
Genetic Variability among Swine Influenza Viruses in Italy: Data Analysis of the Period 2017–2020

 , , , , ,
, , , , ,
Abstract
:1. Introduction
- “Avian-like” swine H1N1 (H1avN1, clade HA-1C) lineage, which appeared in 1979 with genes of avian origin and adapted in swine;
- “Human-like” H3N2 lineage, originating from the reassortment of human-like swine H3N2 and a H1avN1 in 1984;
- “Human-like” H1N2 (H1huN2, clade HA-1B) lineage, originating from a human-like H3N2 and a seasonal human H1N1 reassortment in 1994;
- “Pandemic 2009”-origin H1N1 (H1N1pdm09, clade HA-1A), circulating in the swine population since 2009.
2. Materials and Methods
3. Results
3.1. swIAV Subtypes Circulation over the Four Years (2017–2020)
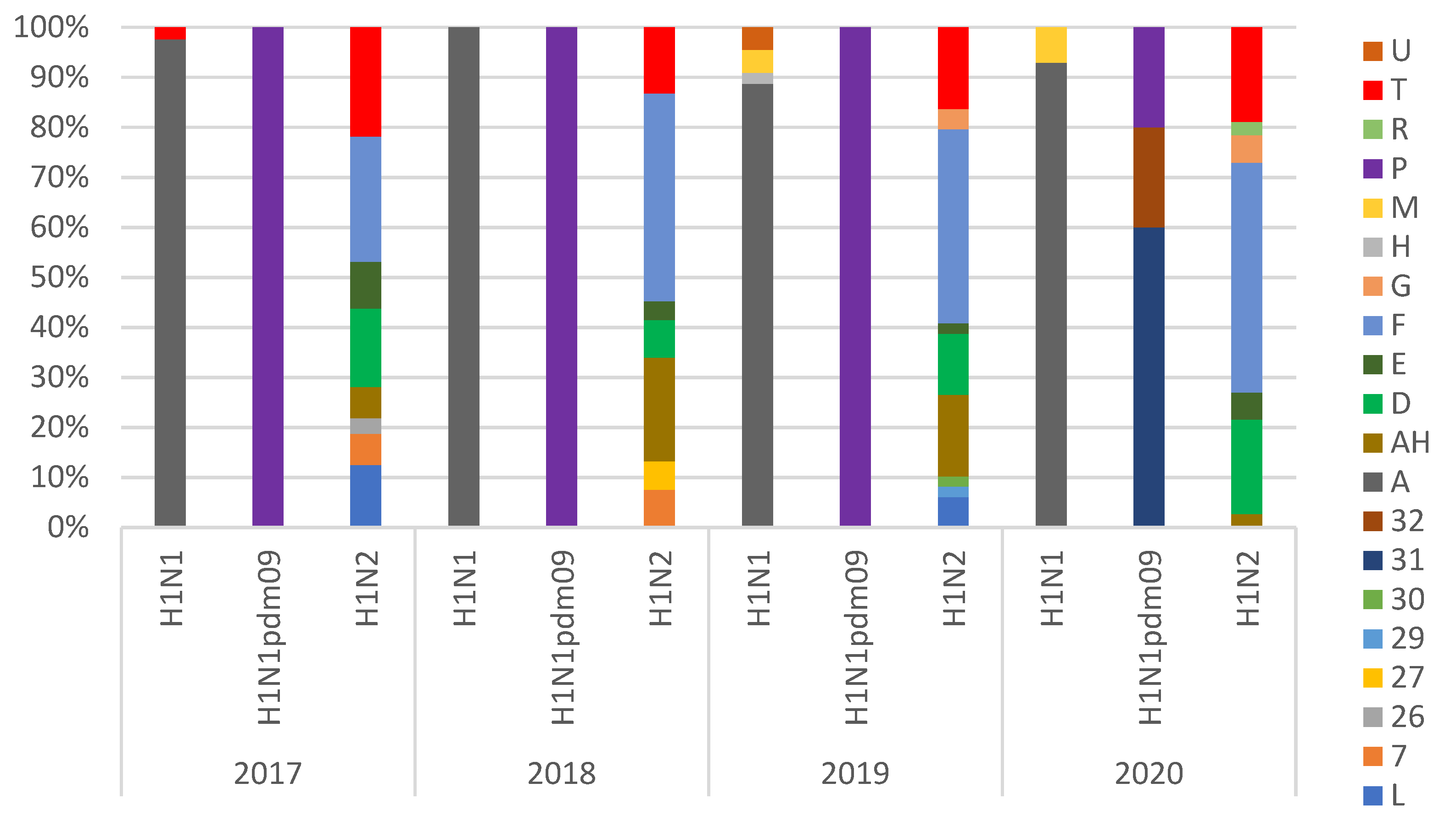
3.2. Genotypes Analysis
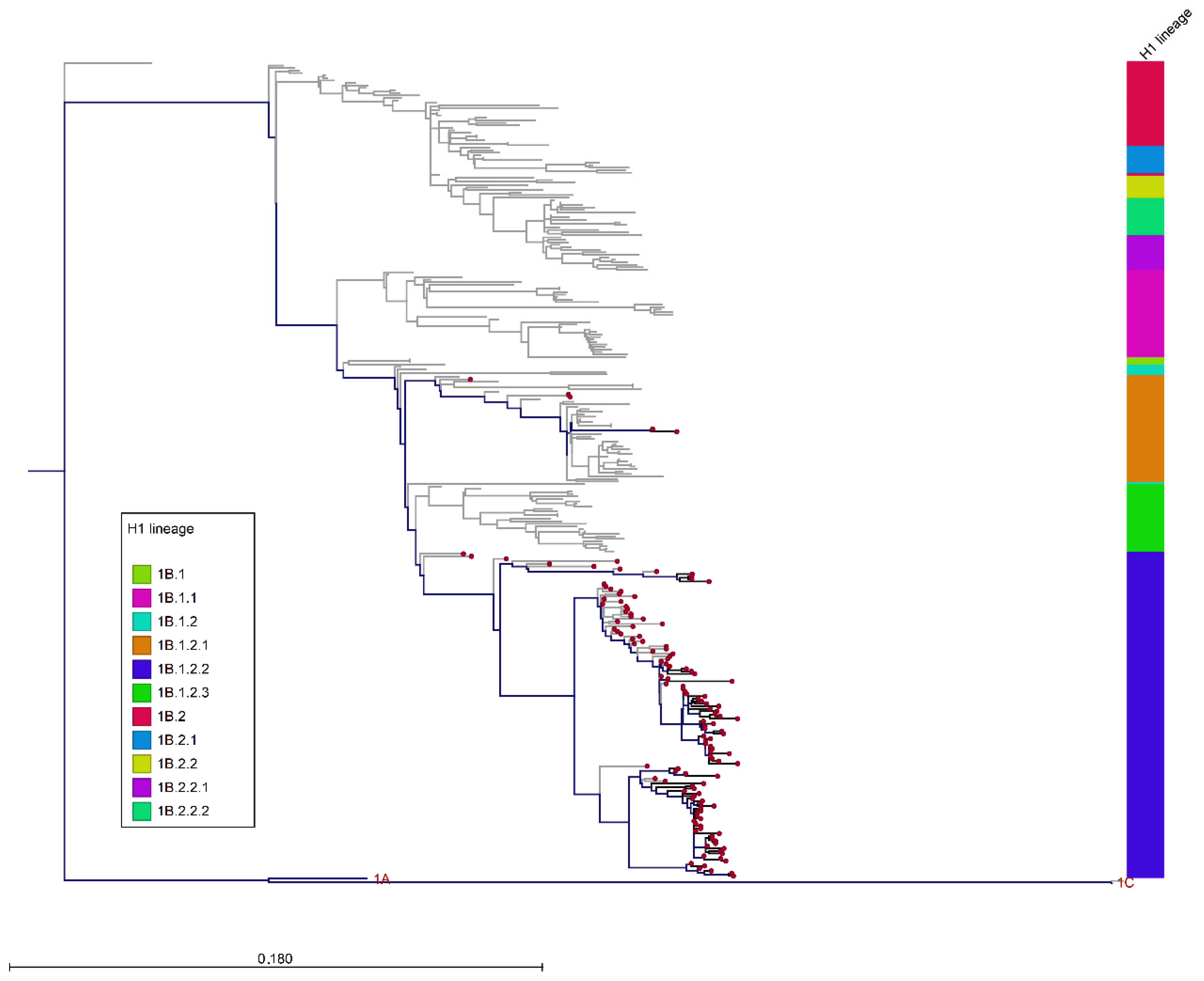
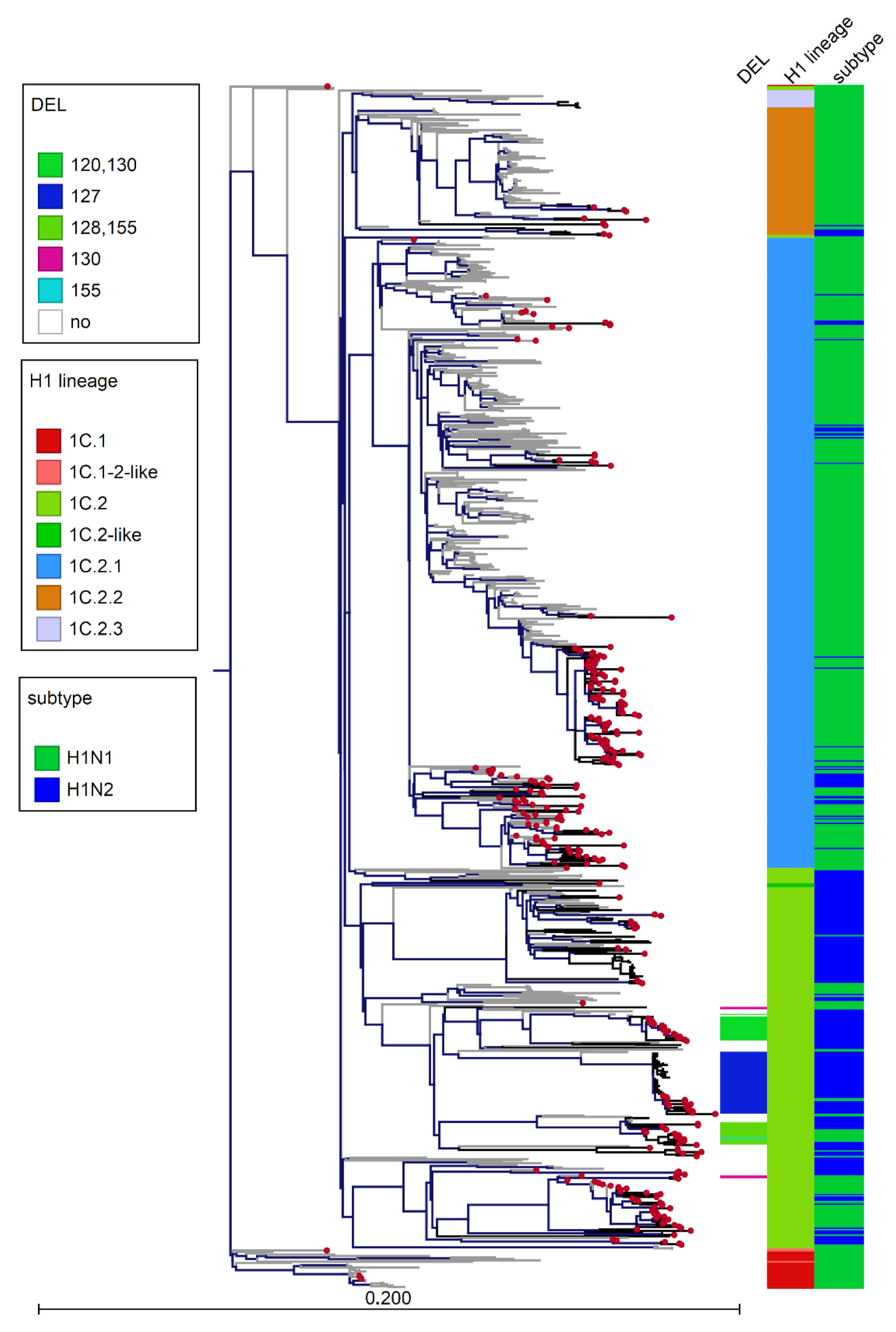
3.3. Phylogenetic Analysis of Italian swIAVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Nelli, R.K.; Kuchipudi, S.V.; White, G.A.; Perez, B.B.; Dunham, S.P.; Chang, K.C. Comparative distribution of human and avian type sialic acid influenza receptors in the pig. BMC Vet. Res. 2010, 6, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mena, I.; Nelson, M.I.; Quezada-Monroy, F.; Dutta, J.; Cortes-Fernández, R.; Lara-Puente, J.H.; Castro-Peralta, F.; Cunha, L.F.; Trovão, N.S.; Lozano-Dubernard, B.; et al. Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. eLife 2016, 5, e16777. [Google Scholar] [CrossRef] [PubMed]
- Zell, R.; Scholtissek, C.; Ludwig, S. Genetics, Evolution, and the Zoonotic Capacity of European Swine Influenza Viruses. Swine Influenza 2013, 370, 29–55. [Google Scholar]
