2 spike glycoprotein drive antibody escape. Science 2021, 371, 1139–1142. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Becavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-Lopez, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Yu, F.; Yan, L.; Wang, N.; Yang, S.; Wang, L.; Tang, Y.; Gao, G.; Wang, S.; Ma, C.; Xie, R.; et al. Quantitative Detection and Viral Load Analysis of SARS-CoV-2 in Infected Patients. Clin. Infect. Dis. 2020, 71, 793–798. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.E.; Li, Z.; Chiew, C.J.; Yong, S.E.; Toh, M.P.; Lee, V.J. Presymptomatic Transmission of SARS-CoV-2-Singapore. Morb. Mortal. Wkly. Rep. 2020, 69, 411–415. [Google Scholar] [CrossRef] [Green Version]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Wang, W.; Liu, Z.; Liang, C.; Wang, W.; Ye, F.; Huang, B.; Zhao, L.; Wang, H.; Zhou, W.; et al. Morphogenesis and cytopathic effect of SARS-CoV-2 infection in human airway epithelial cells. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and Surface Stability of SARS-CoV-2 as Compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction Between Virus-Like Particles (VLPs) and Pattern Recognition Receptors (PRRs) From Dendritic Cells (DCs): Toward Better Engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef]
- Martínez-Flores, D.; Zepeda-Cervantes, J.; Cruz-Reséndiz, A.; Aguirre-Sampieri, S.; Sampieri, A.; Vaca, L. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front. Immunol. 2021, 12, 701501. [Google Scholar] [CrossRef]
- Wong, S.H.; Lui, R.N.; Sung, J.J. COVID-19 and the digestive system. J. Gastroenterol. Hepatol. 2020, 35, 744–748. [Google Scholar] [CrossRef]
- Liu, J.; Li, Y.; Liu, Q.; Yao, Q.; Wang, X.; Zhang, H.; Chen, R.; Ren, L.; Min, J.; Deng, F.; et al. SARS-CoV-2 cell tropism and multiorgan infection. Cell Discov. 2021, 7, 1–4. [Google Scholar] [CrossRef]
- Zhang, B.-Z.; Chu, H.; Han, S.; Shuai, H.; Deng, J.; Hu, Y.-F.; Gong, H.-R.; Lee, A.C.-Y.; Zou, Z.; Yau, T.; et al. SARS-CoV-2 infects human neural progenitor cells and brain organoids. Cell Res. 2020, 30, 928–931. [Google Scholar] [CrossRef]
- Tang, Y.; Liu, J.; Zhang, D.; Xu, Z.; Ji, J.; Wen, C. Cytokine Storm in COVID-19: The Current Evidence and Treatment Strategies. Front. Immunol. 2020, 11, 1708. [Google Scholar] [CrossRef]
- Chen, L.; Liu, H.G.; Liu, W.; Liu, J.; Liu, K.; Shang, J.; Deng, Y.; Wei, S. Analysis of clinical features of 29 patients with 2019 novel coronavirus pneumonia. Chin. J. Tuberc. Respir. Dis. 2020, 43, e005. [Google Scholar] [CrossRef]
- Konig, M.F.; Powell, M.A.; Staedtke, V.; Bai, R.-Y.; Thomas, D.L.; Fischer, N.M.; Huq, S.; Khalafallah, A.M.; Koenecke, A.; Xiong, R.; et al. Preventing cytokine storm syndrome in COVID-19 using α-1 adrenergic receptor antagonists. J. Clin. Investig. 2020, 130, 3345–3347. [Google Scholar] [CrossRef]
- Napoli, C.; Tritto, I.; Mansueto, G.; Coscioni, E.; Ambrosio, G. Immunosenescence exacerbates the COVID-19. Arch. Gerontol. Geriatr. 2020, 90, 104174. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, L.H. Cytokine storm and the prospects for immunotherapy with COVID-19. Clevel. Clin. J. Med. 2020, 87, 389–393. [Google Scholar] [CrossRef]
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The cytokine storm of COVID-19: A spotlight on prevention and protection. Expert Opin. Ther. Targets 2020, 24, 723–730. [Google Scholar] [CrossRef]
- Castelli, V.; Cimini, A.; Ferri, C. Cytokine Storm in COVID-19: “When You Come Out of the Storm, You Won’t Be the Same Person Who Walked in”. Front. Immunol. 2020, 11, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Tipnis, S.R.; Hooper, N.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [Green Version]
- Tikellis, C.; Thomas, M. Angiotensin-Converting Enzyme 2 (ACE2) Is a Key Modulator of the Renin Angiotensin System in Health and Disease. Int. J. Pept. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guy, J.; Lambert, D.; Warner, F.; Hooper, N.; Turner, A. Membrane-associated zinc peptidase families: Comparing ACE and ACE2. Biochim. et Biophys. Acta (BBA) Proteins Proteom. 2005, 1751, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Saponaro, F.; Rutigliano, G.; Sestito, S.; Bandini, L.; Storti, B.; Bizzarri, R.; Zucchi, R. ACE2 in the Era of SARS-CoV-2: Controversies and Novel Perspectives. Front. Mol. Biosci. 2020, 7, 588618. [Google Scholar] [CrossRef] [PubMed]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Mehdipour, A.R.; Hummer, G. Dual nature of human ACE2 glycosylation in binding to SARS-CoV-2 spike. Proc. Natl. Acad. Sci. USA 2021, 118, 2100425118. [Google Scholar] [CrossRef]
