
Bayesian Inference of State-Level COVID-19 Basic Reproduction Numbers across the United States

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Model
2.2. Simulations
2.3. Calculation of Epidemic Parameters and
2.4. Bayesian Inference
3. Results
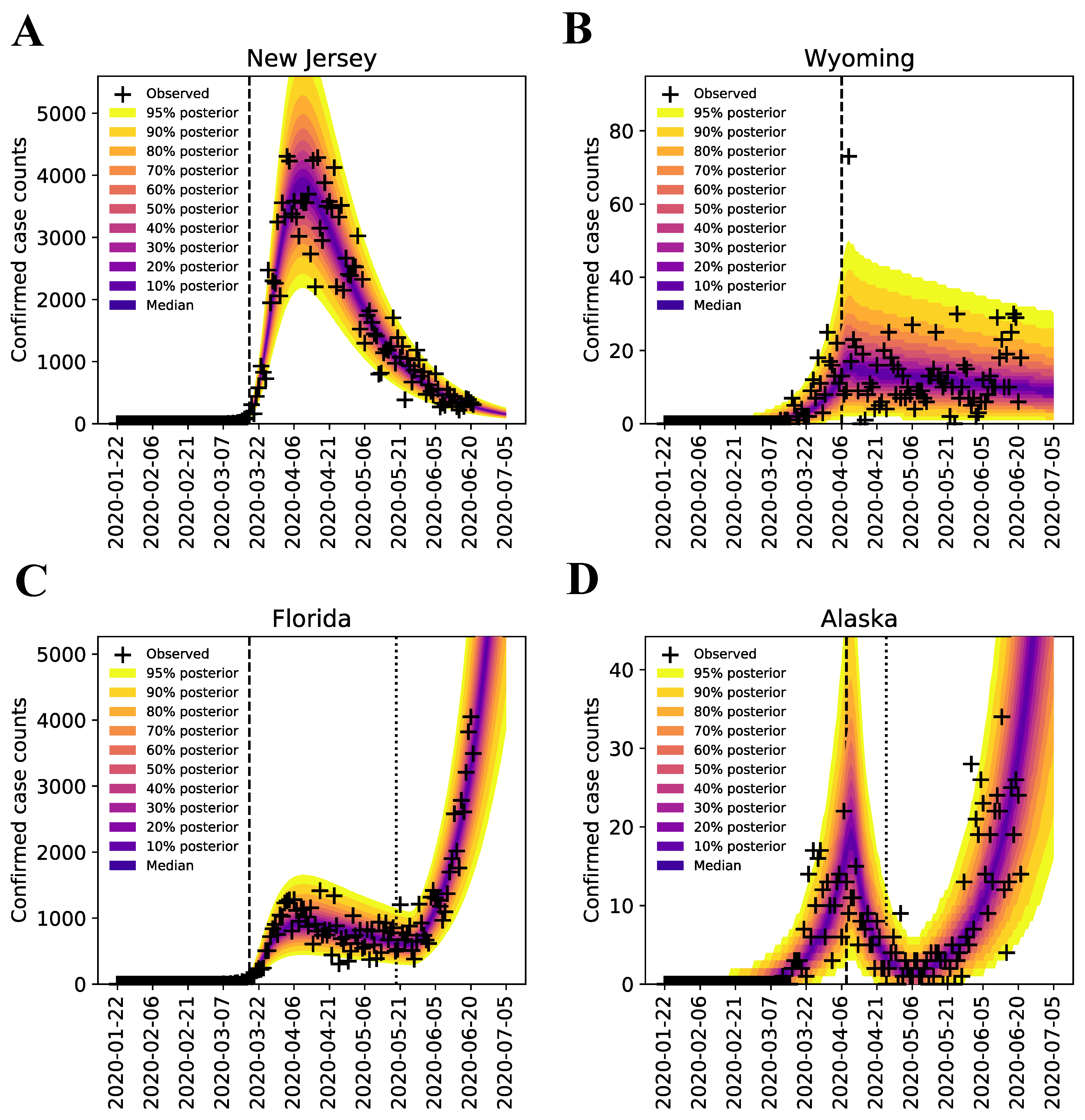
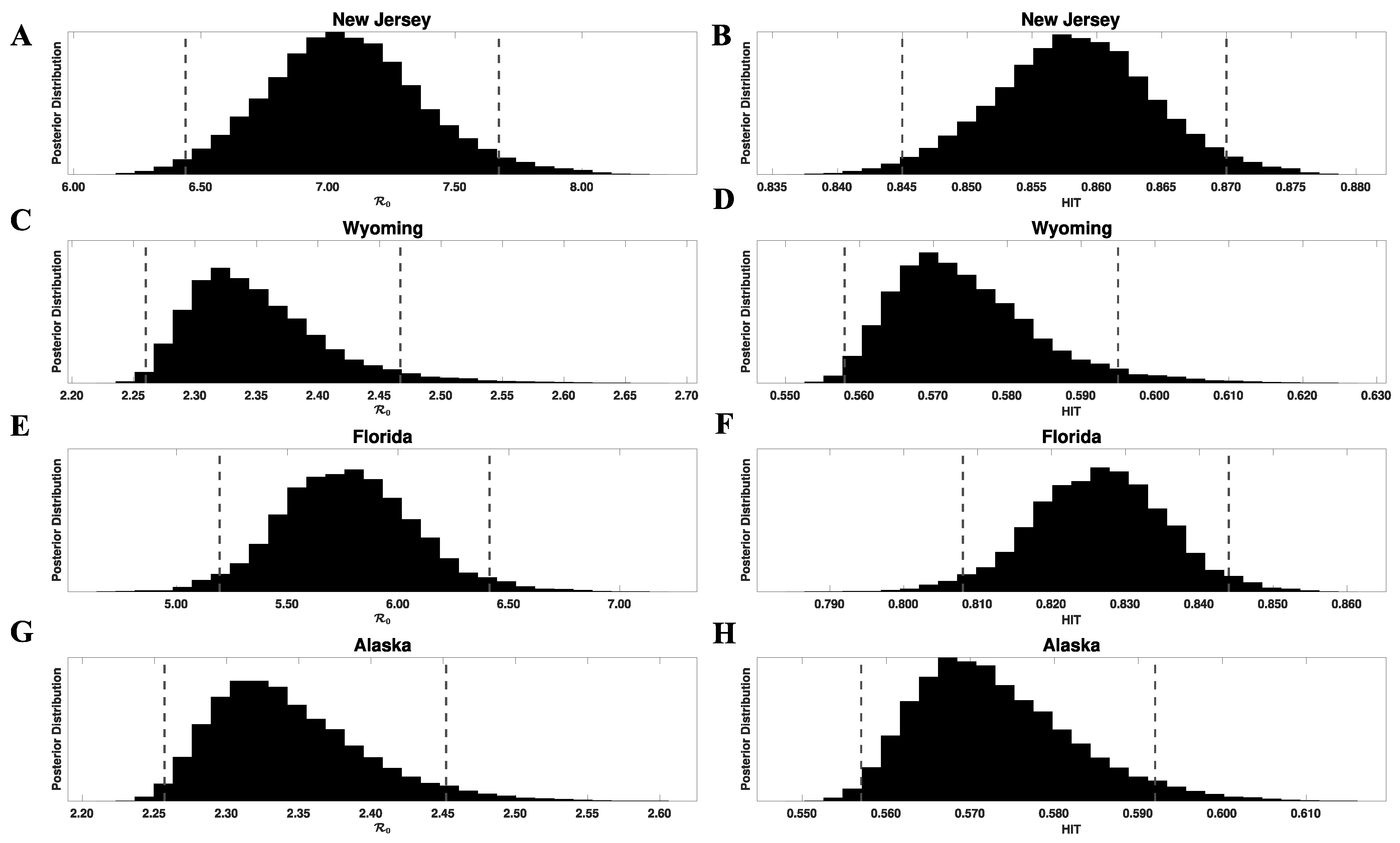
