
Drivers and Distribution of Henipavirus-Induced Syncytia: What Do We Know?

 , , , , , , and
, , , , , , and
Abstract
:1. Introduction
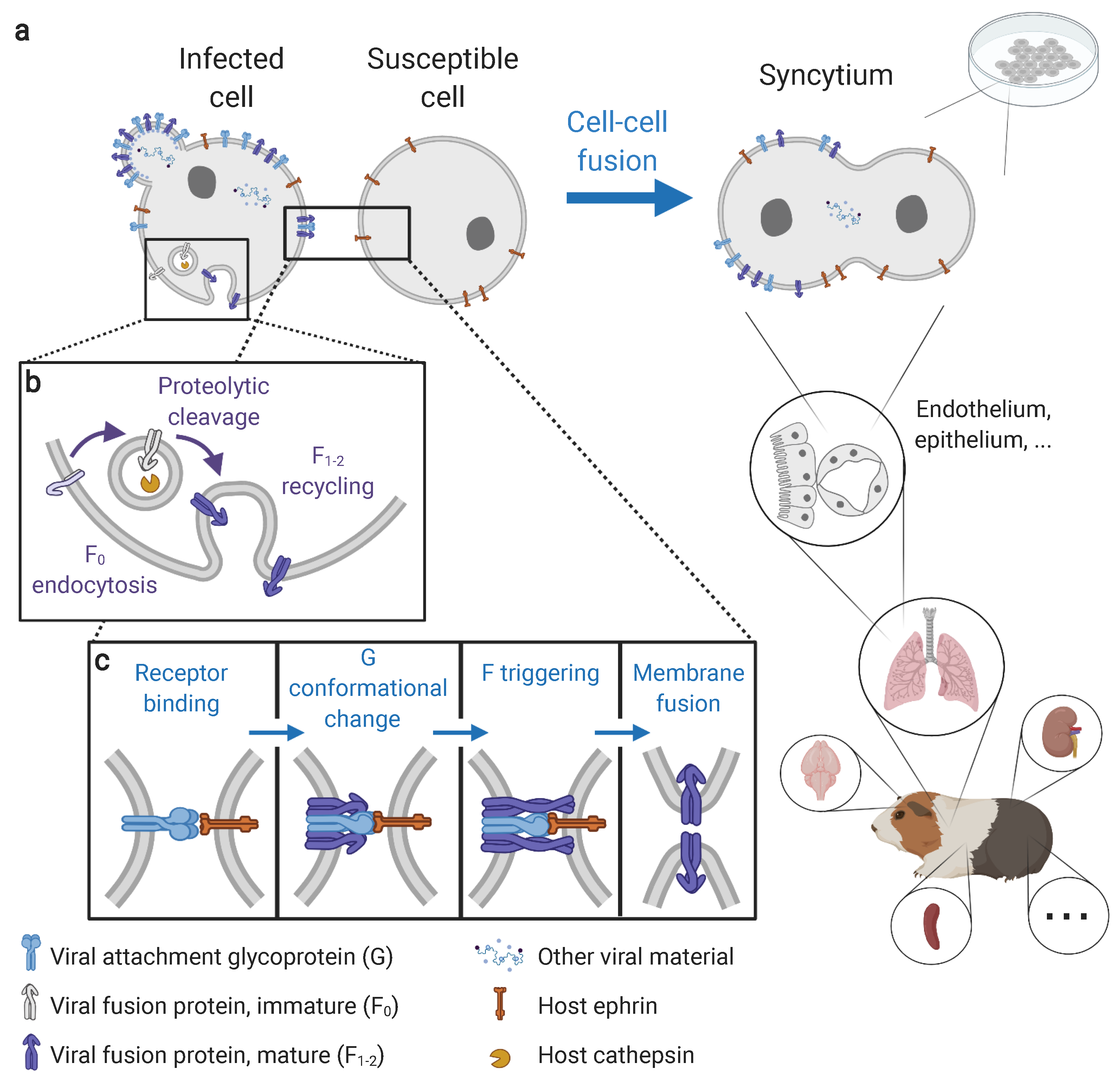
2. Insights on Syncytium Formation from In Vitro Studies
2.1. The Molecular Prerequisites of Syncytium Formation
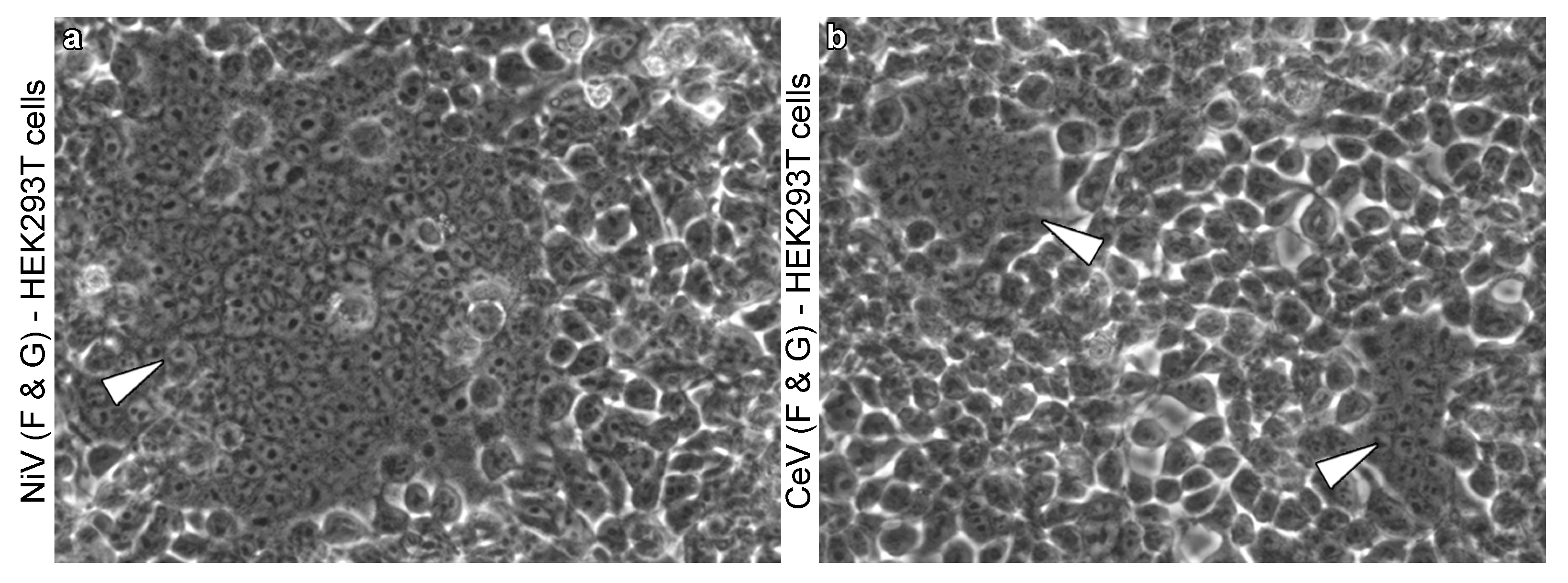
2.2. Occurrence of Henipavirus-Induced Syncytia across Viruses and Cell Lines
3. Mapping Syncytium Formation In Vivo
3.1. Knowledge from Other Paramyxoviruses

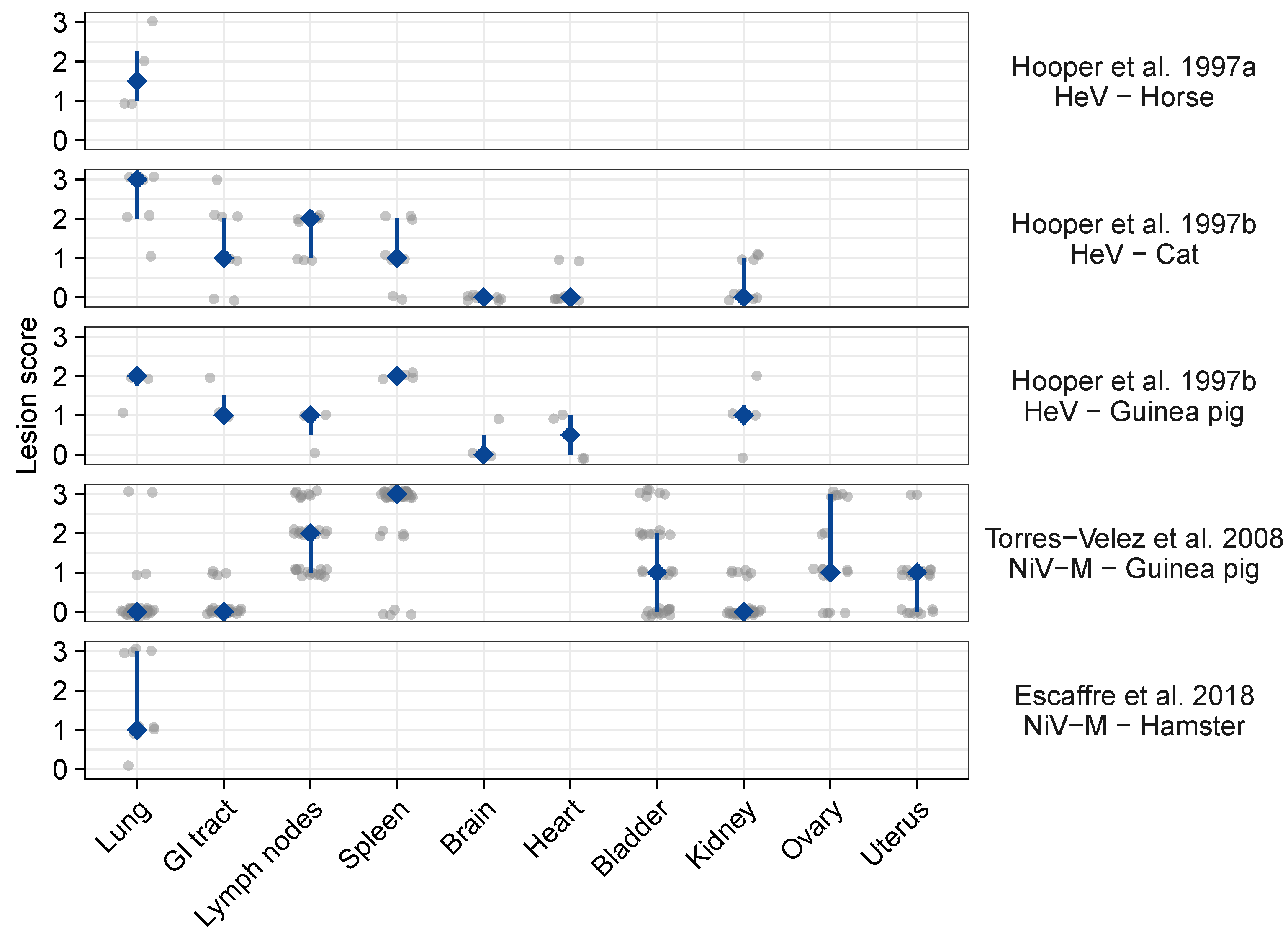
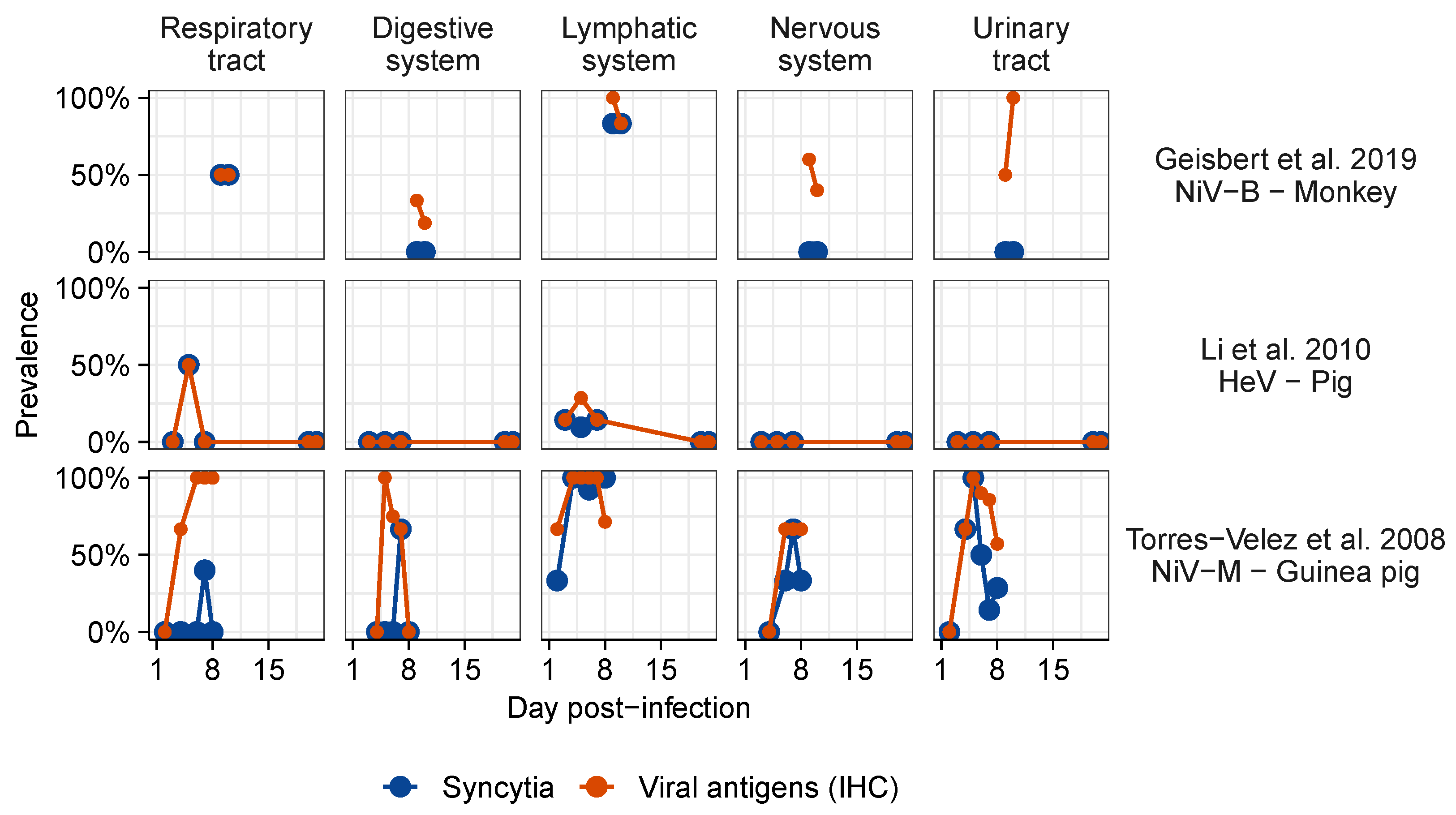
3.2. Meta-Analysis of Henipavirus-Induced Syncytium Occurrence
3.2.1. Data Availability
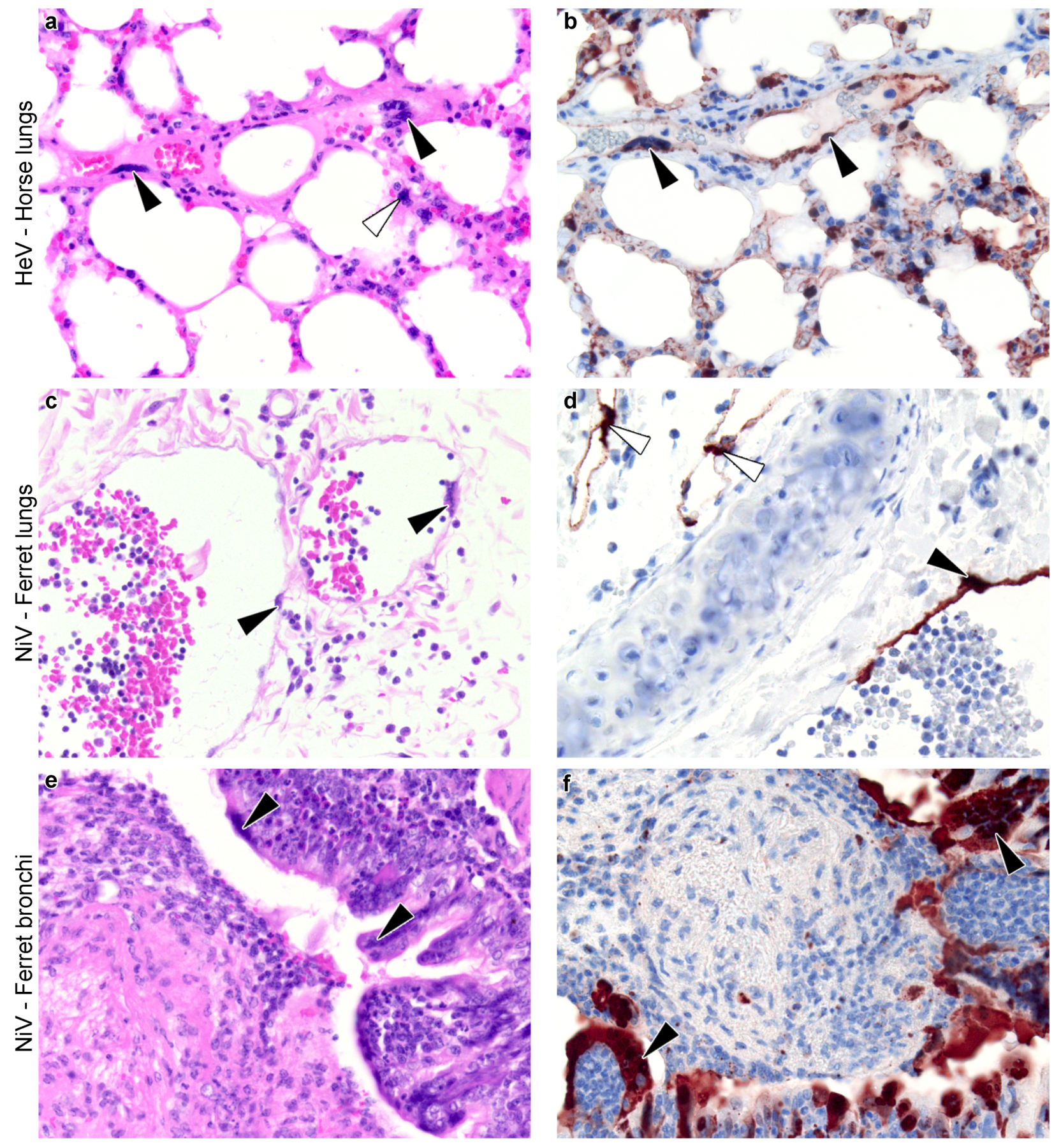
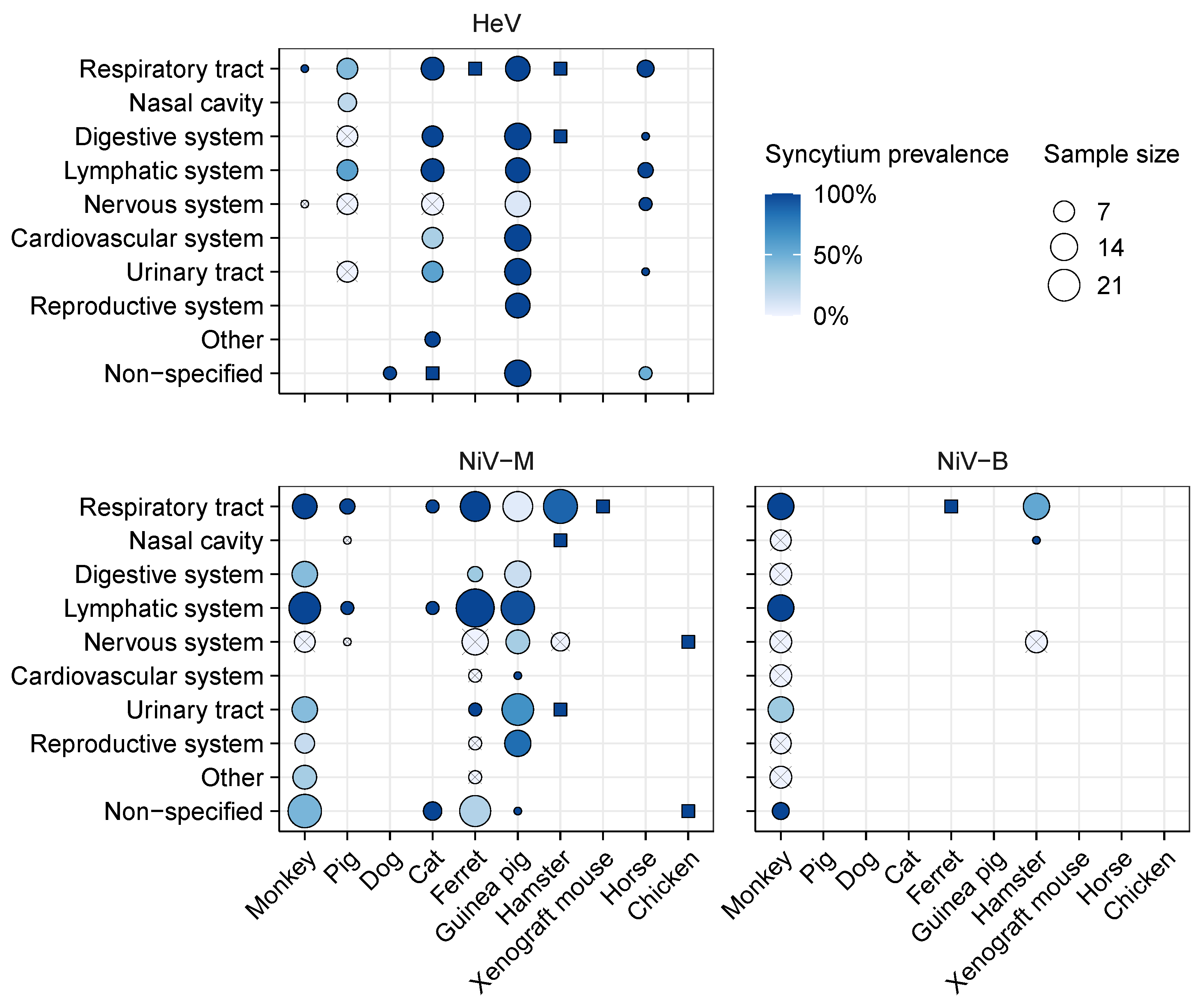
3.2.2. Syncytium Distribution across Tissues, Hosts, and Viruses
4. Discussion
4.1. Identified Patterns
4.2. Methodological Challenges of Syncytium Mapping
4.3. Comparison of Syncytium Occurrence Patterns In Vitro and In Vivo
4.4. Knowns and Unknowns of Syncytium Formation and Future Directions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Connolly, S.A.; Jardetzky, T.S.; Longnecker, R. The structural basis of herpesvirus entry. Nat. Rev. Microbiol. 2021, 19, 110–121. [Google Scholar] [CrossRef]
- Oliver, S.L.; Zhou, M.; Arvin, A.M. Varicella-zoster virus: Molecular controls of cell fusion-dependent pathogenesis. Biochem. Soc. Trans. 2020, 48, 2415–2435. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracq, L.; Xie, M.; Benichou, S.; Bouchet, J. Mechanisms for cell-to-cell transmission of HIV-1. Front. Immunol. 2018, 9, 260. [Google Scholar] [CrossRef]
- Lavi, E.; Wang, Q.; Weiss, S.R.; Gonatas, N.K. Syncytia formation induced by coronavirus infection is associated with fragmentation and rearrangement of the Golgi apparatus. Virology 1996, 221, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef] [Green Version]
- Fenrich, M.; Mrdenovic, S.; Balog, M.; Tomic, S.; Zjalic, M.; Roncevic, A.; Mandic, D.; Debeljak, Z.; Heffer, M. SARS-CoV-2 dissemination through peripheral nerves explains multiple organ injury. Front. Cell. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef]
- Chang, A.; Dutch, R.E. Paramyxovirus fusion and entry: Multiple paths to a common end. Viruses 2012, 4, 613–636. [Google Scholar] [CrossRef]
- Hall, C.B. Respiatory syncytial virus. In Textbook of Pediatric Infectious Diseases; Saunders: Philadelphia, PA, USA, 1992; pp. 1633–1656. [Google Scholar]
- Compton, A.A.; Schwartz, O. They might be giants: Does syncytium formation sink or spread HIV infection? PLoS Pathog. 2017, 13, e1006099. [Google Scholar] [CrossRef]
- Cifuentes-Muñoz, N.; Dutch, R.E.; Cattaneo, R. Direct cell-to-cell transmission of respiratory viruses: The fast lanes. PLoS Pathog. 2018, 14, e1007015. [Google Scholar] [CrossRef] [Green Version]
- Zhong, P.; Agosto, L.M.; Munro, J.B.; Mothes, W. Cell-to-cell transmission of viruses. Curr. Opin. Virol. 2013, 3, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Komarova, N.L.; Anghelina, D.; Voznesensky, I.; Trinité, B.; Levy, D.N.; Wodarz, D. Relative contribution of free-virus and synaptic transmission to the spread of HIV-1 through target cell populations. Biol. Lett. 2013, 9, 20121049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, J.L.; Engel, J.P. Altered pathogenesis in herpes simplex virus type 1 infection due to a syncytial mutation mapping to the carboxy terminus of glycoprotein B. J. Virol. 1991, 65, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engel, J.P.; Boyer, E.P.; Goodman, J.L. Two novel single amino acid syncytial mutations in the carboxy terminus of glycoprotein B of herpes simplex virus type 1 confer a unique pathogenic phenotype. Virology 1993, 192, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Arvin, A.M.; Oliver, S.L. The cytoplasmic domain of varicella-zoster virus glycoprotein H regulates syncytia formation and skin pathogenesis. PLoS Pathog. 2014, 10, e1004173. [Google Scholar] [CrossRef] [Green Version]
- Park, B.H.; Lavi, E.; Blank, K.J.; Gaulton, G.N. Intracerebral hemorrhages and syncytium formation induced by endothelial cell infection with a murine leukemia virus. J. Virol. 1993, 67, 6015–6024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, M.; Kizhatil, K.; Albritton, L.M.; Gaulton, G.N. Induction of syncytia by neuropathogenic murine leukemia viruses depends on receptor density, host cell determinants, and the intrinsic fusion potential of envelope protein. J. Virol. 1999, 73, 9377–9385. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, D.M.P.; Patterson, C.E.; Gales, T.L.; D’Orazio, J.L.; Vaughn, M.M.; Rall, G.F. Measles Virus Spread between Neurons Requires Cell Contact but Not CD46 Expression, Syncytium Formation, or Extracellular Virus Production. J. Virol. 2000, 74, 1908–1918. [Google Scholar] [CrossRef] [Green Version]
- Negrete, O.A.; Levroney, E.L.; Aguilar, H.C.; Bertolotti-Ciarlet, A.; Nazarian, R.; Tajyar, S.; Lee, B. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 2005, 436, 401–405. [Google Scholar] [CrossRef]
- Bonaparte, M.I.; Dimitrov, A.S.; Bossart, K.N.; Crameri, G.; Mungall, B.A.; Bishop, K.A.; Choudhry, V.; Dimitrov, D.S.; Wang, L.F.; Eaton, B.T.; et al. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc. Natl. Acad. Sci. USA 2005, 102, 10652–10657. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, H.C.; Aspericueta, V.; Robinson, L.R.; Aanensen, K.E.; Lee, B. A quantitative and kinetic fusion protein-triggering assay can discern distinct steps in the Nipah virus membrane fusion cascade. J. Virol. 2010, 84, 8033–8041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pernet, O.; Beaty, S.; Lee, B. Functional rectification of the newly described African henipavirus fusion glycoprotein (Gh-M74a). J. Virol. 2014, 88, 5171–5176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradel-Tretheway, B.G.; Zamora, J.L.R.; Stone, J.A.; Liu, Q.; Li, J.; Aguilar, H.C. Nipah and Hendra virus glycoproteins induce comparable homologous but distinct heterologous fusion phenotypes. J. Virol. 2019, 93, e00577-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef]