- Sang, E.R.; Tian, Y.; Gong, Y.; Miller, L.C.; Sang, Y. Integrate structural analysis, isoform diversity, and interferon-inductive propensity of ACE2 to predict SARS-CoV2 susceptibility in vertebrates. Heliyon 2020, 6, e04818. [Google Scholar] [CrossRef] [PubMed]
- Luan, J.; Lu, Y.; Jin, X.; Zhang, L. Spike protein recognition of mammalian ACE2 predicts the host range and an optimized ACE2 for SARS-CoV-2 infection. Biochem. Biophys. Res. Commun. 2020, 526, 165–169. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhao, Y.-B.; Wang, Q.; Li, J.-Y.; Zhou, Z.-J.; Liao, C.-H.; Ge, X.-Y. Predicting the angiotensin converting enzyme 2 (ACE2) utilizing capability as the receptor of SARS-CoV-2. Microbes Infect. 2020, 22, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Damas, J.; Hughes, G.M.; Keough, K.C.; Painter, C.A.; Persky, N.S.; Corbo, M.; Hiller, M.; Koepfli, K.-P.; Pfenning, A.R.; Zhao, H.; et al. Broad host range of SARS-CoV-2 predicted by comparative and structural analysis of ACE2 in vertebrates. Proc. Natl. Acad. Sci. USA 2020, 117, 22311–22322. [Google Scholar] [CrossRef]
- Oude Munnink, B.B.; Sikkema, R.S.; Nieuwenhuijse, D.F.; Molenaar, R.J.; Munger, E.; Molenkamp, R.; van der Spek, A.; Tolsma, P.; Rietveld, A.; Brouwer, M.; et al. Transmission of SARS-CoV-2 on mink farms between humans and mink and back to humans. Science 2020, 371, 172–177. [Google Scholar] [CrossRef]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halfmann, P.J.; Hatta, M.; Chiba, S.; Maemura, T.; Fan, S.; Takeda, M.; Kinoshita, N.; Hattori, S.-I.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; et al. Transmission of SARS-CoV-2 in Domestic Cats. N. Engl. J. Med. 2020, 383, 592–594. [Google Scholar] [CrossRef]
- Deng, J.; Jin, Y.; Liu, Y.; Sun, J.; Hao, L.; Bai, J.; Huang, T.; Lin, D.; Jin, Y.; Tian, K. Serological survey of SARS-CoV-2 for experimental, domestic, companion and wild animals excludes intermediate hosts of 35 different species of animals. Transbound. Emerg. Dis. 2020, 67, 1745–1749. [Google Scholar] [CrossRef] [PubMed]
- Cleary, S.J.; Pitchford, S.C.; Amison, R.T.; Carrington, R.; Cabrera, C.L.R.; Magnen, M.; Looney, M.R.; Gray, E.; Page, C.P. Animal models of mechanisms of SARS-CoV-2 infection and COVID-19 pathology. J. Cereb. Blood Flow Metab. 2020, 177, 4851–4865. [Google Scholar] [CrossRef] [PubMed]
- Patterson, E.I.; Elia, G.; Grassi, A.; Giordano, A.; Desario, C.; Medardo, M.; Smith, S.L.; Anderson, E.R.; Prince, T.; Patterson, G.T.; et al. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. Nat. Commun. 2020, 11, 6231. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, N.N.; Carossino, M.; Morozov, I.; Trujillo, J.D.; Meekins, D.A.; Madden, D.W.; Cool, K.; Artiaga, B.L.; McDowell, C.; Bold, D.; et al. Experimental re-infected cats do not transmit SARS-CoV-2. Emerg. Microbes Infect. 2021, 10, 638–650. [Google Scholar] [CrossRef]
- Mallapaty, S. COVID mink analysis shows mutations are not dangerous—Yet. Nature 2020, 587, 340–341. [Google Scholar] [CrossRef]
- Jiang, R.-D.; Liu, M.-Q.; Chen, Y.; Shan, C.; Zhou, Y.-W.; Shen, X.-R.; Li, Q.; Zhang, L.; Zhu, Y.; Si, H.-R.; et al. Pathogenesis of SARS-CoV-2 in Transgenic Mice Expressing Human Angiotensin-Converting Enzyme 2. Cell 2020, 182, 50–58.e8. [Google Scholar] [CrossRef]
- Stawiski, E.W.; Diwanji, D.; Suryamohan, K.; Gupta, R.; Fellouse, F.A.; Sathirapongsasuti, J.F.; Liu, J.; Jiang, Y.-P.; Ratan, A.; Mis, M.; et al. Human ACE2 receptor polymorphisms predict SARS-CoV-2 susceptibility. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Gracia-Ramos, A.E. Is the ACE2 Overexpression a Risk Factor for COVID-19 Infection? Arch. Med. Res. 2020, 51, 345–346. [Google Scholar] [CrossRef]
- Tang, X.; Yang, M.; Duan, Z.; Liao, Z.; Liu, L.; Cheng, R.; Fang, M.; Wang, G.; Liu, H.; Xu, J.; et al. Transferrin receptor is another receptor for SARS-CoV-2 entry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Loganathan, S.K.; Schleicher, K.; Malik, A.; Quevedo, R.; Langille, E.; Teng, K.; Oh, R.H.; Rathod, B.; Tsai, R.; Samavarchi-Tehrani, P.; et al. Rare driver mutations in head and neck squamous cell carcinomas converge on NOTCH signaling. Science 2020, 367, 1264–1269. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Ismail, A.M.; Elfiky, A.A. SARS-CoV-2 spike behavior in situ: A Cryo-EM images for a better understanding of the COVID-19 pandemic. Signal Transduct. Target. Ther. 2020, 5, 1–2. [Google Scholar] [CrossRef]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.-R.; Mottaghi, S.S.; Mehrabi, M.-R.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. 2021, 90, 104773. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.-P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Gibson, W.T.; Evans, D.M.; An, J.; Jones, S.J.M. ACE 2 Coding Variants: A Potential X-linked Risk Factor for COVID-19 Disease. bioRxiv 2020. [Google Scholar] [CrossRef]
