
Host-Adapted Gene Families Involved in Murine Cytomegalovirus Immune Evasion


Abstract
:1. Introduction
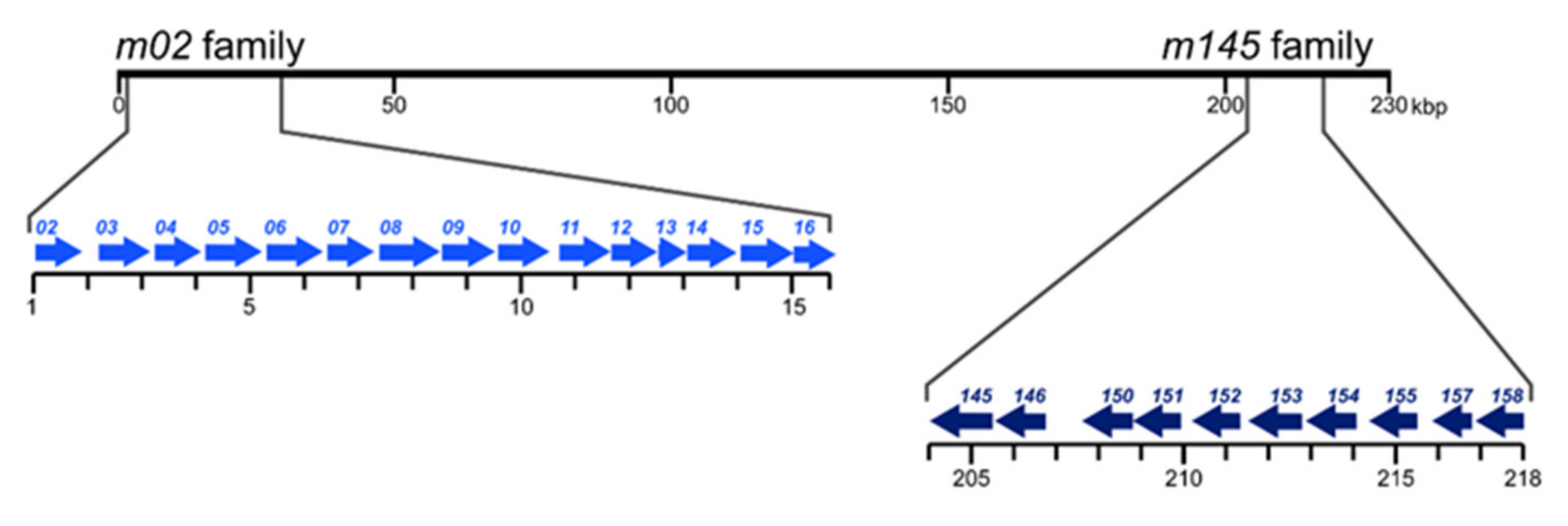
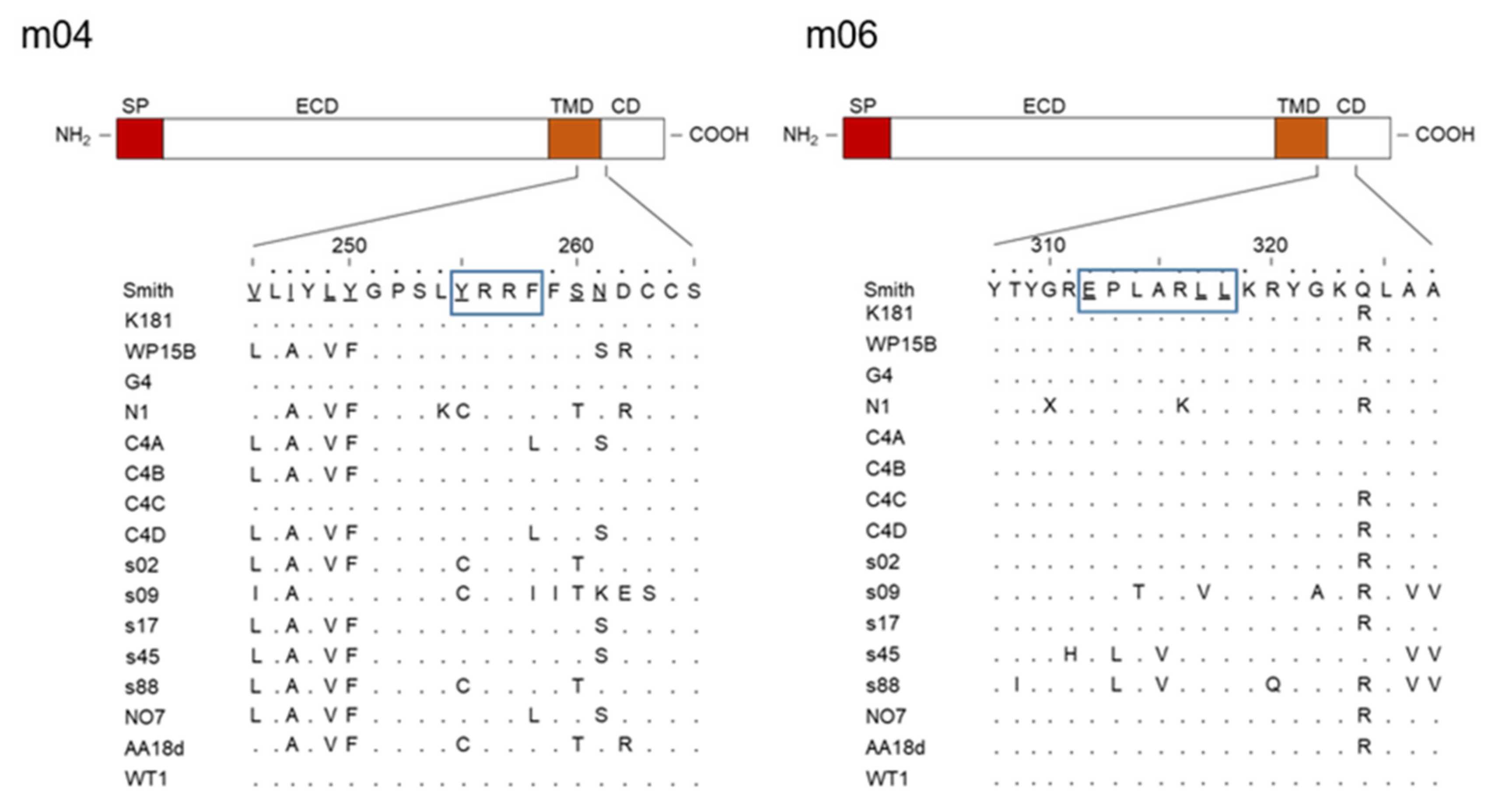
2. The m02 Gene Family
3. The m145 Gene Family
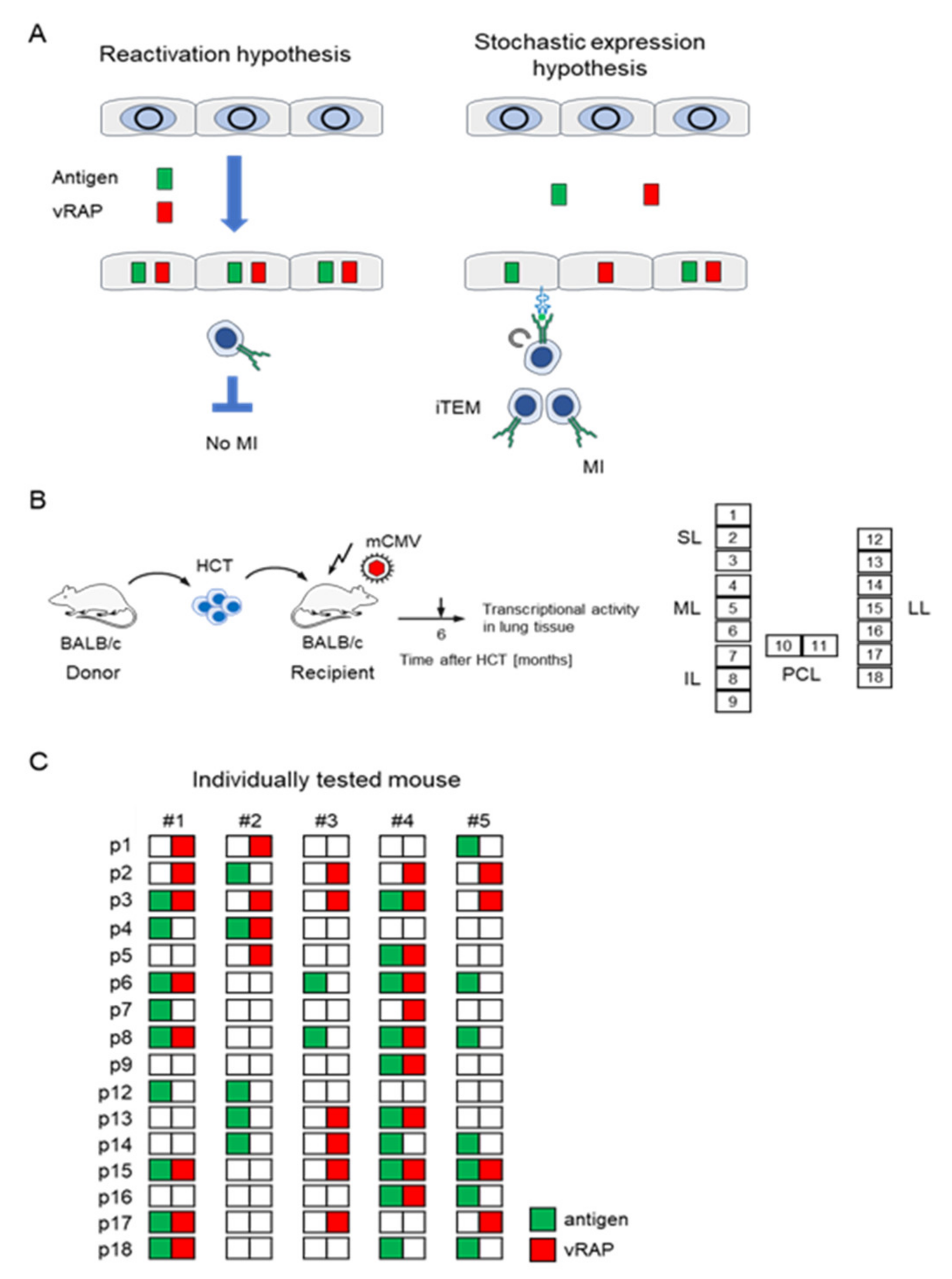
4. The Immune Evasion Enigma in Cytomegalovirus Memory Inflation
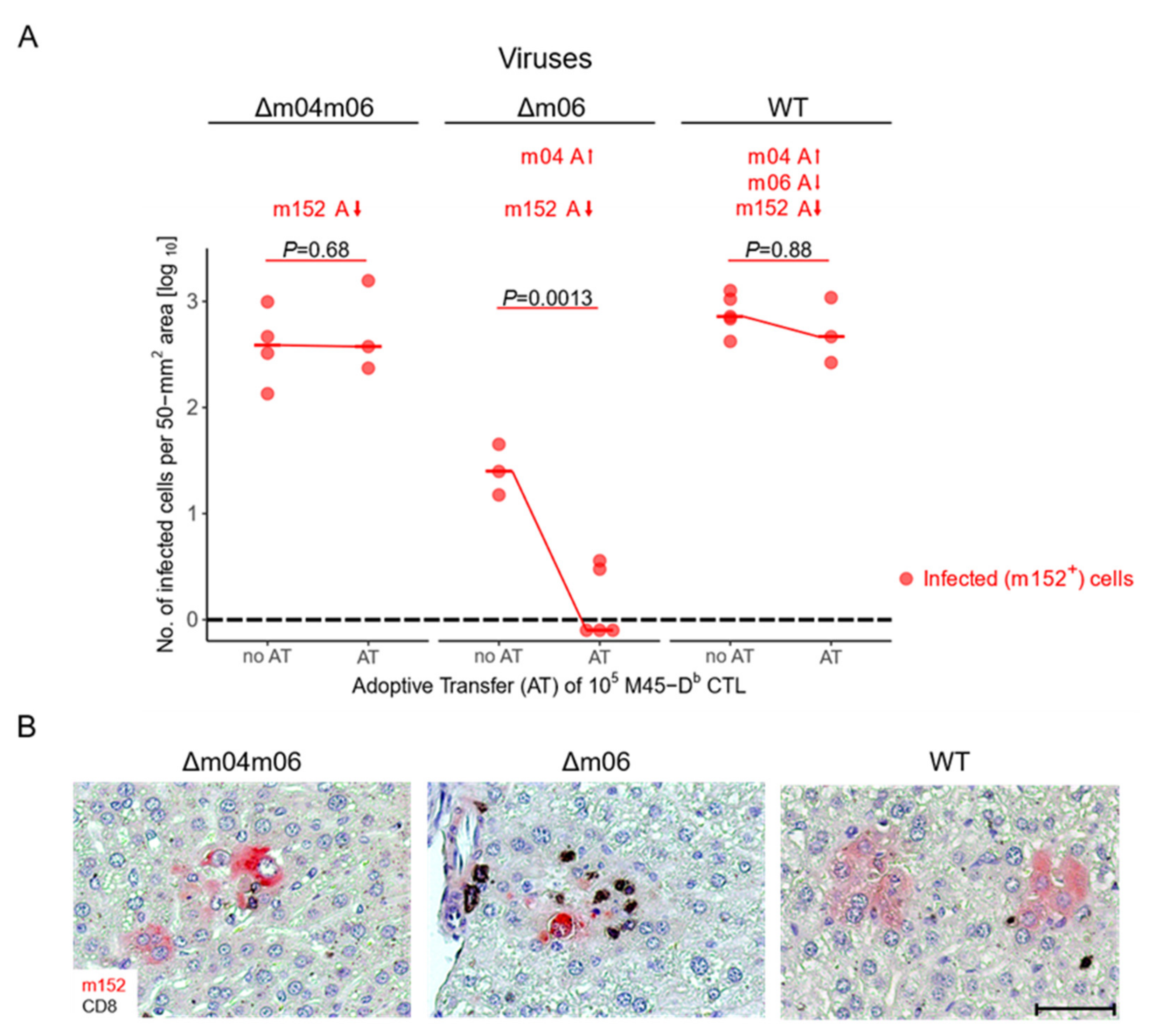
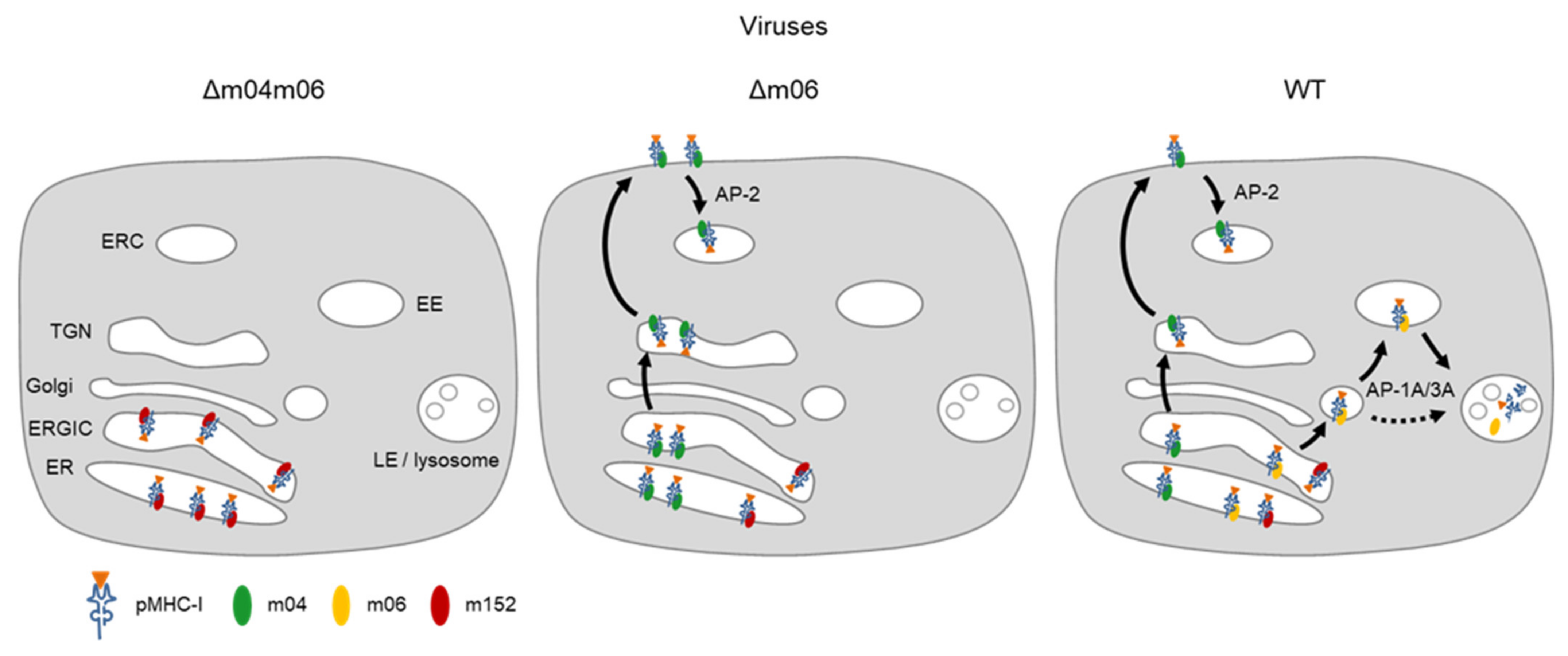
5. Concerted Action of vRAPs in Acute Infection
6. Synopsis of vRAP Interplay in Regulating Antigen Presentation
7. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davison, A.J.; Holton, M.; Dolan, A.; Dargan, D.J.; Gatherer, D.; Hayward, G.S. Comparative genomics of primate cytomegaloviruses. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 1–22. [Google Scholar]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boppana, S.B.; Britt, W.J. Synopsis of clinical aspects of human cytomegalovirus disease. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 1–25. [Google Scholar]
- Cannon, M.J.; Grosse, S.D.; Fowler, K.B. The epidemiology and public health impact of congenital cytomegalovirus infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 26–48. [Google Scholar]
- Adler, S.P.; Nigro, G. Clinical cytomegalovirus research: Congenital infection. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 55–73. [Google Scholar]
- Avery, R.K. Clinical cytomegalovirus research: Thoracic organ transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 286–300. [Google Scholar]
- Emery, V.C.; Milne, R.S.B.; Griffiths, P.D. Clinical cytomegalovirus research: Liver and kidney transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 301–311. [Google Scholar]
- Seo, S.; Boeckh, M. Clinical cytomegalovirus research: Hematopoietic cell transplantation. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 2, pp. 337–353. [Google Scholar]
- Stern, L.; Withers, B.; Avdic, S.; Gottlieb, D.; Abendroth, A.; Blyth, E.; Slobedman, B. Human cytomegalovirus latency and reactivation in allogeneic hematopoietic stem cell transplant recipients. Front. Microbiol. 2019, 10, 1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffiths, P.; Reeves, M. Pathogenesis of human cytomegalovirus in the immunocompromised host. Nat. Rev. Microbiol. 2021, 19, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J. Antigens and immunoevasins: Opponents in cytomegalovirus immune surveillance. Nat. Rev. Immunol. 2002, 2, 831–844. [Google Scholar] [CrossRef]
- Powers, C.; DeFilippis, V.; Malouli, D.; Früh, K. Cytomegalovirus immune evasion. Curr. Top. Microbiol. Immunol. 2008, 325, 333–359. [Google Scholar] [PubMed]
- Lisnić, B.; Lisnić, V.J.; Jonjić, S. NK cell interplay with cytomegaloviruses. Curr. Opin. Virol. 2015, 15, 9–18. [Google Scholar] [CrossRef]
- Stempel, M.; Chan, B.; Brinkmann, M.M. Coevolution pays off: Herpesviruses have the license to escape the DNA sensing pathway. Med. Microbiol. Immunol. 2019, 208, 495–512. [Google Scholar] [CrossRef]
- Berry, R.; Watson, G.M.; Jonjic, S.; Degli-Esposti, M.A.; Rossjohn, J. Modulation of innate and adaptive immunity by cytomegaloviruses. Nat. Rev. Immunol. 2019, 20, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Fielding, C.A.; Weekes, M.P.; Nobre, L.V.; Ruckova, E.; Wilkie, G.S.; Paulo, J.A.; Chang, C.; Suárez, N.M.; Davies, J.A.; Antrobus, R.; et al. Control of immune ligands by members of a cytomegalovirus gene expansion suppresses natural killer cell activation. Elife 2017, 6, e22206. [Google Scholar] [CrossRef] [PubMed]