- Watson, S.J.; Langat, P.; Reid, S.M.; Lam, T.T.; Cotten, M.; Kelly, M.; Van Reeth, K.; Qiu, Y.; Simon, G.; Bonin, E.; et al. Molecular Epidemiology and Evolution of Influenza Viruses Circulating within European Swine between 2009 and 2013. J. Virol. 2015, 89, 9920–9931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henritzi, D.; Petric, P.P.; Lewis, N.S.; Graaf, A.; Pessia, A.; Starick, E.; Breithaupt, A.; Strebelow, G.; Luttermann, C.; Parker, L.M.K.; et al. Surveillance of European Domestic Pig Populations Identifies an Emerging Reservoir of Potentially Zoonotic Swine Influenza A Viruses. Cell Host Microbe 2020, 28, 614–627. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.S.; Russell, C.A.; Langat, P.; Anderson, T.K.; Berger, K.; Bielejec, F.; Burke, D.F.; Dudas, G.; Fonville, J.M.; Fouchier, R.A.; et al. The global antigenic diversity of swine influenza A viruses. eLife 2016, 5, e12217. [Google Scholar] [CrossRef] [Green Version]
- Anderson, T.K.; Macken, C.A.; Lewis, N.S.; Scheuermann, R.H.; Van Reeth, K.; Brown, I.H.; Swenson, S.L.; Simon, G.; Saito, T.; Berhane, Y.; et al. A Phylogeny-Based Global Nomenclature System and Automated Annotation Tool for H1 Hemagglutinin Genes from Swine Influenza A Viruses. mSphere 2016, 1, e00275-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chastagner, A.; Herve, S.; Queguiner, S.; Hirchaud, E.; Lucas, P.; Gorin, S.; Beven, V.; Barbier, N.; Deblanc, C.; Blanchard, Y.; et al. Genetic and Antigenic Evolution of European Swine Influenza A Viruses of HA-1C (Avian-like) and HA-1B (Human-like) Lineages in France from 2000 to 2018. Viruses 2020, 12, 1304. [Google Scholar] [CrossRef] [PubMed]
- Trebbien, R.; Bragstad, K.; Larsen, L.E.; Nielsen, J.; Botner, A.; Heegaard, P.M.; Fomsgaard, A.; Viuff, B.; Hjulsager, C.K. Genetic and biological characterisation of an avian-like H1N2 swine influenza virus generated by reassortment of circulating avian-like H1N1 and H3N2 subtypes in Denmark. Virol. J. 2013, 10, 290. [Google Scholar] [CrossRef]