- Srivastava, A.; Bandopadhyay, A.; Das, D.; Pandey, R.K.; Singh, V.; Khanam, N.; Srivastava, N.; Singh, P.P.; Dubey, P.K.; Pathak, A.; et al. Genetic Association of ACE2 rs2285666 Polymorphism With COVID-19 Spatial Distribution in India. Front. Genet. 2020, 11, 564741. [Google Scholar] [CrossRef] [PubMed]
- Mohaghegh, S.; Motie, P.; Motamedian, S.R. Role of ACE2 polymorphism in COVID-19: Impact of age. Clin. Chem. Lab. Med. 2021, 59, 1623–1627. [Google Scholar] [CrossRef] [PubMed]
- Beacon, M.T.H.; Delcuve, G.P.; Davie, J.R. Epigenetic regulation of ACE2, the receptor of the SARS-CoV-2 virus1. Genome 2021, 64, 386–399. [Google Scholar] [CrossRef]
- Chlamydas, S.; Papavassiliou, A.G.; Piperi, C. Epigenetic mechanisms regulating COVID-19 infection. Epigenetics 2021, 16, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Pinto, B.G.G.; Oliveira, A.E.R.; Singh, Y.; Jimenez, L.; Gonçalves, A.A.N.; Ogava, R.L.T.; Creighton, R.; Peron, J.P.S.; Nakaya, I.H. ACE2 Expression Is Increased in the Lungs of Patients with Comorbidities Associated with Severe COVID-19. J. Infect. Dis. 2020, 222, 556–563. [Google Scholar] [CrossRef]
- Fakhouri, E.W.; Peterson, S.J.; Kothari, J.; Alex, R.; Shapiro, J.I.; Abraham, N.G. Genetic Polymorphisms Complicate COVID-19 Therapy: Pivotal Role of HO-1 in Cytokine Storm. Antioxidants 2020, 9, 636. [Google Scholar] [CrossRef]
- Senapati, S.; Kumar, S.; Singh, A.K.; Banerjee, P.; Bhagavatula, S. Assessment of risk conferred by coding and regulatory variations of TMPRSS2 and CD26 in susceptibility to SARS-CoV-2 infection in human. J. Genet. 2020, 99, 1–5. [Google Scholar] [CrossRef]
- Li, Y.; Ke, Y.; Xia, X.; Wang, Y.; Cheng, F.; Liu, X.; Jin, X.; Li, B.; Xie, C.; Liu, S.; et al. Genome-wide association study of COVID-19 severity among the Chinese population. Cell Discov. 2021, 7, 1–16. [Google Scholar] [CrossRef]
- Paniri, A.; Hosseini, M.M.; Akhavan-Niaki, H. First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J. Biomol. Struct. Dyn. 2021, 39, 3576–3593. [Google Scholar] [CrossRef]
- Matsuura, K.; Sawai, H.; Ikeo, K.; Ogawa, S.; Iio, E.; Isogawa, M.; Shimada, N.; Komori, A.; Toyoda, H.; Kumada, T.; et al. Genome-Wide Association Study Identifies TLL1 Variant Associated With Development of Hepatocellular Carcinoma After Eradication of Hepatitis C Virus Infection. Gastroenterology 2017, 152, 1383–1394. [Google Scholar] [CrossRef] [Green Version]
- Iio, E.; Matsuura, K.; Shimada, N.; Atsukawa, M.; Itokawa, N.; Abe, H.; Kato, K.; Takaguchi, K.; Senoh, T.; Eguchi, Y.; et al. TLL1 variant associated with development of hepatocellular carcinoma after eradication of hepatitis C virus by interferon-free therapy. J. Gastroenterol. 2018, 54, 339–346. [Google Scholar] [CrossRef]
- Ganna, A. COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 1–8. [Google Scholar] [CrossRef]
- Lehrer, S.; Rheinstein, P.H. ABO blood groups, COVID-19 infection and mortality. Blood Cells Mol. Dis. 2021, 89, 102571. [Google Scholar] [CrossRef] [PubMed]
- Fricke-Galindo, I.; Falfán-Valencia, R. Genetics Insight for COVID-19 Susceptibility and Severity: A Review. Front. Immunol. 2021, 12, 1–11. [Google Scholar] [CrossRef]
- Henry, B.M. COVID-19, ECMO, and lymphopenia: A word of caution. Lancet Respir. Med. 2020, 8, e24. [Google Scholar] [CrossRef]
- Battagello, D.S.; Dragunas, G.; Klein, M.O.; Ayub, A.L.; Velloso, F.J.; Correa, R.G. Unpuzzling COVID-19: Tissue-related signaling pathways associated with SARS-CoV-2 infection and transmission. Clin. Sci. 2020, 134, 2137–2160. [Google Scholar] [CrossRef]
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, M.L.V.; Zanchettin, A.C.; de Paula, C.B.V.; Júnior, J.D.S.M.; Malaquias, M.A.S.; Raboni, S.M.; Neto, P.C.; Zeni, R.C.; Prokopenko, A.; Borges, N.H.; et al. Lung Neutrophilic Recruitment and IL-8/IL-17A Tissue Expression in COVID-19. Front. Immunol. 2021, 12, 656350. [Google Scholar] [CrossRef]
- Batur, L.K.; Hekim, N. Correlation between interleukin gene polymorphisms and current prevalence and mortality rates due to novel coronavirus disease 2019 (COVID-2019) in 23 countries. J. Med. Virol. 2021, 93, 5853–5863. [Google Scholar] [CrossRef]
- Paim, A.A.O.; Lopes-Ribeiro, A.; e Silva, D.S.D.; Andrade, L.A.F.; Moraes, T.F.; Barbosa-Stancioli, E.F.; da Fonseca, F.G.; Coelho-Dos-Reis, J.G. Will a little change do you good? A putative role of polymorphisms in COVID-19. Immunol. Lett. 2021, 235, 9–14. [Google Scholar] [CrossRef]