- Esolen, L.M.; Park, S.W.; Hardwick, J.M.; Griffin, D.E. Apoptosis as a cause of death in measles virus-infected cells. J. Virol. 1995, 69, 3955–3958. [Google Scholar] [CrossRef] [Green Version]
- Fugier-Vivier, I.; Servet-Delprat, C.; Rivailler, P.; Rissoan, M.C.; Liu, Y.J.; Rabourdin-Combe, C. Measles virus suppresses cell-mediated immunity by interfering with the survival and functions of dendritic and T cells. J. Exp. Med. 1997, 186, 813–823. [Google Scholar] [CrossRef]
- Ludlow, M.; McQuaid, S.; Milner, D.; de Swart, R.L.; Duprex, W.P. Pathological Consequences of Systemic Measles Virus Infection. J. Pathol. 2015, 235, 253–265. [Google Scholar] [CrossRef]
- Laksono, B.M.; De Vries, R.D.; McQuaid, S.; Duprex, W.P.; De Swart, R.L. Measles Virus Host Invasion and Pathogenesis. Viruses 2016, 8, 210. [Google Scholar] [CrossRef] [Green Version]
- Kumberger, P.; Durso-Cain, K.; Uprichard, S.L.; Dahari, H.; Graw, F. Accounting for space—Quantification of cell-to-cell transmission kinetics using virus dynamics models. Viruses 2018, 10, 200. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Stone, J.A.; Bradel-Tretheway, B.; Dabundo, J.; Montano, J.A.B.; Santos-Montanez, J.; Biering, S.B.; Nicola, A.V.; Iorio, R.M.; Lu, X.; et al. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog. 2013, 9, e1003770. [Google Scholar] [CrossRef]
- Liu, Q.; Bradel-Tretheway, B.; Monreal, A.I.; Saludes, J.P.; Lu, X.; Nicola, A.V.; Aguilar, H.C. Nipah virus attachment glycoprotein stalk C-terminal region links receptor binding to fusion triggering. J. Virol. 2015, 89, 1838–1850. [Google Scholar] [CrossRef] [Green Version]
- Contreras, E.M.; Johnston, G.P.; Buchholz, D.W.; Ortega, V.; Monreal, I.A.; Zamora, J.L.R.; Cheung, T.; Aguilar, H.C. Roles of Cholesterol in Early and Late Steps of the Nipah Virus Membrane Fusion Cascade. J. Virol. 2021, 95, e02323-20. [Google Scholar] [CrossRef]
- Schountz, T.; Campbell, C.; Wagner, K.; Rovnak, J.; Martellaro, C.; DeBuysscher, B.L.; Feldmann, H.; Prescott, J. Differential innate immune responses elicited by Nipah virus and Cedar virus correlate with disparate in vivo pathogenesis in hamsters. Viruses 2019, 11, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seifert, S.N.; Letko, M.C.; Bushmaker, T.; Laing, E.D.; Saturday, G.; Meade-White, K.; van Doremalen, N.; Broder, C.C.; Munster, V.J. Rousettus aegyptiacus bats do not support productive Nipah virus replication. J. Infect. Dis. 2020, 221, S407–S413. [Google Scholar] [CrossRef] [PubMed]
- Marsh, G.A.; Wang, L.F. Hendra and Nipah viruses: Why are they so deadly? Curr. Opin. Virol. 2012, 2, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Drexler, J.; Corman, V.; Müller, M.; Maganga, G.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [Green Version]
- Marsh, G.A.; De Jong, C.; Barr, J.A.; Tachedjian, M.; Smith, C.; Middleton, D.; Yu, M.; Todd, S.; Foord, A.J.; Haring, V.; et al. Cedar virus: A novel Henipavirus isolated from Australian bats. PLoS Pathog. 2012, 8, e1002836. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, L.; Yang, F.; Ren, X.; Jiang, J.; Dong, J.; Sun, L.; Zhu, Y.; Zhou, H.; Jin, Q. Novel henipa-like virus, Mojiang paramyxovirus, in rats, China, 2012. Emerg. Infect. Dis. 2014, 20, 1064. [Google Scholar] [CrossRef]
- Pernet, O.; Schneider, B.S.; Beaty, S.M.; LeBreton, M.; Yun, T.E.; Park, A.; Zachariah, T.T.; Bowden, T.A.; Hitchens, P.; Ramirez, C.M.; et al. Evidence for henipavirus spillover into human populations in Africa. Nat. Commun. 2014, 5, 5342. [Google Scholar] [CrossRef] [Green Version]
- Weingartl, H.M.; Berhane, Y.; Czub, M. Animal models of henipavirus infection: A review. Vet. J. 2009, 181, 211–220. [Google Scholar] [CrossRef]
- Williamson, M.; Torres-Velez, F. Henipavirus: A review of laboratory animal pathology. Vet. Pathol. 2010, 47, 871–880. [Google Scholar] [CrossRef] [Green Version]
- Vigant, F.; Lee, B. Hendra and Nipah infection: Pathology, models and potential therapies. Infect. Disord.-Drug Targets Former. Curr. Drug Targets-Infect. Disord. 2011, 11, 315–336. [Google Scholar] [CrossRef] [Green Version]
- Maisner, A.; Weingartl, H. Organ-and endotheliotropism of Nipah virus infections in vivo and in vitro. Thromb. Haemost. 2009, 102, 1014–1023. [Google Scholar] [PubMed]
- Torres-Velez, F.J.; Shieh, W.J.; Rollin, P.E.; Morken, T.; Brown, C.; Ksiazek, T.G.; Zaki, S.R. Histopathologic and Immunohistochemical Characterization of Nipah Virus Infection in the Guinea Pig. Vet. Pathol. 2008, 45, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Howley, P.M.; Knipe, D.M. Fields Virology: Emerging Viruses; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2020. [Google Scholar]
- Aguilar, H.C.; Iorio, R.M. Henipavirus membrane fusion and viral entry. In Henipavirus; Springer: Berlin/Heidelberg, Germany, 2012; pp. 79–94. [Google Scholar]
- Yeo, Y.Y.; Buchholz, D.W.; Gamble, A.; Jager, M.; Aguilar, H.C. Headless henipaviral receptor binding glycoproteins reveal key post-receptor binding contributions of the globular head and head/stalk interface in fusion promotion. J. Virol. 2021, JVI-00666. [Google Scholar] [CrossRef]
- Aguilar, H.C.; Lee, B. Emerging paramyxoviruses: Molecular mechanisms and antiviral strategies. Expert Rev. Mol. Med. 2011, 13, e6. [Google Scholar] [CrossRef]
- Negrete, O.A.; Wolf, M.C.; Aguilar, H.C.; Enterlein, S.; Wang, W.; Mühlberger, E.; Su, S.V.; Bertolotti-Ciarlet, A.; Flick, R.; Lee, B. Two key residues in ephrinB3 are critical for its use as an alternative receptor for Nipah virus. PLoS Pathog. 2006, 2, e7. [Google Scholar] [CrossRef]