- Krog, J.S.; Hjulsager, C.K.; Larsen, M.A.; Larsen, L.E. Triple-reassortant influenza A virus with H3 of human seasonal origin, NA of swine origin, and internal A(H1N1) pandemic 2009 genes is established in Danish pigs. Influenza Other Respir Viruses 2017, 11, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Zell, R.; Groth, M.; Krumbholz, A.; Lange, J.; Philipps, A.; Durrwald, R. Novel reassortant swine H3N2 influenza A viruses in Germany. Sci. Rep. 2020, 10, 14296. [Google Scholar] [CrossRef]
- Chastagner, A.; Herve, S.; Bonin, E.; Queguiner, S.; Hirchaud, E.; Henritzi, D.; Beven, V.; Gorin, S.; Barbier, N.; Blanchard, Y.; et al. Spatiotemporal Distribution and Evolution of the A/H1N1 2009 Pandemic Influenza Virus in Pigs in France from 2009 to 2017: Identification of a Potential Swine-Specific Lineage. J. Virol. 2018, 92, e00988-18. [Google Scholar] [CrossRef] [Green Version]
- Zell, R.; Groth, M.; Krumbholz, A.; Lange, J.; Philipps, A.; Durrwald, R. Displacement of the Gent/1999 human-like swine H1N2 influenza A virus lineage by novel H1N2 reassortants in Germany. Arch. Virol. 2020, 165, 55–67. [Google Scholar] [CrossRef]
- Harder, T.C.; Grosse Beilage, E.; Lange, E.; Meiners, C.; Dohring, S.; Pesch, S.; Noe, T.; Grund, C.; Beer, M.; Starick, E. Expanded cocirculation of stable subtypes, emerging lineages, and new sporadic reassortants of porcine influenza viruses in swine populations in Northwest Germany. J. Virol. 2013, 87, 10460–10476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zell, R.; Groth, M.; Krumbholz, A.; Lange, J.; Philipps, A.; Durrwald, R. Cocirculation of Swine H1N1 Influenza A Virus Lineages in Germany. Viruses 2020, 12, 762. [Google Scholar] [CrossRef]
- Chepkwony, S.; Parys, A.; Vandoorn, E.; Stadejek, W.; Xie, J.; King, J.; Graaf, A.; Pohlmann, A.; Beer, M.; Harder, T.; et al. Genetic and antigenic evolution of H1 swine influenza A viruses isolated in Belgium and the Netherlands from 2014 through 2019. Sci. Rep. 2021, 11, 11276. [Google Scholar] [CrossRef] [PubMed]
- Ryt-Hansen, P.; Krog, J.S.; Breum, S.O.; Hjulsager, C.K.; Pedersen, A.G.; Trebbien, R.; Larsen, L.E. Co-circulation of multiple influenza A reassortants in swine harboring genes from seasonal human and swine influenza viruses. eLife 2021, 10, e60940. [Google Scholar] [CrossRef] [PubMed]