- Hemann, E.; Gale, M.J.; Savan, R. Interferon Lambda Genetics and Biology in Regulation of Viral Control. Front. Immunol. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Donnelly, R.P.; Kotenko, S.V. Interferon-Lambda: A New Addition to an Old Family. J. Interf. Cytokine Res. 2010, 30, 555–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syedbasha, M.; Egli, A. Interferon Lambda: Modulating Immunity in Infectious Diseases. Front. Immunol. 2017, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011205. [Google Scholar] [CrossRef] [Green Version]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Fukunaga-Kalabis, M.; Martinez, G.; Liu, Z.-J.; Kalabis, J.; Mrass, P.; Weninger, W.; Firth, S.M.; Planque, N.; Perbal, B.; Herlyn, M. CCN3 controls 3D spatial localization of melanocytes in the human skin through DDR1. J. Cell Biol. 2006, 175, 563–569. [Google Scholar] [CrossRef] [Green Version]
- Mehlotra, R.K. Chemokine receptor gene polymorphisms and COVID-19: Could knowledge gained from HIV/AIDS be important? Infect. Genet. Evol. 2020, 85, 104512. [Google Scholar] [CrossRef]
- Ray, P.R.; Wangzhou, A.; Ghneim, N.; Yousuf, M.S.; Paige, C.; Tavares-Ferreira, D.; Mwirigi, J.M.; Shiers, S.; Sankaranarayanan, I.; McFarland, A.J.; et al. A pharmacological interactome between COVID-19 patient samples and human sensory neurons reveals potential drivers of neurogenic pulmonary dysfunction. Brain Behav. Immun. 2020, 89, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; Seethamraju, H.; Dhody, K.; Corley, M.J.; Kazempour, K.; Lalezari, J.P.; Pang, A.P.; Sugai, C.; Francisco, E.B.; Pise, A.; et al. Disruption of the CCL5/RANTES-CCR5 Pathway Restores Immune Homeostasis and Reduces Plasma Viral Load in Critical COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Mummidi, S.; Ahuja, S.S.; McDaniel, B.L.; Ahuja, S.K. The Human CC Chemokine Receptor 5 (CCR5) Gene. J. Biol. Chem. 1997, 272, 30662–30671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, A.K.; Padhi, A.; Prusty, B.A.K. CCR5 Δ32 minorallele is associated with susceptibility to SARS-CoV-2 infection and death: An epidemiological investigation. Clin. Chim. Acta 2020, 510, 60–61. [Google Scholar] [CrossRef]
- Severe COVID-19 GWAS Group. Genomewide Association Study of Severe COVID-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Chemokines and chemokine receptors during COVID-19 infection. Comput. Struct. Biotechnol. J. 2021, 19, 976–988. [Google Scholar] [CrossRef]
- Conte, C. Possible Link between SARS-CoV-2 Infection and Parkinson’s Disease: The Role of Toll-Like Receptor 4. Int. J. Mol. Sci. 2021, 22, 7135. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Toll-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [Green Version]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis, E.; Sousa, C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [Green Version]
- Barton, G.M.; Medzhitov, R. Toll-Like Receptor Signaling Pathways. Science 2003, 300, 1524–1525. [Google Scholar] [CrossRef]
- Teimouri, H.; Maali, A. Pasteur Institute of Iran Single-Nucleotide Polymorphisms in Host Pattern-Recognition Receptors Show Association with Antiviral Responses against SARS-CoV-2, in-silico Trial. J. Med. Microbiol. Infect. Dis. 2020, 8, 65–70. [Google Scholar] [CrossRef]
- Croci, S.; Veneri, M.A.; Mantovani, S.; Fallerini, C.; Benetti, E.; Picchiotti, N.; Campolo, F.; Imperatore, F.; Palimeri, M.; Daga, S.; et al. The polymorphism L412F in TLR3 inhibits autophagy and is a marker of severe COVID-19 in males. mredRxiv 2021. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Bolze, A.; Jouanguy, E.; Zhang, S.-Y.; Cobat, A.; Notarangelo, L.D.; Su, H.C.; Abel, L.; Casanova, J.-L. Life-Threatening COVID-19: Defective Interferons Unleash Excessive Inflammation. Med 2020, 1, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Auto-antibodies against type I IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef]
- Lim, H.K.; Huang, S.X.; Chen, J.; Kerner, G.; Gilliaux, O.; Bastard, P.; Dobbs, K.; Hernandez, N.; Goudin, N.; Hasek, M.L.; et al. Severe influenza pneumonitis in children with inherited TLR3 deficiency. J. Exp. Med. 2019, 216, 2038–2056. [Google Scholar] [CrossRef]