- Reddehase, M.J.; Lemmermann, N.A. Mouse model of cytomegalovirus disease and immunotherapy in the immunocompromised host: Predictions for medical translation that survived the “test of time”. Viruses 2018, 10, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Val, M.; Münch, K.; Reddehase, M.J.; Koszinowski, U.H. Presentation of CMV immediate-early antigen to cytolytic T lymphocytes is selectively prevented by viral genes expressed in the early phase. Cell 1989, 58, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Del Val, M.; Hengel, H.; Häcker, H.; Hartlaub, U.; Ruppert, T.; Lucin, P.; Koszinowski, U.H. Cytomegalovirus prevents antigen presentation by blocking the transport of peptide-loaded major histocompatibility complex class I molecules into the medial-Golgi compartment. J. Exp. Med. 1992, 176, 729–738. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redwood, A.J.; Shellam, G.R.; Smith, L.M. Molecular evolution of murine cytomegalovirus genomes. In Cytomegaloviruses: From Molecular Pathogenesis to Intervention; Reddehase, M.J., Ed.; Caister Academic Press: Norfolk, UK, 2013; Volume 1, pp. 23–37. [Google Scholar]
- Oliveira, S.A.; Park, S.H.; Lee, P.; Bendelac, A.; Shenk, T.E. Murine cytomegalovirus m02 gene family protects against natural killer cell-mediated immune surveillance. J. Virol. 2002, 76, 885–894. [Google Scholar] [CrossRef] [Green Version]
- Corbett, A.J.; Forbes, C.A.; Moro, D.; Scalzo, A.A. Extensive sequence variation exists among isolates of murine cytomegalovirus within members of the m02 family of genes. J. Gen. Virol. 2007, 88, 758–769. [Google Scholar] [CrossRef]
- Berry, R.; Vivian, J.P.; Deuss, F.A.; Balaji, G.R.; Saunders, P.M.; Lin, J.; Littler, D.R.; Brooks, A.G.; Rossjohn, J. The structure of the cytomegalovirus-encoded m04 glycoprotein, a prototypical member of the m02 family of immunoevasins. J. Biol. Chem. 2014, 289, 23753–23763. [Google Scholar] [CrossRef] [Green Version]
- Kleijnen, M.F.; Huppa, J.B.; Lucin, P.; Mukherjee, S.; Farrell, H.; Campbell, A.E.; Koszinowski, U.H.; Hill, A.B.; Ploegh, H.L. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 1997, 16, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, D.G.; Koszinowski, U.H.; Hill, A.B. The murine cytomegalovirus immune evasion protein m4/gp34 forms biochemically distinct complexes with class I MHC at the cell surface and in a pre-Golgi compartment. J. Immunol. 2001, 167, 3894–3902. [Google Scholar] [CrossRef]
- Lu, X.; Kavanagh, D.G.; Hill, A.B. Cellular and molecular requirements for association of the murine cytomegalovirus protein m4/gp34 with major histocompatibility complex class I molecules. J. Virol. 2006, 80, 6048–6055. [Google Scholar] [CrossRef] [Green Version]
- Babić, M.; Pyzik, M.; Zafirova, B.; Mitrović, M.; Butorac, V.; Lanier, L.L.; Krmpotić, A.; Vidal, S.M.; Jonjić, S. Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J. Exp. Med. 2010, 207, 2663–2673. [Google Scholar] [CrossRef] [PubMed]
- Fink, A.; Blaum, F.; Babic Cac, M.; Ebert, S.; Lemmermann, N.A.; Reddehase, M.J. An endocytic YXXΦ (YRRF) cargo sorting motif in the cytoplasmic tail of murine cytomegalovirus AP2 ‘adapter adapter’ protein m04/gp34 antagonizes virus evasion of natural killer cells. Med. Microbiol. Immunol. 2015, 204, 383–394. [Google Scholar] [CrossRef]
- Železnjak, J.; Lisnić, V.J.; Popović, B.; Lisnić, B.; Babić, M.; Halenius, A.; L’Hernault, A.; Roviš, T.L.; Hengel, H.; Erhard, F.; et al. The complex of MCMV proteins and MHC class I evades NK cell control and drives the evolution of virus-specific activating Ly49 receptors. J. Exp. Med. 2019, 216, 1809–1827. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, R.; Gillert-Marien, D.; Thomas, D.; Podlech, J.; Deegen, P.; Herter, S.; Oehrlein-Karpi, S.A.; Strand, D.; Wagner, M.; Reddehase, M.J. Cytomegalovirus encodes a positive regulator of antigen presentation. J. Virol. 2006, 80, 7613–7624. [Google Scholar] [CrossRef] [Green Version]
- Becker, S.; Fink, A.; Podlech, J.; Giese, I.; Schmiedeke, J.K.; Bukur, T.; Reddehase, M.J.; Lemmermann, N.A. Positive role of the MHC class-I antigen presentation regulator m04/gp34 of murine cytomegalovirus in antiviral protection by CD8 T cells. Front. Cell. Infect. Microbiol. 2020, 10, 454. [Google Scholar] [CrossRef] [PubMed]