3.1. Bayesian Uncertainty Quantification
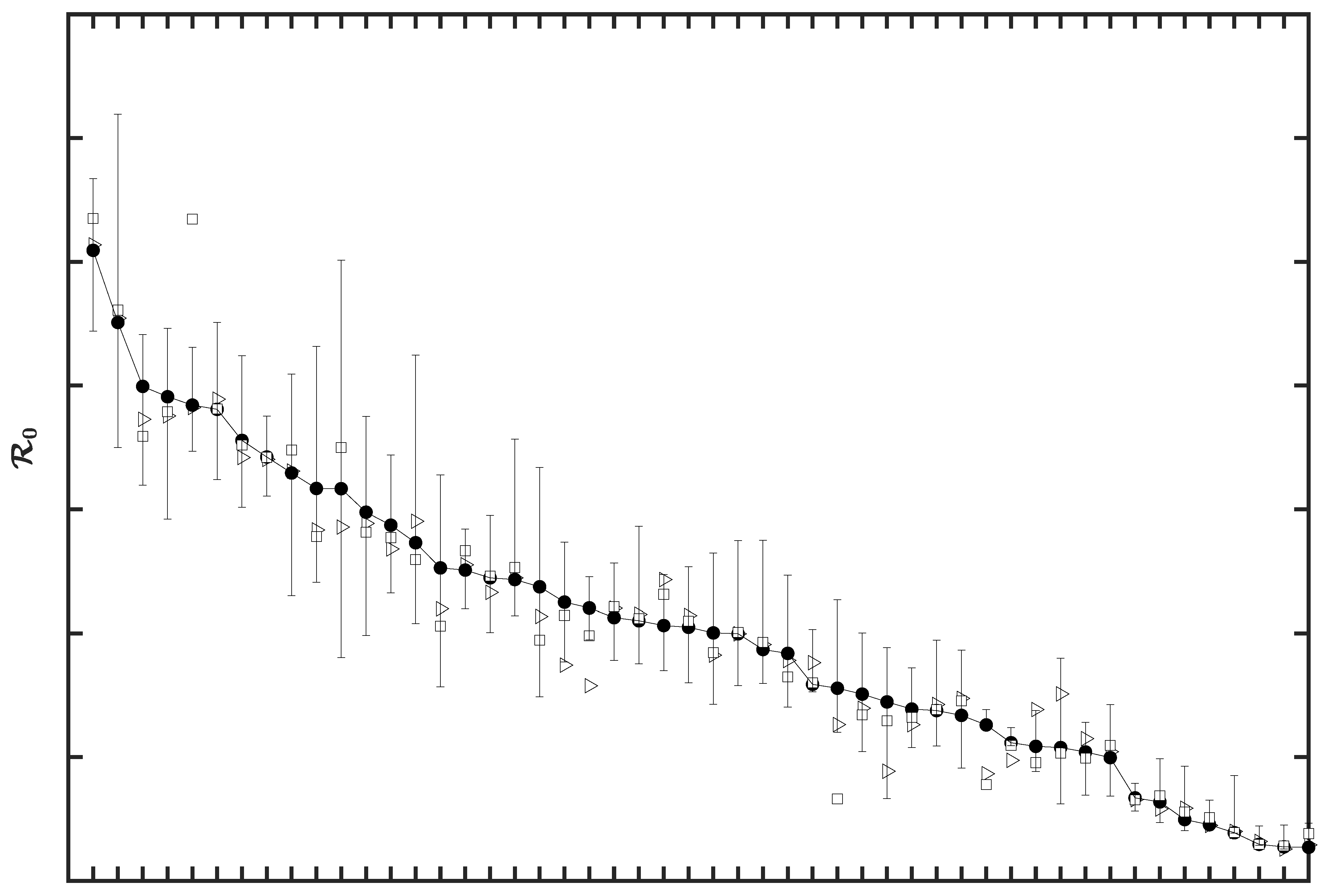
3.2. Region-Specific Basic Reproduction Numbers and Herd Immunity Thresholds
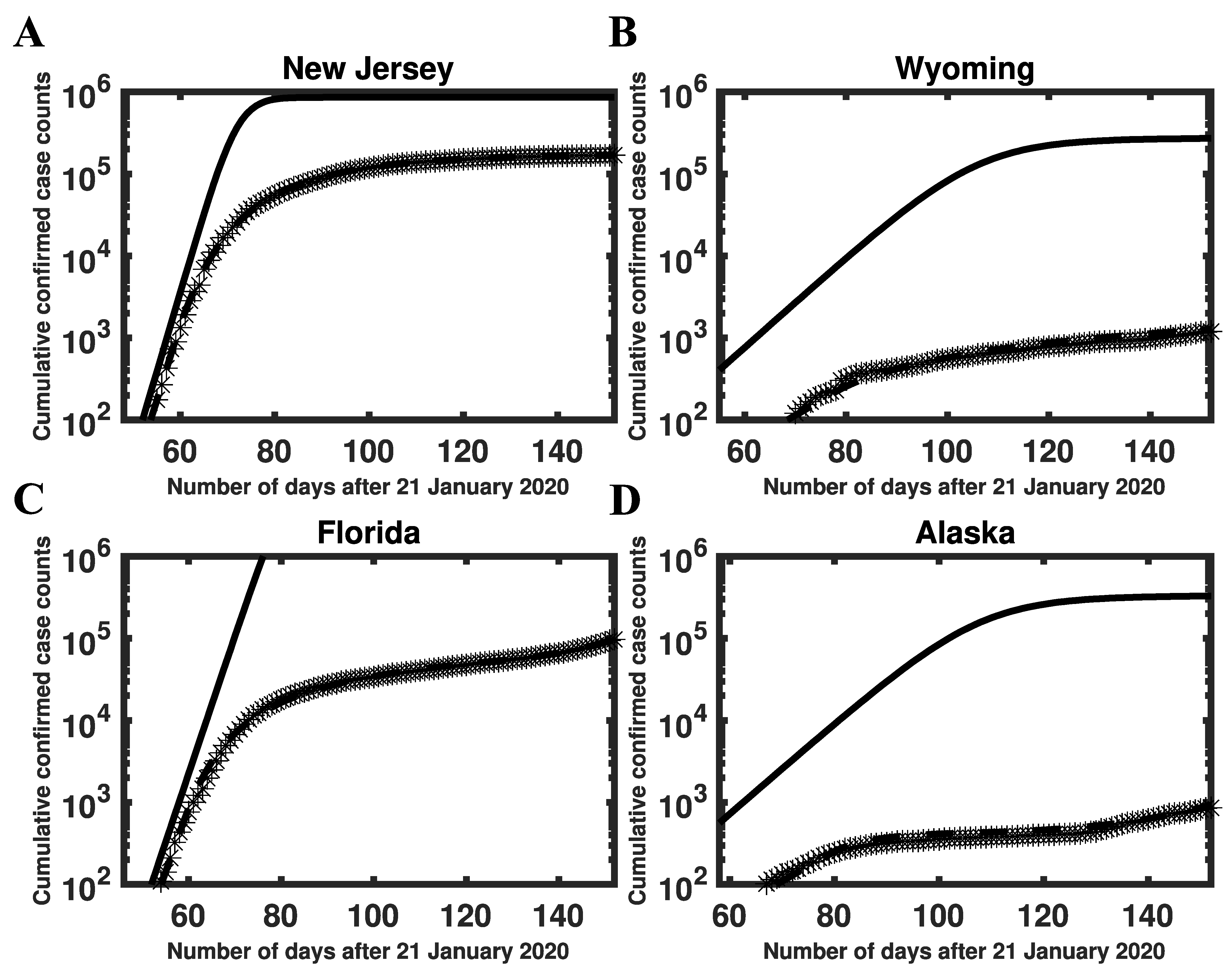
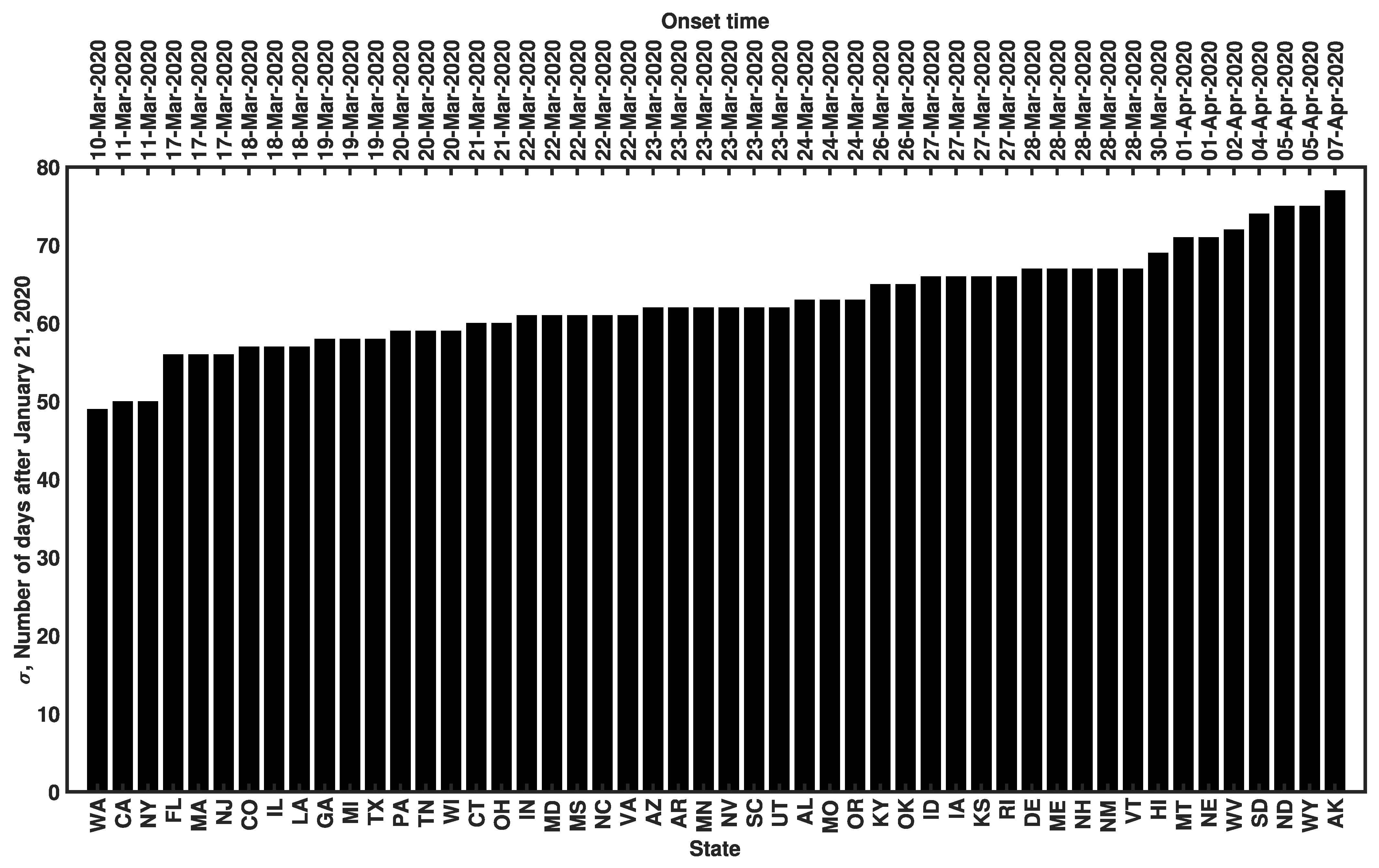
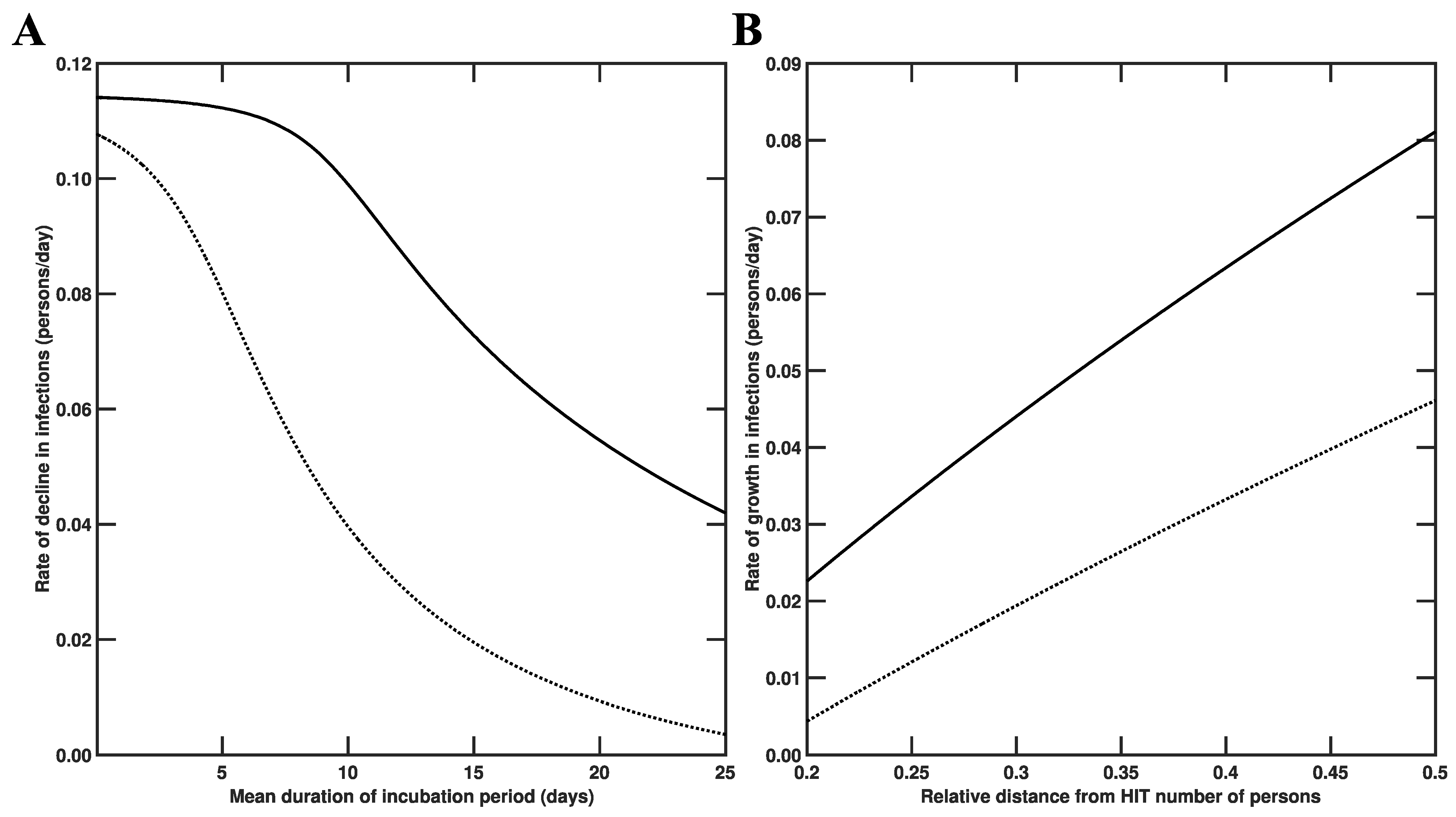