- Thomsen, M.M.; Jørgensen, S.E.; Gad, H.H.; Storgaard, M.; Gjedsted, J.; Christiansen, M.; Hartmann, R.; Mogensen, T.H. Defective interferon priming and impaired antiviral responses in a patient with an IRF7 variant and severe influenza. Med. Microbiol. Immunol. 2019, 208, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.M.; Jørgensen, S.E.; Storgaard, M.; Kristensen, L.S.; Gjedsted, J.; Christiansen, M.; Gad, H.H.; Hartmann, R.; Mogensen, T.H. Identification of an IRF3 variant and defective antiviral interferon responses in a patient with severe influenza. Eur. J. Immunol. 2019, 49, 2111–2114. [Google Scholar] [CrossRef]
- Bhattacharya, M.; Sharma, A.R.; Mallick, B.; Sharma, G.; Lee, S.-S.; Chakraborty, C. Immunoinformatics approach to understand molecular interaction between multi-epitopic regions of SARS-CoV-2 spike-protein with TLR4/MD-2 complex. Infect. Genet. Evol. 2020, 85, 104587. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar] [CrossRef]
- Brandão, S.C.S.; de Oliveira XavierRamos, J.; Dompieri, L.T.; Godoi, E.T.A.M.; Figueiredo, J.L.; Sarinho, E.S.C.; Chelvanambi, S.; Aikawa, M. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2021, 58, 102–110. [Google Scholar] [CrossRef]
- Gubernatorova, E.; Gorshkova, E.; Polinova, A.; Drutskaya, M. IL-6: Relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020, 53, 13–24. [Google Scholar] [CrossRef]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell 2020, 181, 1016–1035.e19. [Google Scholar] [CrossRef]
- Khanolkar, A.; Hartwig, S.M.; Haag, B.A.; Meyerholz, D.K.; Harty, J.; Varga, S.M. Toll-Like Receptor 4 Deficiency Increases Disease and Mortality after Mouse Hepatitis Virus Type 1 Infection of Susceptible C3H Mice. J. Virol. 2009, 83, 8946–8956. [Google Scholar] [CrossRef] [Green Version]
- Puthothu, B.; Forster, J.; Heinzmann, A.; Krueger, M. TLR-4 andCD14 Polymorphisms in Respiratory Syncytial Virus Associated Disease. Dis. Markers 2006, 22, 303–308. [Google Scholar] [CrossRef] [Green Version]
- Van Der Made, C.I.; Simons, A.; Schuurs-Hoeijmakers, J.; Heuvel, G.V.D.; Mantere, T.; Kersten, S.; Van Deuren, R.C.; Steehouwer, M.; Van Reijmersdal, S.V.; Jaeger, M.; et al. Presence of Genetic Variants Among Young Men With Severe COVID-19. JAMA J. Am. Med. Assoc. 2020, 324, 663. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Yan, X.; Yuan, L. Human genetic basis of coronavirus disease 2019. Signal Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Khosroshahi, L.M.; Rezaei, N. Dysregulation of the immune response in coronavirus disease 2019. Cell Biol. Int. 2021, 45, 702–707. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, W.; Zhang, J.; He, J.; Zhu, F. Distribution of HLA allele frequencies in 82 Chinese individuals with coronavirus disease-2019 (COVID-19). HLA 2020, 96, 194–196. [Google Scholar] [CrossRef] [PubMed]
- Lorente, L.; Martín, M.; Franco, A.; Barrios, Y.; Cáceres, J.; Solé-Violán, J.; Perez, A.; Ramos, J.M.Y.; Ramos-Gómez, L.; Ojeda, N.; et al. HLA genetic polymorphisms and prognosis of patients with COVID-19. Med. Intensiv. 2021, 45, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Ikeda, T.; Sato, R.; Sakagami, T. Association between HLA gene polymorphisms and mortality of COVID-19: An in silico analysis. Immun. Inflamm. Dis. 2020, 8, 684–694. [Google Scholar] [CrossRef]
- Rahimi, A.; Mirzazadeh, A.; Tavakolpour, S. Genetics and genomics of SARS-CoV-2: A review of the literature with the special focus on genetic diversity and SARS-CoV-2 genome detection. Genomics 2021, 113, 1221–1232. [Google Scholar] [CrossRef]
- Nguyen, A.; David, J.K.; Maden, S.K.; Wood, M.A.; Weeder, B.R.; Nellore, A.; Thompson, R.F. Human Leukocyte Antigen Susceptibility Map for Severe Acute Respiratory Syndrome Coronavirus 2. J. Virol. 2020, 94, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debnath, M.; Banerjee, M.; Berk, M. Genetic gateways to COVID-19 infection: Implications for risk, severity, and outcomes. FASEB J. 2020, 34, 8787–8795. [Google Scholar] [CrossRef]
- González-Galarza, F.F.; Takeshita, L.Y.; Santos, E.J.; Kempson, F.; Maia, M.H.T.; Da Silva, A.L.S.; Silva, E.A.L.T.; Ghattaoraya, G.; Alfirevic, A.; Jones, A.; et al. Allele frequency net 2015 update: New features for HLA epitopes, KIR and disease and HLA adverse drug reaction associations. Nucleic Acids Res. 2015, 43, D784–D788. [Google Scholar] [CrossRef]
- Ovsyannikova, I.G.; Haralambieva, I.H.; Crooke, S.; Poland, G.A.; Kennedy, R.B. The role of host genetics in the immune response to SARS-CoV-2 and COVID-19 susceptibility and severity. Immunol. Rev. 2020, 296, 205–219. [Google Scholar] [CrossRef]