- Negrete, O.A.; Chu, D.; Aguilar, H.C.; Lee, B. Single amino acid changes in the Nipah and Hendra virus attachment glycoproteins distinguish ephrinB2 from ephrinB3 usage. J. Virol. 2007, 81, 10804–10814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laing, E.D.; Navaratnarajah, C.K.; Da Silva, S.C.; Petzing, S.R.; Xu, Y.; Sterling, S.L.; Marsh, G.A.; Wang, L.F.; Amaya, M.; Nikolov, D.B.; et al. Structural and functional analyses reveal promiscuous and species specific use of ephrin receptors by Cedar virus. Proc. Natl. Acad. Sci. USA 2019, 116, 20707–20715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryce, R.; Azarm, K.; Rissanen, I.; Harlos, K.; Bowden, T.A.; Lee, B. A key region of molecular specificity orchestrates unique ephrin-B1 utilization by Cedar virus. Life Sci. Alliance 2020, 3, e201900578. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.; Pernet, O.; Ahmed, A.A.; Zeltina, A.; Beaty, S.M.; Bowden, T.A. Molecular recognition of human ephrinB2 cell surface receptor by an emergent African henipavirus. Proc. Natl. Acad. Sci. USA 2015, 112, E2156–E2165. [Google Scholar] [CrossRef] [Green Version]
- Thibault, P.A.; Watkinson, R.E.; Moreira-Soto, A.; Drexler, J.F.; Lee, B. Zoonotic potential of emerging paramyxoviruses: Knowns and unknowns. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2017; Volume 98, pp. 1–55. [Google Scholar]
- Pernet, O.; Wang, Y.E.; Lee, B. Henipavirus receptor usage and tropism. In Henipavirus; Springer: Berlin/Heidelberg, Germany, 2012; pp. 59–78. [Google Scholar]
- Rissanen, I.; Ahmed, A.A.; Azarm, K.; Beaty, S.; Hong, P.; Nambulli, S.; Duprex, W.P.; Lee, B.; Bowden, T.A. Idiosyncratic Mòjiāng virus attachment glycoprotein directs a host-cell entry pathway distinct from genetically related henipaviruses. Nat. Commun. 2017, 8, 16060. [Google Scholar] [CrossRef]
- Diederich, S.; Sauerhering, L.; Weis, M.; Altmeppen, H.; Schaschke, N.; Reinheckel, T.; Erbar, S.; Maisner, A. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J. Virol. 2012, 86, 3736–3745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pager, C.T.; Dutch, R.E. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 2005, 79, 12714–12720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pager, C.T.; Craft, W.W., Jr.; Patch, J.; Dutch, R.E. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology 2006, 346, 251–257. [Google Scholar] [CrossRef]
- Johnston, G.P.; Bradel-Tretheway, B.; Piehowski, P.D.; Brewer, H.M.; Lee, B.N.R.; Usher, N.T.; Zamora, J.L.R.; Ortega, V.; Contreras, E.M.; Teuton, J.R.; et al. Nipah virus-like particle egress is modulated by cytoskeletal and vesicular trafficking pathways: A validated particle proteomics analysis. Msystems 2019, 4, e00194-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zamora, J.L.R.; Ortega, V.; Johnston, G.P.; Li, J.; André, N.M.; Monreal, I.A.; Contreras, E.M.; Whittaker, G.R.; Aguilar, H.C. Third Helical Domain of the Nipah Virus Fusion Glycoprotein Modulates both Early and Late Steps in the Membrane Fusion Cascade. J. Virol. 2020, 94, e00644-20. [Google Scholar] [CrossRef] [PubMed]
- Zamora, J.L.R.; Ortega, V.; Johnston, G.P.; Li, J.; Aguilar, H.C. Novel roles of the N1 loop and N4 alpha helical region of the Nipah Virus Fusion glycoprotein in modulating early and late steps of the membrane fusion cascade. J. Virol. 2021, 95, e01707-20. [Google Scholar] [CrossRef] [PubMed]
- Moll, M.; Kaufmann, A.; Maisner, A. Influence of N-glycans on processing and biological activity of the Nipah virus fusion protein. J. Virol. 2004, 78, 7274–7278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, J.R.; Pager, C.T.; Fowler, S.D.; Dutch, R.E. Role of N-linked glycosylation of the Hendra virus fusion protein. J. Virol. 2005, 79, 7922–7925. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, H.C.; Matreyek, K.A.; Filone, C.M.; Hashimi, S.T.; Levroney, E.L.; Negrete, O.A.; Bertolotti-Ciarlet, A.; Choi, D.Y.; McHardy, I.; Fulcher, J.A.; et al. N-glycans on Nipah virus fusion protein protect against neutralization but reduce membrane fusion and viral entry. J. Virol. 2006, 80, 4878–4889. [Google Scholar] [CrossRef] [Green Version]
- Biering, S.B.; Huang, A.; Vu, A.T.; Robinson, L.R.; Bradel-Tretheway, B.; Choi, E.; Lee, B.; Aguilar, H.C. N-Glycans on the Nipah virus attachment glycoprotein modulate fusion and viral entry as they protect against antibody neutralization. J. Virol. 2012, 86, 11991–12002. [Google Scholar] [CrossRef] [Green Version]
- Bradel-Tretheway, B.G.; Liu, Q.; Stone, J.A.; McInally, S.; Aguilar, H.C. Novel functions of Hendra virus G N-glycans and comparisons to Nipah virus. J. Virol. 2015, 89, 7235–7247. [Google Scholar] [CrossRef] [Green Version]
- Stone, J.A.; Nicola, A.V.; Baum, L.G.; Aguilar, H.C. Multiple novel functions of henipavirus O-glycans: The first O-glycan functions identified in the paramyxovirus family. PLoS Pathog. 2016, 12, e1005445. [Google Scholar] [CrossRef] [Green Version]
- Dietzel, E.; Kolesnikova, L.; Sawatsky, B.; Heiner, A.; Weis, M.; Kobinger, G.P.; Becker, S.; Von Messling, V.; Maisner, A. Nipah virus matrix protein influences fusogenicity and is essential for particle infectivity and stability. J. Virol. 2016, 90, 2514–2522. [Google Scholar] [CrossRef] [Green Version]
- Weis, M.; Behner, L.; Hoffmann, M.; Krüger, N.; Herrler, G.; Drosten, C.; Drexler, J.F.; Dietzel, E.; Maisner, A. Characterization of African bat henipavirus GH-M74a glycoproteins. J. Gen. Virol. 2014, 95, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laksono, B.M.; Fortugno, P.; Nijmeijer, B.M.; de Vries, R.D.; Cordisco, S.; Kuiken, T.; Geijtenbeek, T.B.; Duprex, W.P.; Brancati, F.; de Swart, R.L. Measles skin rash: Infection of lymphoid and myeloid cells in the dermis precedes viral dissemination to the epidermis. PLoS Pathog. 2020, 16, e1008253. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Wang, L.F.; Eaton, B.T.; Broder, C.C. Functional expression and membrane fusion tropism of the envelope glycoproteins of Hendra virus. Virology 2001, 290, 121–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakane, S.; Matsuda, Z. Dual split protein (DSP) assay to monitor cell–cell membrane fusion. In Cell Fusion; Springer: Berlin/Heidelberg, Germany, 2015; pp. 229–236. [Google Scholar]
- Summers, B.A.; Appel, M.J.G. Syncytia Formation: An Aid in the Diagnosis of Canine Distemper Encephalomyelitis. J. Comp. Pathol. 1985, 95, 425–435. [Google Scholar] [CrossRef]