- Reusch, U.; Muranyi, W.; Lucin, P.; Burgert, H.G.; Hengel, H.; Koszinowski, U.H. A cytomegalovirus glycoprotein re-routes MHC class I complexes to lysosomes for degradation. EMBO J. 1999, 18, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Bubeck, A.; Reusch, U.; Wagner, M.; Ruppert, T.; Muranyi, W.; Kloetzel, P.M.; Koszinowski, U.H. The glycoprotein gp48 of murine cytomegalovirus: Proteasome-dependent cytosolic dislocation and degradation. J. Biol. Chem. 2002, 277, 2216–2224. [Google Scholar] [CrossRef] [Green Version]
- Reusch, U.; Bernhard, O.; Koszinowski, U.; Schu, P. AP-1A and AP-3A lysosomal sorting functions. Traffic 2002, 3, 752–761. [Google Scholar] [CrossRef]
- Fink, A.; Mikuličić, S.; Blaum, F.; Reddehase, M.J.; Florin, L.; Lemmermann, N.A. Function of the cargo sorting dileucine motif in a cytomegalovirus immune evasion protein. Med. Microbiol. Immunol. 2019, 208, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, O.A.; Berry, R.; Rahim, M.M.A.; Reichel, J.J.; Popović, B.; Tanaka, M.; Fu, Z.; Balaji, G.R.; Lau, T.N.H.; Tu, M.M.; et al. A viral immunoevasin controls innate immunity by targeting the prototypical natural killer cell receptor family. Cell 2017, 169, 58–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, B.; Arapovi, M.; Masters, L.L.; Rwandamuiye, F.; Jonjic, S.; Smith, L.M.; Redwood, A.J. The m15 locus of murine cytomegalovirus modulates natural killer cell responses to promote dissemination to the salivary glands and viral shedding. Pathogens 2021, 10, 866. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Traub, L.M. Signals for sorting of transmembrane proteins to endosomes and lysosomes. Annu. Rev. Biochem. 2003, 72, 395–447. [Google Scholar] [CrossRef] [Green Version]
- Nakatsu, F.; Ohno, H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell. Struct. Funct. 2003, 28, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Guo, X. Adaptor protein complexes and intracellular transport. Biosci. Rep. 2014, 34, e00123. [Google Scholar] [CrossRef]
- Redwood, A.J.; Messerle, M.; Harvey, N.L.; Hardy, C.M.; Koszinowski, U.H.; Lawson, M.A.; Shellam, G.R. Use of a murine cytomegalovirus K181-derived bacterial artificial chromosome as a vaccine vector for immunocontraception. J. Virol. 2005, 79, 2998–3008. [Google Scholar] [CrossRef] [Green Version]
- Smith, L.M.; McWhorter, A.R.; Masters, L.L.; Shellam, G.R.; Redwood, A.J. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 2008, 82, 6689–6696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, L.M.; McWhorter, A.R.; Shellam, G.R.; Redwood, A.J. The genome of murine cytomegalovirus is shaped by purifying selection and extensive recombination. Virology 2013, 435, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Čížková, D.; Baird, S.J.E.; Těšíková, J.; Voigt, S.; Ľudovít, Ď.; Piálek, J.; Goüy de Bellocq, J. Host subspecific viral strains in European house mice: Murine cytomegalovirus in the Eastern (Mus musculus musculus) and Western house mouse (Mus musculus domesticus). Virology 2018, 521, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.P.; Valentine, M.C.; Gao, J.; Pingel, J.T.; Yokoyama, W.M. Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J. Virol. 2010, 84, 2623–2628. [Google Scholar] [CrossRef] [Green Version]
- Sgourakis, N.G.; Natarajan, K.; Ying, J.; Vogeli, B.; Boyd, L.F.; Margulies, D.H.; Bax, A. The structure of mouse cytomegalovirus m04 protein obtained from sparse NMR data reveals a conserved fold of the m02-m06 viral immune modulator family. Structure 2014, 22, 1263–1273. [Google Scholar] [CrossRef] [Green Version]