3.3. Estimates of Initial Region-Specific Epidemic Growth Rates
3.4. Sensitivity of to the Surveillance Data Used in Inference
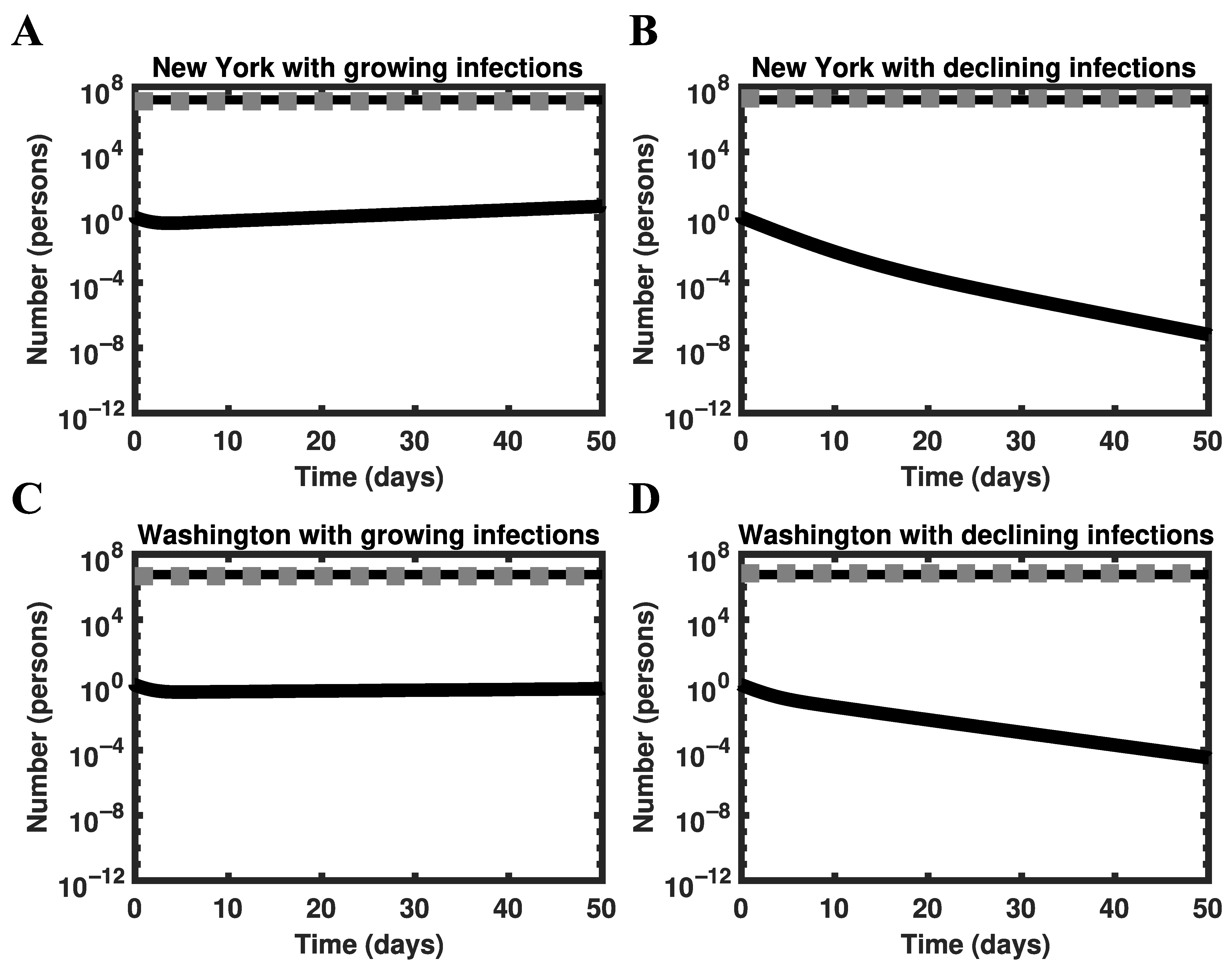
3.5. Global Asymptotic Stability of the Disease-Free Equilibrium
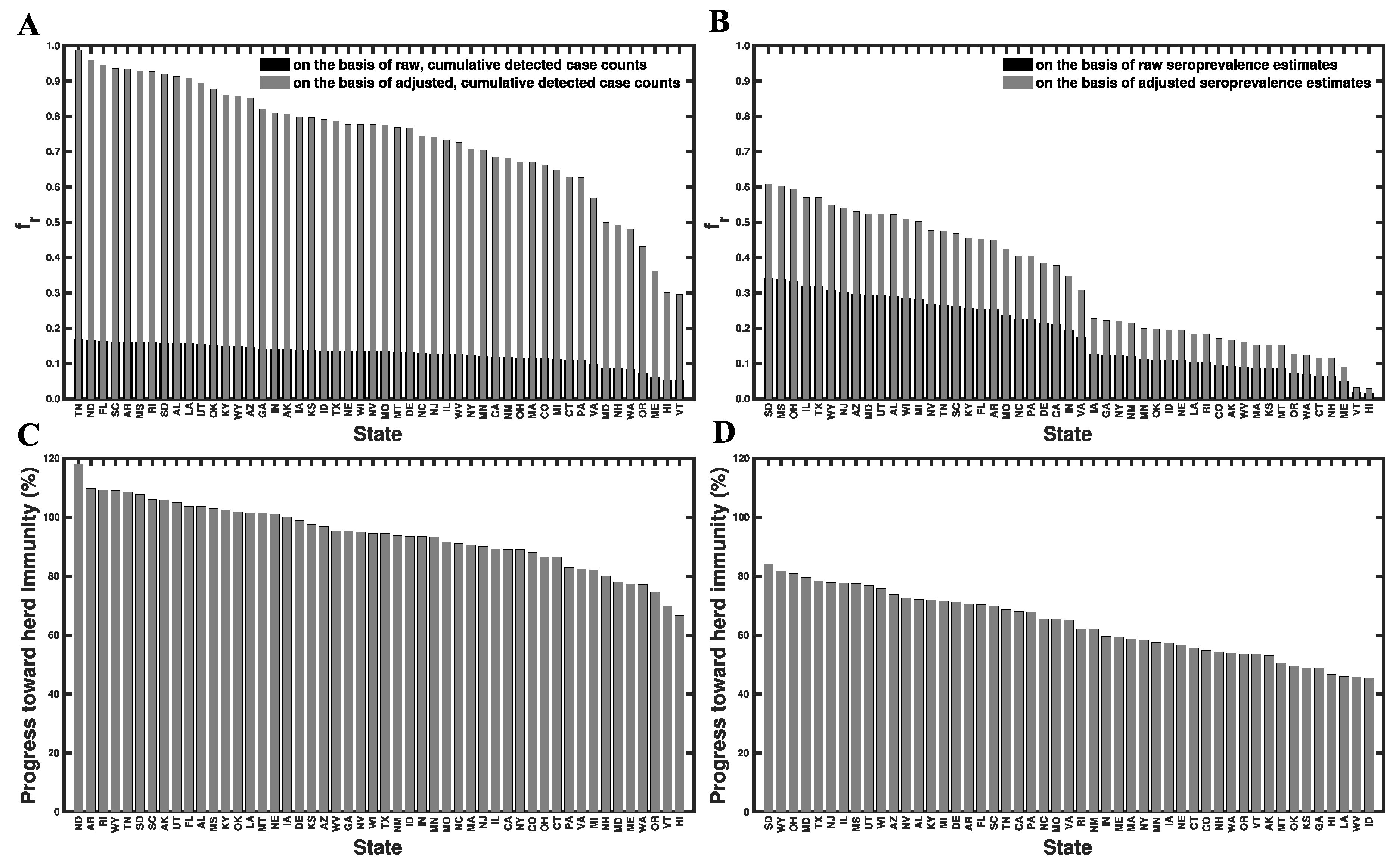
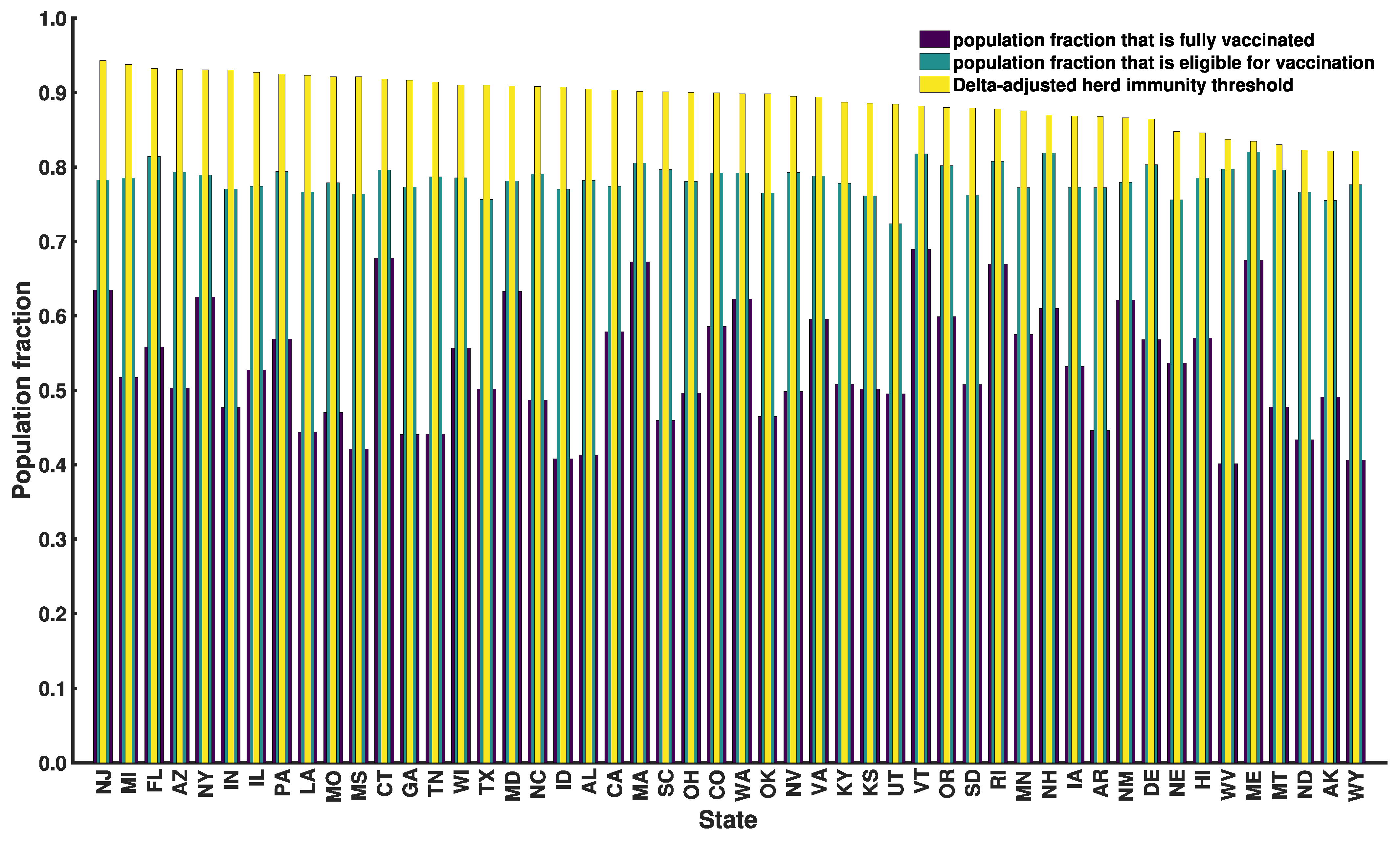
3.6. Progress toward Herd Immunity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gee, J.; Marquez, P.; Su, J.; Calvert, G.M.; Liu, R.; Myers, T.; Nair, N.; Martin, S.; Clark, T.; Markowitz, L.; et al. First month of COVID-19 vaccine safety monitoring—United States, 14 December 2020–13 January 2021. MMWR Morb. Mortal Wkly Rep. 2020, 70, 283–288. [Google Scholar] [CrossRef]
- National Center for Immunization and Respiratory Diseases (NCIRD), Data from Centers for Disease Control and Prevention (CDC). Available online: https://data.cdc.gov/Vaccinations/COVID-19-Vaccinations-in-the-United-States-Jurisdi/unsk-b7fc (accessed on 20 September 2021).