- DeBuysscher, B.L.; de Wit, E.; Munster, V.J.; Scott, D.; Feldmann, H.; Prescott, J. Comparison of the pathogenicity of Nipah virus isolates from Bangladesh and Malaysia in the Syrian hamster. PLoS Negl. Trop. Dis. 2013, 7, e2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Griffin, B.D.; Leung, A.; Chan, M.; Warner, B.M.; Ranadheera, C.; Tierney, K.; Audet, J.; Frost, K.L.; Safronetz, D.; Embury-Hyatt, C.; et al. Establishment of an RNA polymerase II-driven reverse genetics system for Nipah virus strains from Malaysia and Bangladesh. Sci. Rep. 2019, 9, 11171. [Google Scholar] [CrossRef] [Green Version]
- Wynne, J.W.; Shiell, B.J.; Marsh, G.A.; Boyd, V.; Harper, J.A.; Heesom, K.; Monaghan, P.; Zhou, P.; Payne, J.; Klein, R.; et al. Proteomics informed by transcriptomics reveals Hendra virus sensitizes bat cells to TRAIL-mediated apoptosis. Genome Biol. 2014, 15, 1–21. [Google Scholar]
- Cheliout Da Silva, S.; Yan, L.; Dang, H.V.; Xu, K.; Epstein, J.H.; Veesler, D.; Broder, C.C. Functional Analysis of the Fusion and Attachment Glycoproteins of Mojiang Henipavirus. Viruses 2021, 13, 517. [Google Scholar] [CrossRef]
- Voigt, K.; Hoffmann, M.; Drexler, J.F.; Müller, M.A.; Drosten, C.; Herrler, G.; Krüger, N. Fusogenicity of the Ghana Virus (Henipavirus: Ghanaian bat henipavirus) Fusion Protein is Controlled by the Cytoplasmic Domain of the Attachment Glycoprotein. Viruses 2019, 11, 800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escaffre, O.; Borisevich, V.; Carmical, J.R.; Prusak, D.; Prescott, J.; Feldmann, H.; Rockx, B. Henipavirus pathogenesis in human respiratory epithelial cells. J. Virol. 2013, 87, 3284–3294. [Google Scholar] [CrossRef] [Green Version]
- Borisevich, V.; Ozdener, M.H.; Malik, B.; Rockx, B. Hendra and Nipah virus infection in cultured human olfactory epithelial cells. Msphere 2017, 2, e00252-17. [Google Scholar] [CrossRef] [Green Version]
- Bunting, C. The giant-cells of measles. Yale J. Biol. Med. 1950, 22, 513. [Google Scholar]
- Hall, W.C.; Kovatch, R.M.; Herman, P.H.; Fox, J.G. Pathology of Measles in Rhesus Monkeys. Vet. Pathol. 1971, 8, 307–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkerton, H.; Smiley, W.L.; Anderson, W.A.D. Giant Cell Pneumonia with Inclusions. Am. J. Pathol. 1945, 21, 1–23. [Google Scholar]
- De Vries, R.D.; McQuaid, S.; Van Amerongen, G.; Yüksel, S.; Verburgh, R.J.; Osterhaus, A.D.; Duprex, W.P.; De Swart, R.L. Measles immune suppression: Lessons from the macaque model. PLoS Pathog. 2012, 8, e1002885. [Google Scholar] [CrossRef] [PubMed]
- Rima, B.; Collins, P.; Easton, A.; Fouchier, R.; Kurath, G.; Lamb, R.A.; Lee, B.; Maisner, A.; Rota, P.; Wang, L.; et al. ICTV virus taxonomy profile: Pneumoviridae. J. Gen. Virol. 2017, 98, 2912–2913. [Google Scholar] [CrossRef] [PubMed]
- Chanock, R.; Roizman, B.; Myers, R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA): Isolation, properties and characterization. Am. J. Epidemiol. 1957, 66, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Coates, H.V.; Chanock, R.M. Experimental infection with respiratory syncytial virus in several species of animals. Am. J. Hyg. 1962, 76, 302–312. [Google Scholar] [PubMed]
- Middleton, D.; Pallister, J.; Klein, R.; Feng, Y.R.; Haining, J.; Arkinstall, R.; Frazer, L.; Huang, J.A.; Edwards, N.; Wareing, M.; et al. Hendra virus vaccine, a one health approach to protecting horse, human, and environmental health. Emerg. Infect. Dis. 2014, 20, 372. [Google Scholar] [CrossRef] [PubMed]
- Bossart, K.N.; Zhu, Z.; Middleton, D.; Klippel, J.; Crameri, G.; Bingham, J.; McEachern, J.A.; Green, D.; Hancock, T.J.; Chan, Y.P.; et al. A Neutralizing Human Monoclonal Antibody Protects against Lethal Disease in a New Ferret Model of Acute Nipah Virus Infection. PLOS Pathog. 2009, e1000642. [Google Scholar] [CrossRef]
- Beineke, A.; Baumgärtner, W.; Wohlsein, P. Cross-species transmission of canine distemper virus—An update. ONE Health 2015, 1, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.; Hooper, P.; Selleck, P.; Gleeson, L.; Daniels, P.; Westbury, H.; Murray, P. Transmission studies of Hendra virus (equine morbilli-virus) in fruit bats, horses and cats. Aust. Vet. J. 1998, 76, 813–818. [Google Scholar] [CrossRef]
- Woon, A.P.; Boyd, V.; Todd, S.; Smith, I.; Klein, R.; Woodhouse, I.B.; Riddell, S.; Crameri, G.; Bingham, J.; Wang, L.F.; et al. Acute experimental infection of bats and ferrets with Hendra virus: Insights into the early host response of the reservoir host and susceptible model species. PLoS Pathog. 2020, 16, e1008412. [Google Scholar] [CrossRef]
- Escaffre, O.; Hill, T.; Ikegami, T.; Juelich, T.L.; Smith, J.K.; Zhang, L.; Perez, D.E.; Atkins, C.; Park, A.; Lawrence, W.S.; et al. Experimental Infection of Syrian Hamsters With Aerosolized Nipah Virus. J. Infect. Dis. 2018, 218, 1602–1610. [Google Scholar] [CrossRef] [Green Version]
- Westbury, H.; Hooper, P.T.; Selleck, P.; Murray, P. Equine morbillivirus pneumonia: Susceptibility of laboratory animals to the virus. Aust. Vet. J. 1995, 72, 278–279. [Google Scholar] [CrossRef]
- Mastorakos, P.; McGavern, D. The anatomy and immunology of vasculature in the central nervous system. Sci. Immunol. 2019, 4, eaav0492. [Google Scholar] [CrossRef]
- Geisbert, J.B.; Borisevich, V.; Prasad, A.N.; Agans, K.N.; Foster, S.L.; Deer, D.J.; Cross, R.W.; Mire, C.E.; Geisbert, T.W.; Fenton, K.A. An Intranasal Exposure Model of Lethal Nipah Virus Infection in African Green Monkeys. J. Infect. Dis. 2019, jiz391. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.N.; Agans, K.N.; Sivasubramani, S.K.; Geisbert, J.B.; Borisevich, V.; Mire, C.E.; Lawrence, W.S.; Fenton, K.A.; Geisbert, T.W. A Lethal Aerosol Exposure Model of Nipah Virus Strain Bangladesh in African Green Monkeys. J. Infect. Dis. 2019, jiz469. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Daddario-DiCaprio, K.M.; Hickey, A.C.; Smith, M.A.; Chan, Y.P.; Wang, L.F.; Mattapallil, J.J.; Geisbert, J.B.; Bossart, K.N.; Broder, C.C. Development of an Acute and Highly Pathogenic Nonhuman Primate Model of Nipah Virus Infection. PLoS ONE 2010, 5, e10690. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.; Hooper, P.; Selleck, P.; Westbury, H.; Slocombe, R. A Guinea-Pig Model of Hendra Virus Encephalitis. J. Comp. Pathol. 2001, 124, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Mungall, B.A.; Middleton, D.; Crameri, G.; Bingham, J.; Halpin, K.; Russell, G.; Green, D.; McEachern, J.; Pritchard, L.I.; Eaton, B.T.; et al. Feline Model of Acute Nipah Virus Infection and Protection with a Soluble Glycoprotein-Based Subunit Vaccine. J. Virol. 2006, 80, 12293–12302. [Google Scholar] [CrossRef] [Green Version]