- Novelli, A.; Andreani, M.; Biancolella, M.; Liberatoscioli, L.; Passarelli, C.; Colona, V.L.; Rogliani, P.; Leonardis, F.; Campana, A.; Carsetti, R.; et al. HLA allele frequencies and susceptibility to COVID -19 in a group of 99 Italian patients. HLA 2020, 96, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Benlyamani, I.; Venet, F.; Coudereau, R.; Gossez, M.; Monneret, G. Monocyte HLA-DR Measurement by Flow Cytometry in COVID -19 Patients: An Interim Review. Cytom. Part A 2020, 97, 1217–1221. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, H.; Aizawa, N.; Hasegawa, K.; Ikeda, N.; Sakai, Y.; Yoh, K.; Takata, R.; Yuri, Y.; Kishino, K.; Shimono, Y.; et al. Possible Relevance of PNPLA3 and TLL1 Gene Polymorphisms to the Efficacy of PEG-IFN Therapy for HBV-Infected Patients. Int. J. Mol. Sci. 2020, 21, 3089. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, S.; Chen, Z.; Liu, L. Correlation between TICAM1 gene polymorphisms and community-acquired pneumonia in children. J. Biochem. Mol. Toxicol. 2020, 34, e22503. [Google Scholar] [CrossRef]
- Herman, M.; Ciancanelli, M.; Ou, Y.-H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; de Diego, R.P.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef] [Green Version]
- Zidi, I. Puzzling out the COVID-19: Therapy targeting HLA-G and HLA-E. Hum. Immunol. 2020, 81, 697–701. [Google Scholar] [CrossRef]
- Carosella, E.D.; Rouas-Freiss, N.; Le Roux, D.T.; Moreau, P.; LeMaoult, J. HLA-G. An Immune Checkpoint Molecule, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 127. [Google Scholar]
- Zhang, S.; Gan, J.; Chen, B.; Zheng, D.; Zhang, J.; Lin, R.; Zhou, Y.; Yang, W.; Lin, A.; Yan, W. Dynamics of peripheral immune cells and their HLA-G and receptor expressions in a patient suffering from critical COVID-19 pneumonia to convalescence. Clin. Transl. Immunol. 2020, 9, e1128. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, D.; Gentili, V.; Rizzo, S.; Rotola, A.; Rizzo, R. Cell Activation via the HLA-E/NKG2A Pathway. Cells 2020, 9, 1975. [Google Scholar] [CrossRef]
- Vietzen, H.; Zoufaly, A.; Traugott, M.; Aberle, J.; Aberle, S.W.; Puchhammer-Stöckl, E. Deletion of the NKG2C receptor encoding KLRC2 gene and HLA-E variants are risk factors for severe COVID-19. Genet. Med. 2021, 23, 963–967. [Google Scholar] [CrossRef]
- Corley, M.J.; Pang, A.P.; Dody, K.; Mudd, P.A.; Patterson, B.K.; Seethamraju, H.; Bram, Y.; Peluso, M.J.; Torres, L.; Iyer, N.S.; et al. Genome-wide DNA methylation profiling of peripheral blood reveals an epigenetic signature associated with severe COVID-19. J. Leukoc. Biol. 2021, 110, 21–26. [Google Scholar] [CrossRef]
- Geoghegan, J.; Holmes, E.C. The phylogenomics of evolving virus virulence. Nat. Rev. Genet. 2018, 19, 756–769. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Zhu, Y.; Qin, K.; Yu, Z.; Gao, R.; Yu, H.; Zhou, J.; Shu, Y. Mutations in Polymerase Genes Enhanced the Virulence of 2009 Pandemic H1N1 Influenza Virus in Mice. PLoS ONE 2012, 7, e33383. [Google Scholar] [CrossRef]
- Menachery, V.; Graham, R.L.; Baric, R.S. Jumping species—a mechanism for coronavirus persistence and survival. Curr. Opin. Virol. 2017, 23, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Iwata, N.; Hasegawa, H.; Fukushi, S.; Yokoyama, M.; Harashima, A.; Sato, Y.; Saijo, M.; Morikawa, S.; Sata, T. Participation of both Host and Virus Factors in Induction of Severe Acute Respiratory Syndrome (SARS) in F344 Rats Infected with SARS Coronavirus. J. Virol. 2007, 81, 1848–1857. [Google Scholar] [CrossRef] [Green Version]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L., Jr.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted model of SARS-CoV-2 to test COVID-19 countermeasures. Nature 2020, 586, 560–566. [Google Scholar] [CrossRef]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef]
- Lauro, R.; Irrera, N.; Eid, A.; Bitto, A. Could Antigen Presenting Cells Represent a Protective Element during SARS-CoV-2 Infection in Children? Pathogens 2021, 10, 476. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Gui, X.; Xiong, Y. Comparison of Clinical Characteristics of Patients with Asymptomatic vs Symptomatic Coronavirus Disease 2019 in Wuhan, China. JAMA Netw. Open 2020, 3, e2010182. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Lu, Z.; Zhang, L.; Fan, T.; Xiong, R.; Shen, X.; Feng, H.; Meng, H.; Lin, W.; Jiang, W.; et al. The clinical course and its correlated immune status in COVID-19 pneumonia. J. Clin. Virol. 2020, 127, 104361. [Google Scholar] [CrossRef]
- Sekine, T.; Perez-Potti, A.; Rivera-Ballesteros, O.; Strålin, K.; Gorin, J.-B.; Olsson, A.; Llewellyn-Lacey, S.; Kamal, H.; Bogdanovic, G.; Muschiol, S.; et al. Robust T Cell Immunity in Convalescent Individuals with Asymptomatic or Mild COVID-19. Cell 2020, 183, 158–168.e14. [Google Scholar] [CrossRef]