- Fine, P.; Eames, K.; Heymann, D.L. Herd immunity: A rough guide. Clin. Infect. Dis. 2011, 7, 911–916. [Google Scholar] [CrossRef] [PubMed]
- Ridenhour, B.; Kowalik, J.M.; Shay, D.K. Unraveling 0: Considerations for public health applications. Am. J. Public Health 2018, 108, S445–S454. [Google Scholar] [CrossRef]
- Temime, L.; Gustin, M.-P.; Duval, A.; Buetti, N.; Crépey, P.; Guillemot, D.; Thiébaut, R.; Vanhems, P.; Zahar, J.-R.; Smith, D.R.M.; et al. A conceptual discussion about the basic reproduction number of severe acute respiratory syndrome coronavirus 2 in healthcare settings. Clin. Infect. Dis. 2021, 72, 141–143. [Google Scholar] [CrossRef]
- Dan, J.M.; Mateus, J.; Kato, Y.; Hastie, K.M.; Yu, E.D.; Faliti, C.E.; Grifoni, A.; Ramirez, S.I.; Haupt, S.; Frazier, A.; et al. Immunological memory to SARS-CoV-2 assessed for up to 8 months after infection. Science 2021, 371, eabf4063. [Google Scholar] [CrossRef]
- Yu, C.-J.; Wang, Z.-X.; Xu, Y.; Hu, M.-X.; Chen, K.; Qin, G. Assessment of basic reproductive number for COVID-19 at global level: A meta-analysis. Medicine 2021, 100, e25837. [Google Scholar] [CrossRef]
- Kucharski, A.J.; Russell, T.W.; Diamond, C.; Liu, Y.; Edmunds, J.; Funk, S.; Eggo, R.M. Early dynamics of transmission and control of COVID-19: A mathematical modelling study. Lancet 2020, 20, 553–558. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Pei, S.; Chen, B.; Song, Y.; Zhang, T.; Yang, W.; Shaman, J. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science 2020, 368, 489–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, L.; Wymant, C.; Kendall, M.; Zhao, L.; Nurtay, A.; Abeler-Dörner, L.; Parker, M.; Bonsall, D.; Fraser, C. Quantifying SARS-CoV-2 transmission suggests epidemic control with digital contact tracing. Science 2020, 368, eabb6936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arienzo, M.; Coniglio, A. Assessment of the SARS-CoV-2 basic reproduction number, R0, based on the early phase of COVID-19 outbreak in Italy. Biosaf. Health 2020, 2, 57–59. [Google Scholar] [CrossRef] [PubMed]
- Sanche, S.; Lin, Y.T.; Xu, C.; Romero-Severson, E.; Hengartner, N.W.; Ke, R. High contagiousness and rapid spread of severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1470–1477. [Google Scholar] [CrossRef] [PubMed]
- Romero-Severson, O.E.; Hengartner, N.; Meadors, G.; Ke, R. Change in global transmission rates of COVID-19 through May 6 2020. PLoS ONE 2020, 15, e0236776. [Google Scholar] [CrossRef] [PubMed]
- Ke, R.; Romero-Severson, E.O.; Sanche, S.; Hengartner, N. Estimating the reproductive number R0 of SARS-CoV-2 in the United States and eight European countries and implications for vaccination. J. Theor. Biol. 2021, 517, 110621. [Google Scholar] [CrossRef]
- Kong, D.J.; Tekwa, E.W.; Gignoux-Wolfsohn, S.A. Social, economic, and environmental factors influencing the basic reproduction number of COVID-19 across countries. PLoS ONE 2021, 16, e0252373. [Google Scholar] [CrossRef]
- Ives, C.A.; Bozzuto, R. State-by-State estimates of R0 at the start of COVID-19 outbreaks in the USA. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.05.17.20104653v3 (accessed on 4 September 2021).
- Fellows, I.E.; Slayton, R.B.; Hakim, A.J. The COVID-19 pandemic, community mobility and the effectiveness of non-pharmaceutical interventions: The United States of America, February to May 2020. arXiv 2020. Available online: https://arxiv.org/abs/2007.12644 (accessed on 8 September 2021).
- Milicevic, O.; Salom, I.; Rodic, A.; Markovic, S.; Tumbas, M.; Zigic, D.; Djordjevic, M.; Djordjevic, M. PM2.5 as a major predictor of COVID-19 basic reproduction number in the USA. Environ. Res. 2021, 201, 111526. [Google Scholar] [CrossRef]
- Ives, A.R.; Bozzuto, C. Estimating and explaining the spread of COVID-19 at the county level in the USA. Commun. Biol. 2021, 4, 1–9. [Google Scholar] [CrossRef]
- Sy, K.T.; White, L.F.; Nichols, B.E. Population density and basic reproductive number of COVID-19 across United States counties. PLoS ONE 2021, 16, e0249271. [Google Scholar] [CrossRef] [PubMed]
- Weissert, C.S.; Uttermark, M.J.; Mackie, K.R.; Artiles, A. Governors in control: Executive orders, state-local preemption, and the COVID-19 pandemic. Publius 2021, 51, 396–428. [Google Scholar] [CrossRef]
- Lin, Y.T.; Neumann, J.; Miller, E.F.; Posner, R.G.; Mallela, A.; Safta, C.; Ray, J.; Thakur, G.; Chintavali, S.; Hlavacek, W.S. Daily forecasting of regional epidemics of coronavirus disease with bayesian uncertainty quantification. Emerg. Infect. Dis. 2021, 27, 767–778. [Google Scholar] [CrossRef] [PubMed]
- The New York Times COVID-19 Data Team. Data from The New York Times. Available online: https://github.com/nytimes/covid-19-data (accessed on 20 September 2021).