- Sgourakis, N.G.; May, N.A.; Boyd, L.F.; Ying, J.; Bax, A.; Margulies, D.H. A novel MHC-I surface targeted for binding by the MCMV m06 immunoevasin revealed by solution NMR. J. Biol. Chem. 2015, 290, 28857–28868. [Google Scholar] [CrossRef] [Green Version]
- Lefranc, M.P. IMGT, the international ImMunoGeneTics database. Nucleic. Acids. Res. 2003, 31, 307–310. [Google Scholar] [CrossRef]
- Ohno, H.; Stewart, J.; Fournier, M.C.; Bosshart, H.; Rhee, I.; Miyatake, S.; Saito, T.; Gallusser, A.; Kirchhausen, T.; Bonifacino, J.S. Interaction of tyrosine-based sorting signals with clathrin-associated proteins. Science 1995, 269, 1872–1875. [Google Scholar] [CrossRef] [Green Version]
- Traub, L.M. Sorting it out: AP-2 and alternate clathrin adaptors in endocytic cargo selection. J. Cell. Biol. 2003, 163, 203–208. [Google Scholar] [CrossRef]
- Reider, A.; Wendland, B. Endocytic adaptors—social networking at the plasma membrane. J. Cell. Sci. 2011, 124, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielczewska, A.; Pyzik, M.; Sun, T.; Krmpotic, A.; Lodoen, M.B.; Munks, M.W.; Babic, M.; Hill, A.B.; Koszinowski, U.H.; Jonjic, S.; et al. Ly49P recognition of cytomegalovirus-infected cells expressing H2-D k and CMV-encoded m04 correlates with the NK cell antiviral response. J. Exp. Med. 2009, 206, 515–523. [Google Scholar] [CrossRef] [PubMed]
- Pyzik, M.; Charbonneau, B.; Gendron-Pontbriand, E.M.; Babić, M.; Krmpotić, A.; Jonjić, S.; Vidal, S.M. Distinct MHC class I-dependent NK cell-activating receptors control cytomegalovirus infection in different mouse strains. J. Exp. Med. 2011, 208, 1105–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, M.; Gutermann, A.; Podlech, J.; Reddehase, M.J.; Koszinowski, U.H. Major histocompatibility complex class I allele-specific cooperative and competitive interactions between immune evasion proteins of cytomegalovirus. J. Exp. Med. 2002, 196, 805–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krmpotic, A.; Hasan, M.; Loewendorf, A.; Saulig, T.; Halenius, A.; Lenac, T.; Polic, B.; Bubic, I.; Kriegeskorte, A.; Pernjak-Pugel, E.; et al. NK cell activation through the NKG2D ligand MULT-1 is selectively prevented by the glycoprotein encoded by mouse cytomegalovirus gene m145. J. Exp. Med. 2005, 201, 211–220. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Natarajan, K.; Revilleza, M.J.R.; Boyd, L.F.; Zhi, L.; Zhao, H.; Robinson, H.; Margulies, D.H. Structural basis of mouse cytomegalovirus m152/gp40 interaction with RAE1 reveals a paradigm for MHC/MHC interaction in immune evasion. Proc. Natl. Acad. Sci. USA 2012, 109, 3578–3587. [Google Scholar] [CrossRef] [Green Version]
- Ziegler, H.; Thale, R.; Lucin, P.; Muranyi, W.; Flohr, T.; Hengel, H.; Farrell, H.; Rawlinson, W.; Koszinowski, U.H. A mouse cytomegalovirus glycoprotein retains MHC class I complexes in the ERGIC/cis-Golgi compartments. Immunity 1997, 6, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Krmpotic, A.; Messerle, M.; Crnkovic-Mertens, I.; Polic, B.; Jonjic, S.; Koszinowski, U.H. The immunoevasive function encoded by the mouse cytomegalovirus gene m152 protects the virus against T cell control in vivo. J. Exp. Med. 1999, 190, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Holtappels, R.; Podlech, J.; Pahl-Seibert, M.F.; Jülch, M.; Thomas, D.; Simon, C.O.; Wagner, M.; Reddehase, M.J. Cytomegalovirus misleads its host by priming of CD8 T cells specific for an epitope not presented in infected tissues. J. Exp. Med. 2004, 199, 131–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krmpotić, A.; Busch, D.H.; Bubić, I.; Gebhardt, F.; Hengel, H.; Hasan, M.; Scalzo, A.A.; Koszinowski, U.H.; Jonjić, S. MCMV glycoprotein gp40 confers virus resistance to CD8+ T cells and NK cells in vivo. Nat. Immunol. 2002, 3, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Lodoen, M.; Ogasawara, K.; Hamerman, J.A.; Arase, H.; Houchins, J.P.; Mocarski, E.S.; Lanier, L.L. NKG2D-mediated natural killer cell protection against cytomegalovirus is impaired by viral gp40 modulation of retinoic acid early inducible 1 gene molecules. J. Exp. Med. 2003, 197, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Zhi, L.; Mans, J.; Paskow, M.J.; Brown, P.H.; Schuck, P.; Jonjić, S.; Natarajan, K.; Margulies, D.H. Direct interaction of the mouse cytomegalovirus m152/gp40 immunoevasin with RAE-1 isoforms. Biochemistry 2010, 49, 2443–2453. [Google Scholar] [CrossRef] [Green Version]