- Chiapponi, C.; Ebranati, E.; Pariani, E.; Faccini, S.; Luppi, A.; Baioni, L.; Manfredi, R.; Carta, V.; Merenda, M.; Affanni, P.; et al. Genetic analysis of human and swine influenza A viruses isolated in Northern Italy during 2010–2015. Zoonoses Public Health 2017, 65, 114–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dornfeld, D.; Petric, P.P.; Hassan, E.; Zell, R.; Schwemmle, M. Eurasian Avian-like Swine Influenza A Viruses Escape Human MxA Restriction through Distinct Mutations in Their Nucleoprotein. J. Virol. 2019, 93, e00997-18. [Google Scholar] [CrossRef] [Green Version]
- Moreno, A.; Gabanelli, E.; Sozzi, E.; Lelli, D.; Chiapponi, C.; Ciccozzi, M.; Zehender, G.; Cordioli, P. Different evolutionary trends of swine H1N2 influenza viruses in Italy compared to European viruses. Vet. Res. 2013, 44, 112. [Google Scholar] [CrossRef] [Green Version]
- Slomka, M.J.; Densham, A.L.; Coward, V.J.; Essen, S.; Brookes, S.M.; Irvine, R.M.; Spackman, E.; Ridgeon, J.; Gardner, R.; Hanna, A.; et al. Real time reverse transcription (RRT)-polymerase chain reaction (PCR) methods for detection of pandemic (H1N1) 2009 influenza virus and European swine influenza A virus infections in pigs. Influenza Other Respir. Viruses 2010, 4, 277–293. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, B.; Harder, T.; Lange, E.; Kalthoff, D.; Reimann, I.; Grund, C.; Oehme, R.; Vahlenkamp, T.W.; Beer, M. New real-time reverse transcriptase polymerase chain reactions facilitate detection and differentiation of novel A/H1N1 influenza virus in porcine and human samples. Berl. Munch. Tierarztl. Wochenschr. 2010, 123, 286–292. [Google Scholar] [PubMed]