- Newton, J.L.; Harney, S.M.J.; Wordsworth, B.P.; Brown, M.A. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004, 5, 151–157. [Google Scholar] [CrossRef] [Green Version]
- Matzaraki, V.; Kumar, V.; Wijmenga, C.; Zhernakova, A. The MHC locus and genetic susceptibility to autoimmune and infectious diseases. Genome Biol. 2017, 18, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M.; Jamieson, S.E.; Burgner, D. HLA and Infectious Diseases. Clin. Microbiol. Rev. 2009, 22, 370–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luckey, D.; Weaver, E.A.; Osborne, D.G.; Billadeau, D.D.; Taneja, V. Immunity to Influenza is dependent on MHC II polymorphism: Study with 2 HLA transgenic strains. Sci. Rep. 2019, 9, 19061. [Google Scholar] [CrossRef] [Green Version]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Kannan, S.R.; Spratt, A.N.; Sharma, K.; Chand, H.S.; Byrareddy, S.N.; Singh, K. Omicron SARS-CoV-2 variant: Unique features and their impact on pre-existing antibodies. J. Autoimmun. 2021, 126, 102779. [Google Scholar] [CrossRef] [PubMed]
- Ingraham, N.E.; Ingbar, D.H. The omicron variant of SARS-CoV-2: Understanding the known and living with unknowns. Clin. Transl. Med. 2021, 11, e685. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SARS-CoV-2 Variants | Spike Mutations |
|---|---|
| B.1.1.7 | 69-HV-70 del, 144-Y del, N501Y, A570D, D614G, P681H, T716I, S982A, and D1118H |
| B.1.351 | L18F, D80A, D215A, 242-LAL-244 del, R246I, K417N, E484K, N501Y, D614G, and A701V |
| P.1 | L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, and T1027I |
| B.1.617 and sub-lineages | T19R, T95I, G142D, E154K, 156-157 del, R158G, L452R, T478K, E484Q, D614G, P681R, D950N, and Q1071H |
| Gene | Implications in COVID-19 | Susceptibility Polymorphisms | Resistance Polymorphisms |
|---|---|---|---|
| TMPRSS2 | A protease that cleaves glycoprotein S for its priming leading to virus entry. | rs112657409, rs11910678, rs77675406, and rs713400 variants can regulate the expression of TMPRSS2 and could be implicated in SARS-CoV-2 infection [151]. rs430915 allele A has been associated with overexpression of TMPRSS2 in lungs [152]. | No data reported. |
| CTSB (encoding for cathepsin C) | Cysteine protease priming glycoprotein S for viral entry. | rs10831496 is associated to severe COVID-19 [152]. | No data reported |
| KANSL1 | A nuclear protein involved in histone acetylation. | No data reported. | Variant rs1819040:T>A was associated with protection [156]. |
| ABO | Responsible for determining blood type. | rs912805253 variant was suggested as a risk factor for SARS-CoV-2 infection [156]. | No data reported. |
| ACE2 | The SARS-CoV-2 receptor: mediates viral attachment and membrane fusion. Under inflammation, ACE2 is overexpressed. Now, ACE2 is considered an IFN-stimulated gene. | E23K, H378R, I21V, K26R, N64K, Q102P, S19P, T27A, and T92I. K26R and T92I have increased affinity to SARS-CoV-2 glycoprotein S [132]. | D355N, D38V, D509Y E35K, E37K, F72V, G326E, G352V, H34R, K31R, K68E, M62V, N33I, N51S, Q388L Y50F, and Y83H. K31R and E37K have decreased affinity to SARS-CoV-2 S glycoprotein [132]. |
| FOXP4 | A forkhead transcription factor that regulates the specific transcription activity in cells. | Variant rs1886814:A>C is associated with the development of COVID-19 and interstitial lung disease [156]. | No data reported. |
| IL-10 | Related to immune tolerance and antibody response. | rs1800896 [163]. | No data reported. |
| IL-17 | Recruits neutrophils in response to a viral infection. | No data reported. | rs2275913 [163]. |
| IFN-λ | It has antiviral properties and can prompt the expression of IFN-stimulated genes. | rs12979860 (CC genotype) as well as C and A alleles [19]. | No data reported. |
| TLL1 | A metalloprotease implicated in the morphogenesis of the heart. This enzyme can also activate SARS-CoV-2 S glycoprotein. | rs17047200 (AA genotype) [19,212]. | No data reported. |
| DPP9 | A protease that cleaves CXCL10, an antiviral molecule. It is involved in inflammation and antigen presentation. | rs2109069 and rs12610495 are related to critical illness and interstitial lung disease, respectively [14,156,169]. | No data reported. |
| DDR1 | A tyrosine kinase receptor activated by collagen and involved in cytokine production, cell differentiation, and the modulation of adhesion molecules. | rs4618569 [19]. | No data reported. |
| CCR5 | CCR5 and its ligand CCL5 play an important role in the inflammatory response by recruiting leukocytes to eliminate infectious pathogens. | CCR5 Δ32 [171,175]. | No data reported. |
| CXCR6 | Allows homing of CD8+ T cells in the lungs. | rs11385942 [176]. | No data reported. |