- Lis, N.; Hein, Z.; Ghanwat, S.S.; Ramnarayan, V.R.; Chambers, B.J.; Springer, S. The murine cytomegalovirus immunoevasin gp40/m152 inhibits NKG2D receptor RAE-1γ by intracellular retention and cell surface masking. J. Cell. Sci. 2021, 134, jcs257428. [Google Scholar] [CrossRef]
- Stempel, M.; Chan, B.; Juranić Lisnić, V.; Krmpotić, A.; Hartung, J.; Paludan, S.R.; Füllbrunn, N.; Lemmermann, N.A.; Brinkmann, M.M. The herpesviral antagonist m152 reveals differential activation of STING -dependent IRF and NF-κB signaling and STING ’s dual role during MCMV infection. EMBO J. 2019, 38, 1–22. [Google Scholar] [CrossRef]
- Mans, J.; Natarajan, K.; Balbo, A.; Schuck, P.; Eike, D.; Hess, S.; Robinson, H.; Šimić, H.; Jonjić, S.; Tiemessen, C.T.; et al. Cellular expression and crystal structure of the murine cytomegalovirus major histocompatibility complex class I-like glycoprotein, m153. J. Biol. Chem. 2007, 282, 35247–35258. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, O.A.; Mesci, A.; Ma, J.; Chen, P.; Kirkham, C.L.; Hundrieser, J.; Voigt, S.; Allan, D.S.; Carlyle, J.R. Modulation of Clr ligand expression and NKR-P1 receptor function during murine cytomegalovirus infection. J. Innate Immun. 2015, 7, 584–600. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, O.A.; Sampaio, I.S.; Rahim, M.M.A.; Samaniego, J.D.; Tilahun, M.E.; Krishnamoorthy, M.; Popović, B.; Babić, M.; Krmpotić, A.; Treanor, B.; et al. Mouse cytomegalovirus m153 protein stabilizes expression of the inhibitory NKR-P1B ligand Clr-b. J. Virol. 2019, 94, e01220-19. [Google Scholar] [CrossRef]
- Zarama, A.; Pérez-Carmona, N.; Farré, D.; Tomic, A.; Borst, E.M.; Messerle, M.; Jonjic, S.; Engel, P.; Angulo, A. Cytomegalovirus m154 hinders CD48 cell-surface expression and promotes viral escape from host natural killer cell control. PLoS Pathog. 2014, 10, e1004000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geljic, I.S.; Brlic, P.K.; Angulo, G.; Brizic, I.; Lisnic, B.; Jenus, T.; Lisnic, V.J.; Pietri, G.P.; Engel, P.; Kaynan, N.; et al. Cytomegalovirus protein m154 perturbs the adaptor protein-1 compartment mediating broad-spectrum immune evasion. Elife 2020, 9, 1–23. [Google Scholar]
- Lodoen, M.B.; Abenes, G.; Umamoto, S.; Houchins, J.P.; Liu, F.; Lanier, L.L. The cytomegalovirus m155 gene product subverts natural killer cell antiviral protection by disruption of H60-NKG2D interactions. J. Exp. Med. 2004, 200, 1075–1081. [Google Scholar] [CrossRef]
- Hasan, M.; Krmpotic, A.; Ruzsics, Z.; Bubic, I.; Lenac, T.; Halenius, A.; Loewendorf, A.; Messerle, M.; Hengel, H.; Jonjic, S.; et al. Selective down-regulation of the NKG2D ligand H60 by mouse cytomegalovirus m155 glycoprotein. J. Virol. 2005, 79, 2920–2930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loewendorf, A.I.; Steinbrueck, L.; Peter, C.; Busche, A.; Benedict, C.A.; Kay-Jackson, P.C. The mouse cytomegalovirus glycoprotein m155 inhibits CD40 expression and restricts CD4 T cell responses. J. Virol. 2011, 85, 5208–5212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, E.J.; Juo, Z.S.; Venook, R.T.; Boulanger, M.J.; Arase, H.; Lanier, L.L.; Garcia, K.C. Structural elucidation of the m157 mouse cytomegalovirus ligand for Ly49 natural killer cell receptors. Proc. Natl. Acad. Sci. USA 2007, 104, 10128–10133. [Google Scholar] [CrossRef] [Green Version]