- The Covid Act Now COVID-19 Data Team. Data from Covid Act Now. Available online: https://covidactnow.org/data-api (accessed on 20 September 2021).
- Surveillance Review and Response Group, Data from Centers for Disease Control and Prevention (CDC). Available online: https://covid.cdc.gov/covid-data-tracker/#national-lab (accessed on 20 September 2021).
- Bajema, K.L.; Wiegand, R.E.; Cuffe, K.; Patel, S.V.; Iachan, R.; Lim, T.; Lee, A.; Moyse, D.; Havers, F.P.; Harding, L.; et al. Estimated SARS-CoV-2 Seroprevalence in the US as of September 2020. JAMA 2021, 181, 450–460. [Google Scholar] [CrossRef]
- Fort, H. A very simple model to account for the rapid rise of the alpha variant of SARS-CoV-2 in several countries and the world. Virus Res. 2021, 304, 198531. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.; Vusirikala, A.; Flannagan, J.; Twohig, K.A.; Zaidi, A.; Chudasama, D.; Lamagni, T.; Groves, N.; Turner, C.; Rawlinson, C.; et al. Increased Household Transmission of COVID-19 Cases Associated with SARS-CoV-2 Variant of Concern B.1.617.2: A National Case-Control Study. 2021. Available online: https://khub.net/documents/135939561/405676950/Increased+Household+Transmission+of+COVID-19+Cases+-+national+case+study.pdf/7f7764fb-ecb0-da31-77b3-b1a8ef7be9aa (accessed on 9 July 2021).
- Hethcote, H.W. The mathematics of infectious diseases. SIAM Rev. Soc. Ind. Appl. Math. 2000, 42, 599–653. [Google Scholar] [CrossRef] [Green Version]
- White House. Proclamation on Declaring a National Emergency Concerning the Novel Coronavirus Disease (COVID-19) Outbreak. Available online: https://trumpwhitehouse.archives.gov/presidential-actions/proclamation-declaring-national-emergency-concerning-novel-coronavirus-disease-covid-19-outbreak/ (accessed on 29 November 2021).
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petzold, L. Automatic selection of methods for solving stiff and nonstiff systems of ordinary differential equations. SIAM J. Sci. Comput. 1983, 4, 136–148. [Google Scholar] [CrossRef]
- Blinov, M.L.; Faeder, J.R.; Goldstein, B.; Hlavacek, W.S. BioNetGen: Software for rule-based modeling of signal transduction based on the interactions of molecular domains. Bioinformatics 2004, 20, 3289–3291. [Google Scholar] [CrossRef] [PubMed]
- Cohen, S.D. CVODE. A stiff/nonstiff ODE solver in C. Comput. Phys. 1996, 10, 138–143. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.K.; Pitrou, A.; Seibert, S. Numba: A LLVM-based Python JIT compiler. In Proceedings of the Second Workshop on the LLVM Compiler Infrastructure in HPC; Association for Computing Machinery: New York, NY, USA, 2015; pp. 1–6. [Google Scholar]
- Diekmann, O.; Heesterbeek, J.A.; Roberts, M.G. The construction of next-generation matrices for compartmental epidemic models. J. R. Soc. Interface 2010, 7, 873–875. [Google Scholar] [CrossRef] [Green Version]
- Wolfram, S. Mathematica: A System for Doing Mathematics by Computer; Addison Wesley Longman Publishing Co., Inc.: Boston, MA, USA, 1991. [Google Scholar]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Wearing, H.J.; Rohani, P.J.M. Keeling. Appropriate models for the management of infectious diseases. PLoS Med. 2005, 7, e174. [Google Scholar]
- Mitra, E.D.; Suderman, R.; Colvin, J.; Ionkov, A.; Hu, A.; Sauro, H.M.; Posner, R.G.; Hlavacek, W.S. PyBioNetFit and the Biological Property Specification Language. iScience 2019, 19, 1012–1036. [Google Scholar] [CrossRef] [Green Version]
- Massey, F.J. The Kolmogorov-Smirnov test for goodness of fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]
- Shuai, Z.; van den Driessche, P. Global stability of infectious disease models using Lyapunov functions. SIAM J. Appl. Math. 2013, 73, 1513–1532. [Google Scholar] [CrossRef] [Green Version]
- Dorigatti, I.; Lavezzo, E.; Manuto, L.; Ciavarella, C.; Pacenti, M.; Boldrin, C.; Cattai, M.; Saluzzo, F.; Franchin, E.; Del Vecchio, C.; et al. SARS-CoV-2 antibody dynamics and transmission from community-wide serological testing in the Italian municipality of Vo’. Nat. Commun. 2021, 12, 1–11. [Google Scholar]
- Fowlkes, A.; Gaglani, M.; Groover, K.; Thiese, M.S.; Tyner, H.; Ellingson, K. Effectiveness of COVID-19 vaccines in preventing SARS-CoV-2 infection among frontline workers before and during B.1.617.2 (Delta) variant predominance—Eight US locations, December 2020-August 2021. MMWR Morb. Mortal Wkly Rep. 2021, 70, 1167–1169. [Google Scholar] [CrossRef]
- Kalish, H.; Klumpp-Thomas, C.; Hunsberger, S.; Baus, H.A.; Fay, M.P.; Siripong, N.; Wang, J.; Hicks, J.; Mehalko, J.; Travers, J.; et al. Undiagnosed SARS-CoV-2 seropositivity during the first 6 months of the COVID-19 pandemic in the United States. Sci. Transl. Med. 2021, 13, eabh3826. [Google Scholar] [CrossRef]
- Takahashi, S.; Greenhouse, B.; Rodríguez-Barraquer, I. Are seroprevalence estimates for severe acute respiratory syndrome coronavirus 2 biased? J. Infect. Dis. 2020, 222, 1772–1775. [Google Scholar] [CrossRef]
- U.S. Census Bureau, Population Division. 2020. Available online: https://www2.census.gov/programs-surveys/popest/tables/2010-2020/national/asrh/sc-est2020-18+pop-res.xlsx (accessed on 29 November 2021).