- Cong, Y.; Lentz, M.R.; Lara, A.; Alexander, I.; Bartos, C.; Bohannon, J.K.; Hammoud, D.; Huzella, L.; Jahrling, P.B.; Janosko, K.; et al. Loss in Lung Volume and Changes in the Immune Response Demonstrate Disease Progression in African Green Monkeys Infected by Small-Particle Aerosol and Intratracheal Exposure to Nipah Virus. PLoS Neglected Trop. Dis. 2017, 11, e0005532. [Google Scholar] [CrossRef]
- Mire, C.E.; Satterfield, B.A.; Geisbert, J.B.; Agans, K.N.; Borisevich, V.; Yan, L.; Chan, Y.P.; Cross, R.W.; Fenton, K.A.; Broder, C.C.; et al. Pathogenic Differences between Nipah Virus Bangladesh and Malaysia Strains in Primates: Implications for Antibody Therapy. Sci. Rep. 2016, 6, 30916. [Google Scholar] [CrossRef]
- Baseler, L.; de Wit, E.; Scott, D.P.; Munster, V.J.; Feldmann, H. Syrian hamsters (Mesocricetus auratus) oronasally inoculated with a Nipah virus isolate from Bangladesh or Malaysia develop similar respiratory tract lesions. Vet. Pathol. 2015, 52, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Embury-Hyatt, C.; Weingartl, H.M. Experimental inoculation study indicates swine as a potential host for Hendra virus. Vet. Res. 2010, 41, 33. [Google Scholar] [CrossRef] [Green Version]
- Munster, V.J.; Prescott, J.B.; Bushmaker, T.; Long, D.; Rosenke, R.; Thomas, T.; Scott, D.; Fischer, E.R.; Feldmann, H.; De Wit, E. Rapid Nipah virus entry into the central nervous system of hamsters via the olfactory route. Sci. Rep. 2012, 2, 736. [Google Scholar] [CrossRef]
- Hooper, P.; Westbury, H.; Russell, G. The lesions of experimental equine morbillivirus disease in cats and guinea pigs. Vet. Pathol. 1997, 34, 323–329. [Google Scholar] [CrossRef]
- Middleton, D.; Riddell, S.; Klein, R.; Arkinstall, R.; Haining, J.; Frazer, L.; Mottley, C.; Evans, R.; Johnson, D.; Pallister, J. Experimental Hendra Virus Infection of Dogs: Virus Replication, Shedding and Potential for Transmission. Aust. Vet. J. 2017, 95, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hooper, P.; Ketterer, P.; Hyatt, A.; Russell, G. Lesions of experimental equine morbillivirus pneumonia in horses. Vet. Pathol. 1997, 34, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.; Ong, K. Pathology of acute henipavirus infection in humans and animals. Pathol. Res. Int. 2011, 2011, 567248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimura, N.; Imada, T.; Kashiwazaki, Y.; Sharifah, S. Distribution of viral antigens and development of lesions in chicken embryos inoculated with Nipah virus. J. Comp. Pathol. 2006, 135, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Hammoud, D.A.; Cong, Y.; Huzella, L.M.; Castro, M.A.; Solomon, J.; Laux, J.; Lackemeyer, M.; Bohannon, J.K.; Rojas, O.; et al. The Use of Large-Particle Aerosol Exposure to Nipah Virus to Mimic Human Neurological Disease Manifestations in the African Green Monkey. J. Infect. Dis. 2020, 221, S419–S430. [Google Scholar] [CrossRef] [Green Version]
- Guillaume, V.; Wong, K.T.; Looi, R.; Georges-Courbot, M.C.; Barrot, L.; Buckland, R.; Wild, T.F.; Horvat, B. Acute Hendra virus infection: Analysis of the pathogenesis and passive antibody protection in the hamster model. Virology 2009, 387, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Rockx, B.; Bossart, K.N.; Feldmann, F.; Geisbert, J.B.; Hickey, A.C.; Brining, D.; Callison, J.; Safronetz, D.; Marzi, A.; Kercher, L.; et al. A Novel Model of Lethal Hendra Virus Infection in African Green Monkeys and the Effectiveness of Ribavirin Treatment. J. Virol. 2010, 84, 9831–9839. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.T.; Grosjean, I.; Brisson, C.; Blanquier, B.; Fevre-Montange, M.; Bernard, A.; Loth, P.; Georges-Courbot, M.C.; Chevallier, M.; Akaoka, H.; et al. A golden hamster model for human acute Nipah virus infection. Am. J. Pathol. 2003, 163, 2127–2137. [Google Scholar] [CrossRef] [Green Version]
- Mungall, B.A.; Middleton, D.; Crameri, G.; Halpin, K.; Bingham, J.; Eaton, B.T.; Broder, C.C. Vertical Transmission and Fetal Replication of Nipah Virus in an Experimentally Infected Cat. J. Infect. Dis. 2007, 196, 812–816. [Google Scholar] [CrossRef] [Green Version]
- Pallister, J.; Middleton, D.; Crameri, G.; Yamada, M.; Klein, R.; Hancock, T.J.; Foord, A.; Shiell, B.; Michalski, W.; Broder, C.C.; et al. Chloroquine Administration Does Not Prevent Nipah Virus Infection and Disease in Ferrets. J. Virol. 2009, 83, 11979–11982. [Google Scholar] [CrossRef] [Green Version]
- Clayton, B.A.; Middleton, D.; Arkinstall, R.; Frazer, L.; Wang, L.F.; Marsh, G.A. The nature of exposure drives transmission of Nipah viruses from Malaysia and Bangladesh in ferrets. PLoS Neglected Trop. Dis. 2016, 10, e0004775. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Georges-Courbot, M.C.; Ikeda, F.; Ishii, M.; Nagata, N.; Jacquot, F.; Raoul, H.; Sato, H.; Kai, C. Recombinant Measles Virus Vaccine Expressing the Nipah Virus Glycoprotein Protects against Lethal Nipah Virus Challenge. PLoS ONE 2013, 8, e58414. [Google Scholar] [CrossRef]
- Geisbert, T.W.; Mire, C.E.; Geisbert, J.B.; Chan, Y.P.; Agans, K.N.; Feldmann, F.; Fenton, K.A.; Zhu, Z.; Dimitrov, D.S.; Scott, D.P.; et al. Therapeutic Treatment of Nipah Virus Infection in Nonhuman Primates with a Neutralizing Human Monoclonal Antibody. Sci. Transl. Med. 2014, 6, 242ra82. [Google Scholar] [CrossRef] [Green Version]
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. The Immunomodulating V and W Proteins of Nipah Virus Determine Disease Course. Nat. Commun. 2015, 6, 7483. [Google Scholar] [CrossRef]