- Berry, R.; Ng, N.; Saunders, P.M.; Vivian, J.P.; Lin, J.; Deuss, F.A.; Corbett, A.J.; Forbes, C.A.; Widjaja, J.M.; Sullivan, L.C.; et al. Targeting of a natural killer cell receptor family by a viral immunoevasin. Nat. Immunol. 2013, 14, 699–705. [Google Scholar] [CrossRef]
- Brown, M.G.; Dokun, A.O.; Heusel, J.W.; Smith, H.R.; Beckman, D.L.; Blattenberger, E.A.; Dubbelde, C.E.; Stone, L.R.; Scalzo, A.A.; Yokoyama, W.M. Vital involvement of a natural killer cell activation receptor in resistance to viral infection. Science 2001, 292, 934–937. [Google Scholar] [CrossRef]
- Arase, H.; Mocarski, E.S.; Campbell, A.E.; Hill, A.B.; Lanier, L.L. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science 2002, 296, 1323–1326. [Google Scholar] [CrossRef]
- Smith, H.R.C.; Heusel, J.W.; Mehta, I.K.; Kim, S.; Dorner, B.G.; Naidenko, O.V.; Iizuka, K.; Furukawa, H.; Beckman, D.L.; Pingel, J.T.; et al. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 8826–8831. [Google Scholar] [CrossRef] [Green Version]
- Voigt, V.; Forbes, C.A.; Tonkin, J.N.; Degli-Esposti, M.A.; Smith, H.R.C.; Yokoyama, W.M.; Scalzo, A.A. Murine cytomegalovirus m157 mutation and variation leads to immune evasion of natural killer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 13483–13488. [Google Scholar] [CrossRef] [Green Version]
- Bubić, I.; Wagner, M.; Krmpotić, A.; Saulig, T.; Kim, S.; Yokoyama, W.M.; Jonjić, S.; Koszinowski, U.H. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J. Virol. 2004, 78, 7536–7544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, A.H.; Guseva, N.V.; Ball, B.L.; Heusel, J.W. Characterization of murine cytomegalovirus m157 from infected cells and identification of critical residues mediating recognition by the NK cell receptor Ly49H. J. Immunol. 2008, 181, 265–275. [Google Scholar] [CrossRef] [Green Version]
- McWhorter, A.R.; Smith, L.M.; Masters, L.L.; Chan, B.; Shellam, G.R.; Redwood, A.J. Natural killer cell dependent within-host competition arises during multiple MCMV infection: Consequences for viral transmission and evolution. PLoS Pathog. 2013, 9, e1003111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, H.; Muranyi, W.; Burgert, H.G.; Kremmer, E.; Koszinowski, U.H. The luminal part of the murine cytomegalovirus glycoprotein gp40 catalyzes the retention of MHC class I molecules. EMBO J. 2000, 19, 870–881. [Google Scholar] [CrossRef]
- Fink, A.; Renzaho, A.; Reddehase, M.J.; Lemmermann, N.A. The p36 isoform of murine cytomegalovirus m152 protein suffices for mediating innate and adaptive immune evasion. Viruses 2013, 5, 3171–3191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janßen, L.; Ramnarayan, V.R.; Aboelmagd, M.; Iliopoulou, M.; Hein, Z.; Majoul, I.; Fritzsche, S.; Halenius, A.; Springer, S. The murine cytomegalovirus immunoevasin gp40 binds MHC class I molecules to retain them in the early secretory pathway. J. Cell Sci. 2016, 129, 219–227. [Google Scholar] [CrossRef] [Green Version]
- Ramnarayan, V.R.; Hein, Z.; Janßen, L.; Lis, N.; Ghanwat, S.; Springer, S. Cytomegalovirus gp40/m152 Uses TMED10 as ER Anchor to Retain MHC Class I. Cell Rep. 2018, 23, 3068–3077. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Lemmermann, N.A. Cellular reservoirs of latent cytomegaloviruses. Med. Microbiol. Immunol. 2019, 208, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Seckert, C.K.; Griessl, M.; Büttner, J.K.; Scheller, S.; Simon, C.O.; Kropp, K.A.; Renzaho, A.; Kühnapfel, B.; Grzimek, N.K.A.; Reddehase, M.J. Viral latency drives ‘memory inflation’: A unifying hypothesis linking two hallmarks of cytomegalovirus infection. Med. Microbiol. Immunol. 2012, 201, 551–566. [Google Scholar] [CrossRef]