| TYK2 | A member of Janus kinases protein families. It is associated with cytoplasmic domains of cytokine receptors prompting their signaling though phosphorylation. | rs74956615:T>A variant confers risk for COVID-19, whereas the missense variant rs34536443:G>C (also p.Pro1104Ala) has been correlated with risk of hospitalization (but it is protective against autoimmune diseases) [156]. | No data reported. |
| TLR-3 | Detects intermediate dsRNA during viral replication. | rs3775291 and rs3775290 [14,15,183,185]. Other variants are p.Ser339fs, p.Pro554Ser, p.Trp769*, and p.Met870Val [17]. In another work, the TLR-3 variant 12-56744928-GA was associated with mild COVID-19, while no association was found with the variants mentioned above [18]. | No data reported. |
| TLR-7 | Detects ssRNA from viruses prompting IFN production. | 4 young male patients were reported to have developed severe COVID-19. These patients were identified with loss-of-function variants of TLR-7, including a 4-nucleotide deletion (c.2129_2132del; p.(Gln710Argfs*18)) and a missense variant (c.2383G>T; p.(Val795Phe)) [152,198]. | No data reported. |
| IRF3 | As its name implies, it is an interferon regulatory transcription factor (IRF). IRF3 includes phosphorylation sites at its C-terminal, a DNA-binding domain, and a nuclear localization signal. | p.Glu49del and p.Asn146Lys variants [17]. | No data reported. |
| IRF7 | Interacts with IRF3, and together, they regulate the IFN-α genes. | p.Pro364fs/p.Pro364fs, p.Met371Val/p.Asp117Asn, p.Arg7fs, p.Gln185*, p.Pro246fs, p.Arg369Gln, and p.Phe95Ser variants [17]. | No data reported. |
| IFNAR1/IFNAR2 | A receptor found in the cell membrane, and it contains both IFNAR1 and IFNAR2. | p.Trp73Cys/Trp73Cys, p.Ser422Arg/Ser422Arg, and p.Pro335del variant from IFNAR1 as well as p.Glu140fs variant from IFNAR2 [17]. | No data reported. |
| TICAM1 | Also known as TLR adaptor molecule 1. Its function is to mediate the interaction between TLR-3 and signal transduction proteins activating NFκB. | p.Thr4Ile, p.Ser60Cys, and p.Gln392Lys variants [17]. Other TICAM1 variants have been related to pneumonia in Chinese children [213]. | No data reported. |
| TBK1 | It is a protein kinase that phosphorylates IRF3, causing its nuclear translocation to induce the transcription of type-1 IFN genes. | p.Phe24Ser and p.Arg308* [17]. TBK1 mutations are also found in children with encephalitis caused by herpesvirus infection [214]. | No data reported. |
| STAT2 | Signal transducer and activator of transcription 2 is associated with IRF9. Upon phosphorylation, STAT2 forms a multimeric complex, which binds to a specific DNA sequence to activate type-1 IFN genes. | STAT2 variant 12-56744928-GA has been associated with severe COVID-19 [18]. | No data reported. |
| HLA class I | A protein used for binding to processed peptides after antigen processing. HLA class I bound to SARS-CoV-2 epitopes to stimulate anti-SARS-CoV-2 CD8+ cells prompting lysis of infected cells. | HLA-A*11 HLA-C*01 HLA-C*07:29 HLA-B*15:27 HLA-B*46:01 HLA-B*07:03 HLA-Cw*08:01 HLA-B*46:01 HLA-A*24:02 [203,204,206,209] | HLA-B*15:03 HLA-Cw*15:02 HLA-A*02:01 [209] |
| HLA class II | Presents epitopes to CD4+ lymphocytes to enhance the cytotoxic effect of CD8+ T lymphocytes (Th1) or enhance antibody production (Th2). Some HLA polymorphisms have low binding capacity, predisposing to COVID-19. | HLA-DQB1*04 HLA-DRB4*01 HLA-DRB1*12:02 HLA-DRB1*15:01 HLA-DQB1*06:02 [203,206,210] | HLA-DRB1*03:01 [209] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zepeda-Cervantes, J.; Martínez-Flores, D.; Ramírez-Jarquín, J.O.; Tecalco-Cruz, Á.C.; Alavez-Pérez, N.S.; Vaca, L.; Sarmiento-Silva, R.E. Implications of the Immune Polymorphisms of the Host and the Genetic Variability of SARS-CoV-2 in the Development of COVID-19. Viruses 2022, 14, 94. https://doi.org/10.3390/v14010094
Zepeda-Cervantes J, Martínez-Flores D, Ramírez-Jarquín JO, Tecalco-Cruz ÁC, Alavez-Pérez NS, Vaca L, Sarmiento-Silva RE. Implications of the Immune Polymorphisms of the Host and the Genetic Variability of SARS-CoV-2 in the Development of COVID-19. Viruses. 2022; 14(1):94. https://doi.org/10.3390/v14010094
Chicago/Turabian StyleZepeda-Cervantes, Jesús, Daniel Martínez-Flores, Josué Orlando Ramírez-Jarquín, Ángeles C. Tecalco-Cruz, Noé Santiago Alavez-Pérez, Luis Vaca, and Rosa Elena Sarmiento-Silva. 2022. "Implications of the Immune Polymorphisms of the Host and the Genetic Variability of SARS-CoV-2 in the Development of COVID-19" Viruses 14, no. 1: 94. https://doi.org/10.3390/v14010094