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Borisevich, V.; Agans, K.N.; Deer, D.J.; Graber, J.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. Nipah Virus C and W Proteins Contribute to Respiratory Disease in Ferrets. J. Virol. 2016, 90, 6326–6343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillaume-Vasselin, V.; Lemaitre, L.; Dhondt, K.P.; Tedeschi, L.; Poulard, A.; Charreyre, C.; Horvat, B. Protection from Hendra virus infection with Canarypox recombinant vaccine. Npj. Vaccines 2016, 1, 16003. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.T.; Shieh, W.J.; Kumar, S.; Norain, K.; Abdullah, W.; Guarner, J.; Goldsmith, C.S.; Chua, K.B.; Lam, S.K.; Tan, C.T.; et al. Nipah virus infection: Pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am. J. Pathol. 2002, 161, 2153–2167. [Google Scholar] [CrossRef]
- Pasquale, E.B. Eph-ephrin bidirectional signaling in physiology and disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genzer, S.C.; Welch, S.R.; Scholte, F.E.M.; Harmon, J.R.; Coleman-McCray, J.D.; Lo, M.K.; Montgomery, J.M.; Nichol, S.T.; Spiropoulou, C.F.; Spengler, J.R. Alterations in Blood Chemistry Levels Associated with Nipah Virus Disease in the Syrian Hamster Model. J. Infect. Dis. 2019, 221, S454–S459. [Google Scholar] [CrossRef]
- Weingartl, H.; Czub, S.; Copps, J.; Berhane, Y.; Middleton, D.; Marszal, P.; Gren, J.; Smith, G.; Ganske, S.; Manning, L.; et al. Invasion of the Central Nervous System in a Porcine Host by Nipah Virus. J. Virol. 2005, 79, 7528–7534. [Google Scholar] [CrossRef] [Green Version]
- Middleton, D.; Westbury, H.; Morrissy, C.; Van der Heide, B.; Russell, G.; Braun, M.; Hyatt, A. Experimental Nipah virus infection in pigs and cats. J. Comp. Pathol. 2002, 126, 124–136. [Google Scholar] [CrossRef]
- Weingartl, H.M.; Berhane, Y.; Caswell, J.L.; Loosmore, S.; Audonnet, J.C.; Roth, J.A.; Czub, M. Recombinant Nipah Virus Vaccines Protect Pigs against Challenge. J. Virol. 2006, 80, 7929–7938. [Google Scholar] [CrossRef] [Green Version]
- Berhane, Y.; Weingartl, H.; Lopez, J.; Neufeld, J.; Czub, S.; Embury-Hyatt, C.; Goolia, M.; Copps, J.; Czub, M. Bacterial infections in pigs experimentally infected with Nipah virus. Transbound. Emerg. Dis. 2008, 55, 165–174. [Google Scholar] [CrossRef]
- Kasloff, S.; Leung, A.; Pickering, B.; Smith, G.; Moffat, E.; Collignon, B.; Embury-Hyatt, C.; Kobasa, D.; Weingartl, H. Pathogenicity of Nipah henipavirus Bangladesh in a swine host. Sci. Rep. 2019, 9, 5230. [Google Scholar] [CrossRef] [Green Version]
- Pallister, J.; Middleton, D.; Wang, L.F.; Klein, R.; Haining, J.; Robinson, R.; Yamada, M.; White, J.; Payne, J.; Feng, Y.R.; et al. A recombinant Hendra virus G glycoprotein-based subunit vaccine protects ferrets from lethal Hendra virus challenge. Vaccine 2011, 29, 5623–5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, B.A.; Middleton, D.; Bergfeld, J.; Haining, J.; Arkinstall, R.; Wang, L.; Marsh, G.A. Transmission routes for Nipah virus from Malaysia and Bangladesh. Emerg. Infect. Dis. 2012, 18, 1983. [Google Scholar] [CrossRef] [PubMed]
- Williamson, M.; Hooper, P.; Selleck, P.; Westbury, H.; Slocombe, R. Experimental hendra virus infectionin pregnant guinea-pigs and fruit Bats (Pteropus poliocephalus). J. Comp. Pathol. 2000, 122, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Middleton, D.; Morrissy, C.; van der Heide, B.; Russell, G.; Braun, M.; Westbury, H.; Halpin, K.; Daniels, P. Experimental Nipah Virus Infection in Pteropid Bats (Pteropus Poliocephalus). J. Comp. Pathol. 2007, 136, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Freiberg, A.N.; Worthy, M.N.; Lee, B.; Holbrook, M.R. Combined chloroquine and ribavirin treatment does not prevent death in a hamster model of Nipah and Hendra virus infection. J. Gen. Virol. 2010, 91, 765. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Lambe, T.; Sebastian, S.; Bushmaker, T.; Fischer, R.; Feldmann, F.; Haddock, E.; Letko, M.; Avanzato, V.A.; Rissanen, I.; et al. A single-dose ChAdOx1-vectored vaccine provides complete protection against Nipah Bangladesh and Malaysia in Syrian golden hamsters. PLoS Neglected Trop. Dis. 2019, 13, e0007462. [Google Scholar] [CrossRef]
- Welch, S.R.; Scholte, F.E.; Harmon, J.R.; Coleman-McCray, J.D.; Lo, M.K.; Montgomery, J.M.; Nichol, S.T.; Spiropoulou, C.F.; Spengler, J.R. In situ imaging of fluorescent Nipah virus respiratory and neurological tissue tropism in the Syrian hamster model. J. Infect. Dis. 2020, 221, S448–S453. [Google Scholar] [CrossRef] [PubMed]
- DeBuysscher, B.L.; Scott, D.; Marzi, A.; Prescott, J.; Feldmann, H. Single-dose live-attenuated Nipah virus vaccines confer complete protection by eliciting antibodies directed against surface glycoproteins. Vaccine 2014, 32, 2637–2644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Bushmaker, T.; Scott, D.; Feldmann, H.; Munster, V.J. Nipah virus transmission in a hamster model. PLoS Negl. Trop. Dis. 2011, 5, e1432. [Google Scholar] [CrossRef] [Green Version]
- Dawes, B.E.; Kalveram, B.; Ikegami, T.; Juelich, T.; Smith, J.K.; Zhang, L.; Park, A.; Lee, B.; Komeno, T.; Furuta, Y.; et al. Favipiravir (T-705) protects against Nipah virus infection in the hamster model. Sci. Rep. 2018, 8, 7604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, M.K.; Bird, B.H.; Chattopadhyay, A.; Drew, C.P.; Martin, B.E.; Coleman, J.D.; Rose, J.K.; Nichol, S.T.; Spiropoulou, C.F. Single-dose replication-defective VSV-based Nipah virus vaccines provide protection from lethal challenge in Syrian hamsters. Antivir. Res. 2014, 101, 26–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dups, J.; Middleton, D.; Yamada, M.; Monaghan, P.; Long, F.; Robinson, R.; Marsh, G.A.; Wang, L.F. A New Model for Hendra Virus Encephalitis in the Mouse. PLoS ONE 2012, 7, e40308. [Google Scholar] [CrossRef] [Green Version]
- Dups, J.; Middleton, D.; Long, F.; Arkinstall, R.; Marsh, G.A.; Wang, L.F. Subclinical infection without encephalitis in mice following intranasal exposure to Nipah virus-Malaysia and Nipah virus-Bangladesh. Virol. J. 2014, 11, 102. [Google Scholar] [CrossRef] [Green Version]