- Klenerman, P.; Oxenius, A. T cell responses to cytomegalovirus. Nat. Rev. Immunol. 2016, 16, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; Van Dommelen, S.; Shellam, G.R.; Hill, A.B. Memory inflation during chronic viral infection is maintained by continuous production of short-lived, functional T cells. Immunity 2008, 29, 650–659. [Google Scholar] [CrossRef] [Green Version]
- Thimme, R.; Appay, V.; Koschella, M.; Panther, E.; Roth, E.; Hislop, A.D.; Rickinson, A.B.; Rowland-Jones, S.L.; Blum, H.E.; Pircher, H. Increased expression of the NK cell receptor KLRG1 by virus-specific CD8 T cells during persistent antigen stimulation. J. Virol. 2005, 79, 12112–12116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torti, N.; Walton, S.M.; Brocker, T.; Rülicke, T.; Oxenius, A. Non-hematopoietic cells in lymph nodes drive memory CD8 T cell inflation during murine cytomegalovirus infection. PLoS Pathog. 2011, 7, e1002313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welten, S.P.M.; Baumann, N.S.; Oxenius, A. Fuel and brake of memory T cell inflation. Med. Microbiol. Immunol. 2019, 208, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Lemmermann, N.A.; Reddehase, M.J. Direct evidence for viral antigen presentation during latent cytomegalovirus infection. Pathogens 2021, 10, 731. [Google Scholar] [CrossRef]
- Griessl, M.; Renzaho, A.; Freitag, K.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A. Stochastic episodes of latent cytomegalovirus transcription drive CD8 T-cell “memory inflation” and avoid immune evasion. Front. Immunol. 2021, 12, 668885. [Google Scholar] [CrossRef]
- Gabel, M.; Baumann, N.S.; Oxenius, A.; Graw, F. Investigating the dynamics of MCMV-specific CD8+ T cell responses in individual hosts. Front. Immunol. 2019, 10, 1358. [Google Scholar] [CrossRef]
- Snyder, C.M.; Cho, K.S.; Bonnett, E.L.; Allan, J.E.; Hill, A.B. Sustained CD8+ T cell memory inflation after infection with a single-cycle cytomegalovirus. PLoS Pathog. 2011, 7, e1002295. [Google Scholar] [CrossRef] [Green Version]
- Gold, M.C.; Munks, M.W.; Wagner, M.; Koszinowski, U.H.; Hill, A.B.; Fling, S.P. The murine cytomegalovirus immunomodulatory gene m152 prevents recognition of infected cells by M45-specific CTL but does not alter the immunodominance of the M45-specific CD8 T cell response in vivo. J. Immunol. 2002, 169, 359–365. [Google Scholar] [CrossRef] [Green Version]
- Holtappels, R.; Thomas, D.; Reddehase, M.J. The efficacy of antigen processing is critical for protection against cytomegalovirus disease in the presence of viral immune evasion proteins. J. Virol. 2009, 83, 9611–9615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, A.; Lemmermann, N.A.; Gillert-Marien, D.; Thomas, D.; Freitag, K.; Böhm, V.; Wilhelmi, V.; Reifenberg, K.; Reddehase, M.J.; Holtappels, R. Antigen presentation under the influence of ‘immune evasion’ proteins and its modulation by interferon-gamma: Implications for immunotherapy of cytomegalovirus infection with antiviral CD8 T cells. Med. Microbiol. Immunol. 2012, 201, 513–525. [Google Scholar] [CrossRef]
- Böhm, V.; Podlech, J.; Thomas, D.; Deegen, P.; Pahl-Seibert, M.F.; Lemmermann, N.A.; Grzimek, N.K.; Oehrlein-Karpi, S.A.; Reddehase, M.J.; Holtappels, R. Epitope-specific in vivo protection against cytomegalovirus disease by CD8 T cells in the murine model of preemptive immunotherapy. Med. Microbiol. Immunol. 2008, 197, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Lemmermann, N.A.; Gergely, K.; Böhm, V.; Deegen, P.; Däubner, T.; Reddehase, M.J. Immune evasion proteins of murine cytomegalovirus preferentially affect cell surface display of recently generated peptide presentation complexes. J. Virol. 2010, 84, 1221–1236. [Google Scholar] [CrossRef] [Green Version]