- Randolph, H.E.; Barreiro, L.B. Herd immunity: Understanding COVID-19. Immunity 2020, 5, 737–741. [Google Scholar] [CrossRef]
- Moghadas, S.M.; Sah, P.; Shoukat, A.; Meyers, L.A.; Galvani, A.P. Population immunity against COVID-19 in the United States. Ann. Intern. Med. 2021, 174, 1586–1591. [Google Scholar] [CrossRef]
- Science Brief: COVID-19 Vaccines and Vaccination. 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/science/science-briefs/fully-vaccinated-people.html (accessed on 8 September 2021).
- Callaway, E.; Ledford, H. How bad is Omicron? What scientists know so far. Nature 2021, 600, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Nishiura, H.; Ito, K.; Anzai, A.; Kobayashi, T.; Piantham, C.; Rodríguez-Morales, A.J. Relative reproduction number of SARS-CoV-2 Omicron (B.1.1.529) compared with Delta variant in South Africa. J. Clin. Med. 2022, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Piantham, C. Nishiura, Relative instantaneous reproduction number of Omicron SARS-CoV-2 variant with respect to the Delta variant in Denmark. J. Med. Virol. 2021. [Google Scholar] [CrossRef]
- Sofonea, M.T.; Roquebert, B.; Foulongne, V.; Verdurme, L.; Trombert-Paolantoni, S.; Roussel, M.; Haim-Boukobza, S.; Alizon, S. From Delta to Omicron: Analysing the SARS-CoV-2 epidemic in France using variant-specific screening tests (September 1 to December 18, 2021). medRxiv 2022. Available online: https://www.medrxiv.org/content/10.1101/2021.12.31.21268583v1 (accessed on 29 November 2021).
- Tartof, S.Y.; Slezak, J.M.; Fischer, H.; Hong, V.; Ackerson, B.K.; Ranasinghe, O.N.; Frankland, T.B.; Ogun, O.A.; Zamparo, J.M.; Gray, S.; et al. Six-month effectiveness of BNT162B2 mRNA COVID-19 vaccine in a large US Integrated health system: A retrospective cohort study. Lancet 2021, 398, 1407–1416. [Google Scholar] [CrossRef]
- Chemaitelly, H.; Tang, P.; Hasan, M.R.; AlMukdad, S.; Yassine, H.M.; Benslimane, F.M.; Al Khatib, H.A.; Coyle, P.; Ayoub, H.H.; Al Kanaani, Z.; et al. Waning of BNT162b2 vaccine protection against SARS-CoV-2 infection in Qatar. N. Engl. J. Med. 2021, 385, e83. [Google Scholar] [CrossRef]
- UK Health Security Agency, Technical Briefing 33. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/1043807/technical-briefing-33.pdf (accessed on 29 November 2021).
- Delamater, P.L.; Street, E.J.; Leslie, T.F.; Yang, Y.T.; Jacobsen, K.H. Complexity of the basic reproduction number (R0). Emerg. Infect. Dis. 2019, 25, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Stadnytskyi, V.; Bax, C.E.; Bax, A.; Anfinrud, P. The airborne lifetime of small speech droplets and their potential importance in SARS-CoV-2 transmission. Proc. Natl. Acad. Sci. USA 2020, 117, 11875–11877. [Google Scholar] [CrossRef]
- Echternach, M.; Gantner, S.; Peters, G.; Westphalen, C.; Benthaus, T.; Jakubaβ, B.; Kuranova, L.; Döllinger, M.; Kniesburges, S. Impulse dispersion of aerosols during singing and speaking: A potential COVID-19 transmission pathway. Am. J. Respir. Crit. Care Med. 2020, 202, 1584–1587. [Google Scholar] [CrossRef]
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; del Buono, R.; Costa, F.; Scalata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Falzone, L.; Gattuso, G.; Tsatsakis, A.; Spandidos, D.A.; Libra, M. Current and innovative methods for the diagnosis of COVID-19 infection. Int. J. Mol. Med. 2021, 47, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Van der Toorn, W.; Oh, D.Y.; von Kleist, M. COVID StrategyCalculator: A software to assess testing and quarantine strategies for incoming travelers, contact management, and de-isolation. Patterns 2021, 2, 100262. [Google Scholar] [CrossRef] [PubMed]
- Larremore, D.B.; Fosdick, B.K.; Zhang, S.; Grad, Y.H. Jointly modeling prevalence, sensitivity and specificity for optimal sample allocation. bioRxiv 2020. Available online: https://www.biorxiv.org/content/10.1101/2020.05.23.112649v1 (accessed on 8 September 2021).
- Gelman, A.; Carpenter, B. Bayesian analysis of tests with unknown specificity and sensitivity. J. R. Stat. Soc. Ser. C Appl. Stat. 2020, 69, 1269–1283. [Google Scholar] [CrossRef]
- Bendavid, E.; Mulaney, B.; Sood, N.; Shah, S.; Bromley-Dulfano, R.; Lai, C.; Weissberg, Z.; Saavedra-Walker, R.; Tedrow, J.; Bogan, A.; et al. COVID-19 antibody seroprevalence in Santa Clara County, California. Int. J. Epidemiol. 2021, 50, 410–419. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| State | HIT *** | Delta-Adjusted HIT **** | |||
|---|---|---|---|---|---|
| New Jersey | 0.65 (0.59–0.71) | 0.45 (0.41–0.48) | 7.1 (6.4–7.7) | 0.86 (0.84–0.87) | 0.94 (0.94–0.95) |
| Wyoming | 0.21 (0.21–0.23) | 0.13 (0.13–0.15) | 2.3 (2.3–2.5) | 0.56 (0.56–0.59) | 0.82 (0.82–0.84) |
| Florida | 0.55 (0.48–0.59) | 0.39 (0.34–0.41) | 6.0 (5.2–6.4) | 0.83 (0.81–0.84) | 0.93 (0.92–0.94) |
| Alaska | 0.21 (0.21–0.23) | 0.13 (0.13–0.14) | 2.3 (2.3–2.5) | 0.56 (0.56–0.59) | 0.82 (0.82–0.84) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mallela, A.; Neumann, J.; Miller, E.F.; Chen, Y.; Posner, R.G.; Lin, Y.T.; Hlavacek, W.S. Bayesian Inference of State-Level COVID-19 Basic Reproduction Numbers across the United States. Viruses 2022, 14, 157. https://doi.org/10.3390/v14010157
Mallela A, Neumann J, Miller EF, Chen Y, Posner RG, Lin YT, Hlavacek WS. Bayesian Inference of State-Level COVID-19 Basic Reproduction Numbers across the United States. Viruses. 2022; 14(1):157. https://doi.org/10.3390/v14010157
Chicago/Turabian StyleMallela, Abhishek, Jacob Neumann, Ely F. Miller, Ye Chen, Richard G. Posner, Yen Ting Lin, and William S. Hlavacek. 2022. "Bayesian Inference of State-Level COVID-19 Basic Reproduction Numbers across the United States" Viruses 14, no. 1: 157. https://doi.org/10.3390/v14010157