- Murray, K.; Selleck, P.; Hooper, P.; Hyatt, A.; Gould, A.; Gleeson, L.; Westbury, H.; Hiley, L.; Selvey, L.; Rodwell, B.; et al. A morbillivirus that caused fatal disease in horses and humans. Science 1995, 268, 94–97. [Google Scholar] [CrossRef]
- Hanna, J.N.; McBride, W.J.; Brookes, D.L.; Shield, J.; Taylor, C.T.; Smith, I.L.; Craig, S.B.; Smith, G.A. Hendra virus infection in a veterinarian. Med. J. Aust. 2006, 185, 562. [Google Scholar] [CrossRef]
- Tanimura, N.; Imada, T.; Kashiwazaki, Y.; Shahirudin, S.; Sharifah, S.; Aziz, A. Monoclonal antibody-based immunohistochemical diagnosis of Malaysian Nipah virus infection in pigs. J. Comp. Pathol. 2004, 131, 199–206. [Google Scholar] [CrossRef]
- Selvey, L.A.; Wells, R.M.; McCormack, J.G.; Ansford, A.J.; Murray, K.; Rogers, R.J.; Lavercombe, P.S.; Selleck, P.; Sheridan, J.W. Infection of humans and horses by a newly described morbillivirus. Med. J. Aust. 1995, 162, 642–644. [Google Scholar] [CrossRef]
- Wong, K.; Robertson, T.; Ong, B.; Chong, J.; Yaiw, K.; Wang, L.; Ansford, A.; Tannenberg, A. Human Hendra virus infection causes acute and relapsing encephalitis. Neuropathol. Appl. Neurobiol. 2009, 35, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Hooijmans, C.; Ritskes-Hoitinga, M. Progress in using systematic reviews of animal studies to improve translational research. PLoS Med. 2013, 10, e1001482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonapersona, V.; Hoijtink, H.; Sarabdjitsingh, R.; Joëls, M. Increasing the statistical power of animal experiments with historical control data. Nat. Neurosci. 2021, 24, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Chretien, J.P.; Rivers, C.M.; Johansson, M.A. Make data sharing routine to prepare for public health emergencies. PLoS Med. 2016, 13, e1002109. [Google Scholar] [CrossRef] [Green Version]
- Roche, D.G.; Kruuk, L.E.; Lanfear, R.; Binning, S.A. Public data archiving in ecology and evolution: How well are we doing? PLoS Biol. 2015, 13, e1002295. [Google Scholar] [CrossRef] [Green Version]
- Colvin, M.E.; Peterson, J.T.; Kent, M.L.; Schreck, C.B. Occupancy modeling for improved accuracy and understanding of pathogen prevalence and dynamics. PLoS ONE 2015, 10, e0116605. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, D.I.; Royle, J.A. Designing occupancy studies: General advice and allocating survey effort. J. Appl. Ecol. 2005, 42, 1105–1114. [Google Scholar] [CrossRef]
- Pacifici, K.; Dorazio, R.M.; Conroy, M.J. A two-phase sampling design for increasing detections of rare species in occupancy surveys. Methods Ecol. Evol. 2012, 3, 721–730. [Google Scholar] [CrossRef]
- DiRenzo, G.V.; Campbell Grant, E.H.; Longo, A.V.; Che-Castaldo, C.; Zamudio, K.R.; Lips, K.R. Imperfect pathogen detection from non-invasive skin swabs biases disease inference. Methods Ecol. Evol. 2018, 9, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Chua, K.; Bellini, W.; Rota, P.; Harcourt, B.; Tamin, A.; Lam, S.; Ksiazek, T.; Rollin, P.; Zaki, S.; Shieh, W.J.; et al. Nipah virus: A recently emergent deadly paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Young, D.; Andrejeva, L.; Livingstone, A.; Goodbourn, S.; Lamb, R.; Collins, P.; Elliott, R.; Randall, R. Virus replication in engineered human cells that do not respond to interferons. J. Virol. 2003, 77, 2174–2181. [Google Scholar] [CrossRef] [Green Version]
- Crameri, G.; Todd, S.; Grimley, S.; McEachern, J.A.; Marsh, G.A.; Smith, C.; Tachedjian, M.; De Jong, C.; Virtue, E.R.; Yu, M.; et al. Establishment, immortalisation and characterisation of pteropid bat cell lines. PLoS ONE 2009, 4, e8266. [Google Scholar] [CrossRef]
- Duncan, R. Fusogenic reoviruses and their fusion-associated small transmembrane (FAST) proteins. Annu. Rev. Virol. 2019, 6, 341–363. [Google Scholar] [CrossRef]
- Bracq, L.; Xie, M.; Lambelé, M.; Vu, L.T.; Matz, J.; Schmitt, A.; Delon, J.; Zhou, P.; Randriamampita, C.; Bouchet, J.; et al. T cell-macrophage fusion triggers multinucleated giant cell formation for HIV-1 spreading. J. Virol. 2017, 91, e01237-17. [Google Scholar] [CrossRef] [Green Version]
- Dedera, D.; Ratner, L. Demonstration of two distinct cytopathic effects with syncytium formation-defective human immunodeficiency virus type 1 mutants. J. Virol. 1991, 65, 6129–6136. [Google Scholar] [CrossRef] [Green Version]
- Nardacci, R.; Perfettini, J.; Grieco, L.; Thieffry, D.; Kroemer, G.; Piacentini, M. Syncytial apoptosis signaling network induced by the HIV-1 envelope glycoprotein complex: An overview. Cell Death Dis. 2015, 6, e1846. [Google Scholar] [CrossRef] [Green Version]
- Kuny, C.V.; Bowen, C.D.; Renner, D.W.; Johnston, C.M.; Szpara, M.L. In vitro evolution of herpes simplex virus 1 (HSV-1) reveals selection for syncytia and other minor variants in cell culture. Virus Evol. 2020, 6, veaa013. [Google Scholar] [CrossRef]
- Annand, E.J.; Horsburgh, B.A.; Xu, K.; Reid, P.A.; Poole, B.; de Kantzow, M.C.; Brown, N.; Tweedie, A.; Michie, M.; Grewar, J.D.; et al. Novel Hendra virus variant detected by sentinel surveillance of Australian horses. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wang, J.; Anderson, D.E.; Halpin, K.; Hong, X.; Chen, H.; Walker, S.; Valdeter, S.; van der Heide, B.; Neave, M.J.; Bingham, J.; et al. A New Hendra Virus Genotype Found in Australian Flying Foxes. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Gibson, L.; Rebas, M.P.; Kemp, J.; Restif, O.; Suu-Ire, R.D.; Wood, J.L.; Cunningham, A.A. Persistence of Multiple Paramyxoviruses in a Closed Captive Colony of Fruit Bats (Eidolon helvum). Viruses 2021, 13, 1659. [Google Scholar] [CrossRef]
- Rockx, B.; Brining, D.; Kramer, J.; Callison, J.; Ebihara, H.; Mansfield, K.; Feldmann, H. Clinical outcome of henipavirus infection in hamsters is determined by the route and dose of infection. J. Virol. 2011, 85, 7658–7671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erbar, S.; Maisner, A. Nipah virus infection and glycoprotein targeting in endothelial cells. Virol. J. 2010, 7, 305. [Google Scholar] [CrossRef] [Green Version]