- Henritzi, D.; Zhao, N.; Starick, E.; Simon, G.; Krog, J.S.; Larsen, L.E.; Reid, S.M.; Brown, I.H.; Chiapponi, C.; Foni, E.; et al. Rapid detection and subtyping of European swine influenza viruses in porcine clinical samples by haemagglutinin- and neuraminidase-specific tetra- and triplex real-time RT-PCRs. Influenza Other Respir. Viruses 2016, 10, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Chiapponi, C.; Zanni, I.; Garbarino, C.; Barigazzi, G.; Foni, E. Comparison of the usefulness of the CACO-2 cell line with standard substrates for isolation of swine influenza A viruses. J. Virol. Methods 2010, 163, 162–165. [Google Scholar] [CrossRef]
- OIE. Manual of Diagnostic Tests and Vaccines for Terrestrial Animals Part 2, Section 3.8, Chapter 3.8.7, 2018th ed.; World Organization for Animal Health: Paris, France, 2018. [Google Scholar]
- Lycett, S.J.; Baillie, G.; Coulter, E.; Bhatt, S.; Kellam, P.; McCauley, J.W.; Wood, J.L.; Brown, I.H.; Pybus, O.G.; Leigh Brown, A.J.; et al. Estimating reassortment rates in co-circulating Eurasian swine influenza viruses. J. Gen. Virol. 2012, 93, 2326–2336. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Manz, B.; Dornfeld, D.; Gotz, V.; Zell, R.; Zimmermann, P.; Haller, O.; Kochs, G.; Schwemmle, M. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog. 2013, 9, e1003279. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Lam, T.T.; Fan, X.; Chen, X.; Zeng, Y.; Zhou, J.; Duan, L.; Tse, M.; Chan, C.H.; Li, L.; et al. Expansion of genotypic diversity and establishment of 2009 H1N1 pandemic-origin internal genes in pigs in China. J. Virol. 2014, 88, 10864–10874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sriwilaijaroen, N.; Suzuki, Y. Molecular basis of the structure and function of H1 hemagglutinin of influenza virus. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2012, 88, 226–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, D.F.; Smith, D.J. A recommended numbering scheme for influenza A HA subtypes. PLoS ONE 2014, 9, e112302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trade Map. Available online: https://www.trademap.org/ (accessed on 30 June 2021).