- Lučin, P.; Mahmutefendić, H.; Blagojević Zagorac, G.; Ilić Tomaš, M. Cytomegalovirus immune evasion by perturbation of endosomal trafficking. Cell. Mol. Immunol. 2015, 12, 154–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holtappels, R.; Schader, S.I.; Oettel, O.; Podlech, J.; Seckert, C.K.; Reddehase, M.J.; Lemmermann, N.A. Insufficient antigen presentation due to viral immune evasion explains lethal cytomegalovirus organ disease after allogeneic hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gezinir, E.; Podlech, J.; Gergely, K.M.; Becker, S.; Reddehase, M.J.; Lemmermann, N.A. Enhancement of antigen presentation by deletion of viral immune evasion genes prevents lethal cytomegalovirus disease in minor histocompatibility antigen-mismatched hematopoietic cell transplantation. Front. Cell. Infect. Microbiol. 2020, 10, 279. [Google Scholar] [CrossRef]
- Reddehase, M.J.; Holtappels, R.; Lemmermann, N.A. Consequence of histoincompatibility beyond GvH-reaction in cytomegalovirus disease associated with allogeneic hematopoietic cell transplantation: Change of paradigm. Viruses 2021, 13, 1530. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Protein | Molecular Mass | Protein Structure | Sorting Motif | Binding Partner (Cargo) | Receptor of Cell Surface Ligand | Role in Immunity | Ref. |
|---|---|---|---|---|---|---|---|
| m02 | ‒ | ‒ | YRDL | ‒ | ‒ | none found | [22] |
| m03;m03.5 | ‒ | ‒ | none | ‒ | not identified | ‒ | [23] |
| m04/gp34 | 34 kDa | PDB ID 4PN6 [24] | YRRF | MHC-I | Ly49-family NK cell receptors Ly49A, C, G2 | NK cell evasion | [25,26,27,28,29,30] |
| TCR | Presentation of pMHC-I | [31,32] | |||||
| m05 | ‒ | ‒ | YICL | ‒ | ‒ | ‒ | |
| m06/gp48 | 48 kDa | ‒ | EPLARLL | MHC-I | ‒ | CD8+ T cell evasion | [33,34,35,36] |
| m07 | ‒ | ‒ | YGFF | ‒ | ‒ | ‒ | |
| m08 | ‒ | ‒ | YGFL | ‒ | ‒ | ‒ | |
| m09 | ‒ | ‒ | YGFL | ‒ | ‒ | ‒ | |
| m10 | ‒ | ‒ | YGFL | ‒ | ‒ | ‒ | |
| m11 | ‒ | ‒ | none | ‒ | ‒ | ‒ | |
| m12 | 40 kDa | PDB ID 5TZN | YRRRGF | NKR-P1B and isoforms | Clr-b or unknown | NK cell evasion | [37] |
| m13 | ‒ | ‒ | none | ‒ | ‒ | ‒ | |
| m14 | ‒ | ‒ | none | ‒ | ‒ | ‒ | |
| m15 locus | ‒ | ‒ | none | ‒ | ‒ | NK cell evasion | [38] |
| m16 | ‒ | ‒ | YAIL | ‒ | ‒ | ‒ |
| Gene/Protein | Molecular Mass | Protein Structure | Binding Partner | Receptor of Cell Surface Ligand | Role in Immunity | Ref. |
|---|---|---|---|---|---|---|
| m145 | 53 kDa | ‒ | MULT-1 | NKG2D | NK cell evasion | [56] |
| m146 | ‒ | ‒ | ‒ | ‒ | ‒ | |
| m150 | ‒ | ‒ | ‒ | ‒ | ‒ | |
| m151 | ‒ | ‒ | ‒ | ‒ | ‒ | |
| m152/gp40 | 48 kDa/ 40 kDa | PDB ID 4G59 [57] | MHC-I | TCR | T cell evasion | [19,58,59,60] |
| RAE-1 | NKG2D | NK cell evasion; | [61,62,63,64] | |||
| STING | ‒ | Innate immunity | [65] | |||
| m153 | 80 kDa | PDB ID 2O5N [66] | Clr-b | NKR-P1B | NK cell evasion | [67,68] |
| m154 | 60 kDa | ‒ | AP-1 cargo: CD18, CD47, CD48, CD54, CD84, CD155, CD162, CD229, CD262, CD270 | CD244 (for CD48) | Broad immune evasion NK cells and CD8+ T cells | [69,70] |
| m155 | 60 kDa | ‒ | H60 | NKG2D | NK cell evasion | [71,72] |
| CD40 | CD40L | CD4+ T cell evasion | [73] | |||
| m157 | 42-50 kDa | PDB ID 2NYK [74]; 4JO8 [75] | Ly49H | Activation of NK cells | [76,77,78,79,80,81,82] | |
| m158 | ‒ | ‒ | ‒ | ‒ | ‒ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Becker, S.; Fink, A.; Podlech, J.; Reddehase, M.J.; Lemmermann, N.A. Host-Adapted Gene Families Involved in Murine Cytomegalovirus Immune Evasion. Viruses 2022, 14, 128. https://doi.org/10.3390/v14010128
Becker S, Fink A, Podlech J, Reddehase MJ, Lemmermann NA. Host-Adapted Gene Families Involved in Murine Cytomegalovirus Immune Evasion. Viruses. 2022; 14(1):128. https://doi.org/10.3390/v14010128
Chicago/Turabian StyleBecker, Sara, Annette Fink, Jürgen Podlech, Matthias J. Reddehase, and Niels A. Lemmermann. 2022. "Host-Adapted Gene Families Involved in Murine Cytomegalovirus Immune Evasion" Viruses 14, no. 1: 128. https://doi.org/10.3390/v14010128