{kind=link}
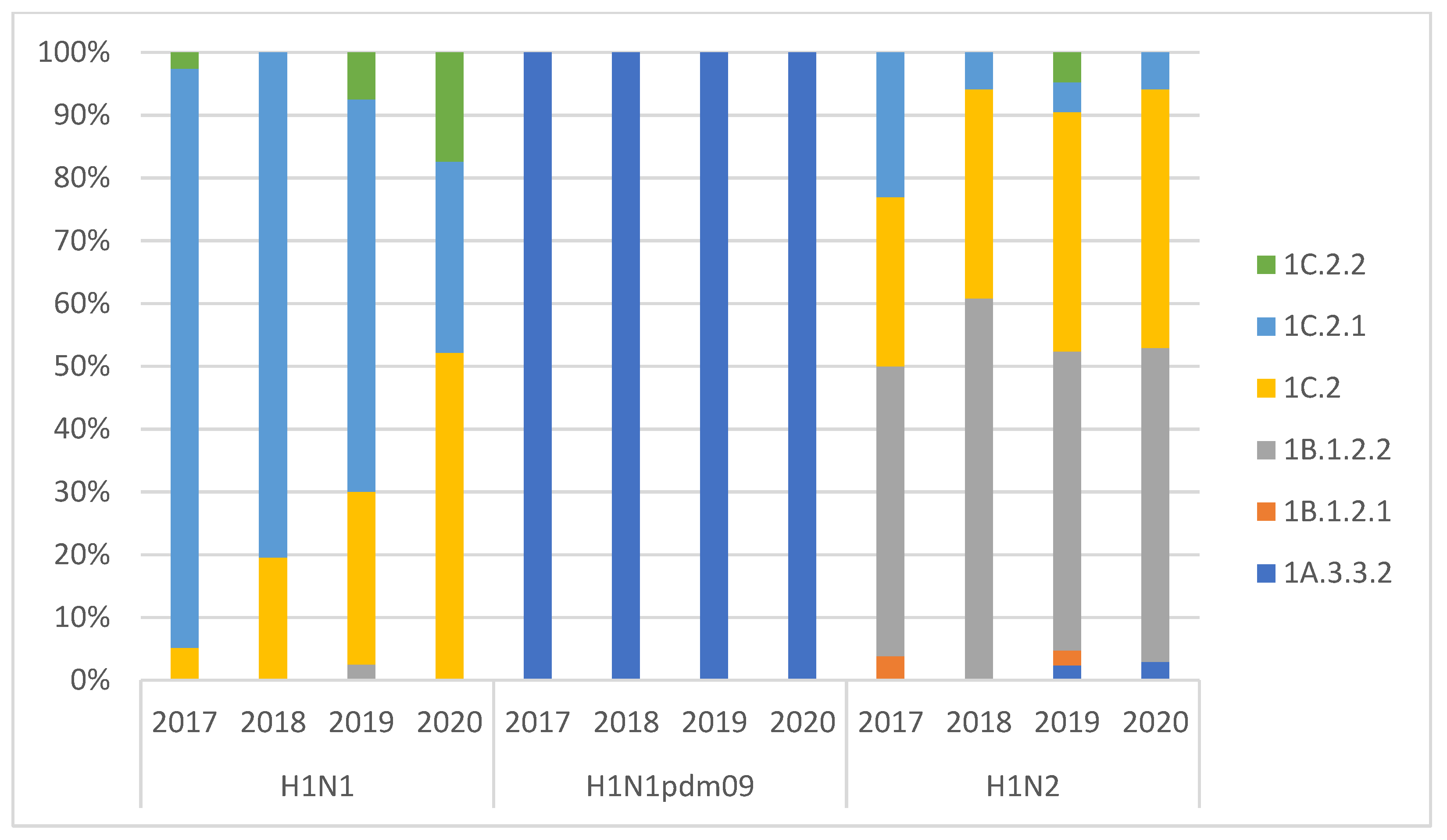{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence 5′-3′ | Subtype-Ref | PCR Product Expected Size |
|---|---|---|---|
| H1N2_for (H1-1B) | GCTACCATGCGAACAATTCA | H1 [19] | 241 |
| H1N2_rev (H1-1B) | TCAGCATTTGGTGTTTCTGC | ||
| H1N1_for (H1-1C) | CTGCACTGAAAGCTGACACC | H1 [19] | 327 |
| H1N1_rev (H1-1C) | GCTGCTCCCTTAATTCCTCA | ||
| SW-H1 FOR 107 (H1-1A) | CAGACACTGTAGACACAGTAC | H1pdm09 [20] | 516 |
| SW H1 REV 623 (H1-1A) | CTAGTAGATGGATGGTGAATGC | ||
| H3_for | CARATTGARGTGACHAATGC | H3 [19] | 722 |
| H3_rev | GGTGCATCTGAYCTCATTA | ||
| N1_for | TGAAATACAATGGCATAATAAC | N1 [19] | 514 |
| N1_rev | GGATCCCAAATCATCTCAAA | ||
| N2_for | GGAAAAGCATGGCTGCAT | N2 [19] | 791 |
| N2_rev | GTGCCACAAAACACAACAAT | ||
| R = A/G, Y = C/T, D = G/A/T, H = A/C/T. | |||
| n. of Subtyped Outbreaks | H1N1 | H1N2 | H3N2 | H3N1 | H1N1 pdm09 | Mixed Infection | |
|---|---|---|---|---|---|---|---|
| 2017 | 196 | 41% | 41% | 15% | 0% | 1% | 2% |
| 2018 | 164 | 36% | 48% | 12% | 1% | 3% | 1% |
| 2019 | 149 | 31% | 44% | 17% | 1% | 5% | 2% |
| 2020 | 163 | 31% | 47% | 5% | 0% | 15% | 3% |
| 2017–2020 | 672 | 35% | 45% | 12% | 0% | 6% | 2% |
| Subtype | Nomenclature | HA | NA | PB2 | PB1 | PA | NP | M | NS | % |
|---|---|---|---|---|---|---|---|---|---|---|
| H1N2 | F | 1B.1.2.2 | It-N2 | av | av | av | av | av | av | 38.6% |
| AH | 1C.2 | N2g | pdm | pdm | pdm | pdm | pdm | av | 12.9% | |
| T | 1C.2.1 | N2g | pdm | pdm | pdm | pdm | pdm | pdm | 0.6% | |
| 1C.2 | N2g | pdm | pdm | pdm | pdm | pdm | pdm | 17.5% | ||
| L | 1C.2.1 | It-N2 | av | av | av | av | av | av | 1.8% | |
| 1C.2 | It-N2 | av | av | av | av | av | av | 2.3% | ||
| D | 1C.2.1 | N2g | av | av | av | av | av | av | 5.8% | |
| 1C.2 | N2g | av | av | av | av | av | av | 7.0% | ||
| E | 1B.1.2.1 | N2g | av | av | av | av | av | av | 1.2% | |
| 1B.1.2.2 | N2g | av | av | av | av | av | av | 2.3% | ||
| 7 * | 1B.1.2.2 | It-N2 | pdm | pdm | pdm | pdm | pdm | pdm | 3.5% | |
| 26 * | 1C.2 | N2g | pdm | pdm | pdm | pdm | pdm | av | 0.6% | |
| 27 * | 1B.1.2.2 | It-N2 | av | av | av | av | pdm | av | 1.8% | |
| 29 * | 1B.1.2.2 | It-N2 | av | av | av | pdm | pdm | av | 0.6% | |
| 30 * | 1A.3.3.2 | It-N2 | av | av | av | av | av | av | 0.6% | |
| G | 1C.2.2 | N2s | av | av | av | av | av | av | 1.2% | |
| 1C.2 | N2s | av | av | av | av | av | av | 1.2% | ||
| R | 1A.3.3.2 | N2g | pdm | pdm | pdm | pdm | pdm | pdm | 0.6% | |
| H3N2 | H3(84) | N2g | av | av | av | av | av | av | 100% |
| Subtype | Nomenclature | HA | NA | PB2 | PB1 | PA | NP | M | NS | % |
|---|---|---|---|---|---|---|---|---|---|---|
| H1N1 | A | 1C.2.1 | av | av | av | av | av | av | av | 58.3% |
| 1C.2 | av | av | av | av | av | av | av | 19.3% | ||
| 1C.2.2 | av | av | av | av | av | av | av | 4.3% | ||
| M | 1C.2.1 | av | av | av | av | av | pdm | av | 0.5% | |
| 1C.2 | av | av | av | av | av | pdm | av | 1.6% | ||
| U | 1C.2.1 | av | pdm | pdm | pdm | pdm | pdm | pdm | 1.1% | |
| P | 1A.3.3.2 | pdm | pdm | pdm | pdm | pdm | pdm | pdm | 8.6% | |
| 31 * | 1A.3.3.2 | av | pdm | pdm | pdm | pdm | pdm | av | 4.3% | |
| 32 * | 1A.3.3.2 | av | pdm | pdm | pdm | pdm | pdm | pdm | 1.6% | |
| H | 1B.1.2.2 | av | av | av | av | av | av | av | 0.5% |
| HI TEST RESULTS | |||||
|---|---|---|---|---|---|
| VIRUS SERUM | A/Swine/Italy/ 284922/2009 H1N2 | A/Swine/Italy/ 311368/2013 H1N1 | A/Swine/Italy/ 311349/2013 H3N2 | A/Swine/Italy/ 282866/2013 H1N1 | |
| LINEAGE | 1B.1.2.2 | 1C.2.1 | H3 | 1A.3.3.2 | |
| A/swine/Italy/284922/2009 H1N2 | 1B.1.2.2 | 640 | <20 | <20 | <20 |
| A/swine/Italy/311368/2013 H1N1 | 1C.2.1 | <20 | 1280 | <20 | <20 |
| A/swine/Italy/311349/2013 H3N2 | H3 | <20 | <20 | 640 | <20 |
| A/swine/Italy/282866/2013 H1N1 sub-cluster (b) | 1A.3.3.2 | <20 | <20 | <20 | 5120 |
| A/swine/Italy/326417/2020 H1N1 | 1C.2 del 128,155 | <20 | <20 | <20 | <20 |
| A/swine/Italy/340406/2020 H1N2 | 1C.2 del 127 | <20 | 20 | <20 | <20 |
| A/swine/Italy/381442/2020 H1N2 | 1C.2 del 128,155 | <20 | <20 | <20 | <20 |
| A/swine/Italy/37307/2020 H1N2 | 1C.2 del 120,130 | <20 | 20 | <20 | <20 |
| A/swine/Italy/31684/2020 H1N2 | 1C.2.1 | <20 | 2560 | <20 | <20 |
| A/swine/Italy/30190/2020 H1N2 | 1C.2 | <20 | 320 | <20 | 160 |
| A/swine/Italy/187080/2020 H1N1 sub-cluster (a) | 1A.3.3.2 | <20 | <20 | <20 | <20 |
| A/swine/Italy/297520/2018 H1N1 sub-cluster (b) | 1A.3.3.2 | <20 | 20 | <20 | 640 |
| A/swine/Italy/60023/2019 H1N1 sub-cluster (c) | 1A.3.3.2 | <20 | 320 | <20 | 5120 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiapponi, C.; Prosperi, A.; Moreno, A.; Baioni, L.; Faccini, S.; Manfredi, R.; Zanni, I.; Gabbi, V.; Calanchi, I.; Fusaro, A.; et al. Genetic Variability among Swine Influenza Viruses in Italy: Data Analysis of the Period 2017–2020. Viruses 2022, 14, 47. https://doi.org/10.3390/v14010047
Chiapponi C, Prosperi A, Moreno A, Baioni L, Faccini S, Manfredi R, Zanni I, Gabbi V, Calanchi I, Fusaro A, et al. Genetic Variability among Swine Influenza Viruses in Italy: Data Analysis of the Period 2017–2020. Viruses. 2022; 14(1):47. https://doi.org/10.3390/v14010047
Chicago/Turabian StyleChiapponi, Chiara, Alice Prosperi, Ana Moreno, Laura Baioni, Silvia Faccini, Roberta Manfredi, Irene Zanni, Valentina Gabbi, Irene Calanchi, Alice Fusaro, and et al. 2022. "Genetic Variability among Swine Influenza Viruses in Italy: Data Analysis of the Period 2017–2020" Viruses 14, no. 1: 47. https://doi.org/10.3390/v14010047