- Zamora, J.L.R.; Aguilar, H.C. Flow virometry as a tool to study viruses. Methods 2018, 134, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, C.; Dhondt, K.P.; Châlons, M.; Mély, S.; Raoul, H.; Negre, D.; Cosset, F.L.; Gerlier, D.; Vivès, R.R.; Horvat, B. Heparan sulfate-dependent enhancement of henipavirus infection. MBio 2015, 6, e02427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, C.; Bartee, E. Syncytia formation in oncolytic virotherapy. Mol. Ther.-Oncolytics 2019, 15, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Fujii, M.; Sato, T. Somatic cell-derived organoids as prototypes of human epithelial tissues and diseases. Nat. Mater. 2020, 20, 156–169. [Google Scholar] [CrossRef]
- Tang, H.; Abouleila, Y.; Si, L.; Ortega-Prieto, A.M.; Mummery, C.L.; Ingber, D.E.; Mashaghi, A. Human organs-on-chips for virology. Trends Microbiol. 2020, 28, 934–946. [Google Scholar] [CrossRef]
- Sachs, N.; Papaspyropoulos, A.; Zomer-van Ommen, D.D.; Heo, I.; Böttinger, L.; Klay, D.; Weeber, F.; Huelsz-Prince, G.; Iakobachvili, N.; Amatngalim, G.D.; et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019, 38, e100300. [Google Scholar] [CrossRef] [PubMed]
- Russell, C.A.; Kasson, P.M.; Donis, R.O.; Riley, S.; Dunbar, J.; Rambaut, A.; Asher, J.; Burke, S.; Davis, C.T.; Garten, R.J.; et al. Science Forum: Improving pandemic influenza risk assessment. Elife 2014, 3, e03883. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med. 2009, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2008; ISBN 3-900051-07-0. [Google Scholar]
- Wickham, H. Tidyr: Tidy Messy Data. R Package Version 1.1.2. 2020. Available online: https://github.com/tidyverse/tidyr (accessed on 21 August 2021).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Wilke, C.O. Cowplot: Streamlined Plot Theme and Plot Annotations for “ggplot2”. R Package Version 0.9. 4. 2019. Available online: https://wilkelab.org/cowplot/ (accessed on 21 August 2021).
- de Wit, E.; Prescott, J.; Falzarano, D.; Bushmaker, T.; Scott, D.; Feldmann, H.; Munster, V.J. Foodborne Transmission of Nipah Virus in Syrian Hamsters. PLoS Pathog. 2014, 10, e1004001. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, D.A.; Lentz, M.R.; Lara, A.; Bohannon, J.K.; Feuerstein, I.; Huzella, L.; Jahrling, P.B.; Lackemeyer, M.; Laux, J.; Rojas, O.; et al. Aerosol Exposure to Intermediate Size Nipah Virus Particles Induces Neurological Disease in African Green Monkeys. PLoS Neglected Trop. Dis. 2018, 12, e0006978. [Google Scholar] [CrossRef]
- Johnston, S.C.; Briese, T.; Bell, T.M.; Pratt, W.D.; Shamblin, J.D.; Esham, H.L.; Donnelly, G.C.; Johnson, J.C.; Hensley, L.E.; Lipkin, W.I.; et al. Detailed Analysis of the African Green Monkey Model of Nipah Virus Disease. PLoS ONE 2015, 10, e0117817. [Google Scholar] [CrossRef]
- McEachern, J.A.; Bingham, J.; Crameri, G.; Green, D.J.; Hancock, T.J.; Middleton, D.; Feng, Y.R.; Broder, C.C.; Wang, L.F.; Bossart, K.N. A Recombinant Subunit Vaccine Formulation Protects against Lethal Nipah Virus Challenge in Cats. Vaccine 2008, 26, 3842–3852. [Google Scholar] [CrossRef]
- Mire, C.E.; Versteeg, K.M.; Cross, R.W.; Agans, K.N.; Fenton, K.A.; Whitt, M.A.; Geisbert, T.W. Single Injection Recombinant Vesicular Stomatitis Virus Vaccines Protect Ferrets against Lethal Nipah Virus Disease. Virol. J. 2013, 10, 353. [Google Scholar] [CrossRef] [Green Version]
- Mire, C.E.; Geisbert, J.B.; Agans, K.N.; Versteeg, K.M.; Deer, D.J.; Satterfield, B.A.; Fenton, K.A.; Geisbert, T.W. Use of single-injection recombinant vesicular stomatitis virus vaccine to protect nonhuman primates against lethal Nipah virus disease. Emerg. Infect. Dis. 2019, 25, 1144. [Google Scholar] [CrossRef] [Green Version]
- Satterfield, B.A.; Borisevich, V.; Foster, S.L.; Rodriguez, S.E.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. Antagonism of STAT1 by Nipah virus P gene products modulates disease course but not lethal outcome in the ferret model. Sci. Rep. 2019, 9, 16710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valbuena, G.; Halliday, H.; Borisevich, V.; Goez, Y.; Rockx, B. A human lung xenograft mouse model of Nipah virus infection. PLoS Pathog. 2014, 10, e1004063. [Google Scholar] [CrossRef] [PubMed]
- Westbury, H.; Hooper, P.; Brouwer, S.; Selleck, P. Susceptibility of cats to equine morbillivirus. Aust. Vet. J. 1996, 74, 132–134. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Guillaume, V.; Ikeda, F.; Sakuma, Y.; Sato, H.; Wild, T.F.; Kai, C. Establishment of a Nipah virus rescue system. Proc. Natl. Acad. Sci. USA 2006, 103, 16508–16513. [Google Scholar] [CrossRef] [Green Version]
- Gamble, A.; Yeo, Y.Y.; Butler, A.; Tang, H.; Snedden, C.; Mason, C.; Buchholz, D.; Binghman, J.; Aguilar, H.; Lloyd-Smith, J. Data from “Drivers and distribution of henipavirus-induced syncytia: What do we know?”. Zenodo 2021. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | HeV | NiV | CedV | KuV | MojV | References | |||
|---|---|---|---|---|---|---|---|---|---|
| live | transf. | live | transf. | live | transf. | transf. | transf. | ||
| Vero (epithelial) | + | + | + | + | + | + | - | + | [20,79,80] |
| HEK293T (epithelial) | + | + | + | + | + | [20,79] | |||
| A549 (epithelial) | + | + | + | + | + | [70,79] | |||
| Raji B (lymphoblast) | - | [20] | |||||||
| PK13 (epithelial) | – | - | [20,31] | ||||||
| BHK (fibroblast) | + | + | + | + | + | + | – | + | [79,80] |
| CHO (epithelial) | – | – | – | – | [48,50,79] | ||||
| L2 (epithelial) | + | + | + | [79] | |||||
| Rat2 (fibroblast) | + | + | + | [79] | |||||
| EidNi (epithelial) | + | [80] | |||||||
| NHBE (epithelial) | + | + | [81] | ||||||
| SAEC (epithelial) | + | + | [81] | ||||||
| hOE (epithelial) | + | + | [82] | ||||||
| HMVEC (endothelial) | + | [20] | |||||||
| Hamster | Cat | Horse | Guinea Pig | Pig | Dog | Ferret | Monkey | Chicken | ||
|---|---|---|---|---|---|---|---|---|---|---|
| HeV | Endothelial | + [110] | + [108] | + [115] | ||||||
| Epithelial | + [106] | + [109] | + [94] | |||||||
| Macrophage | + [110] | |||||||||
| Neuron | ||||||||||
| Smooth muscle | + [110] | |||||||||
| NiV-M | Endothelial | + [116] | + [102,117] | + [118,119] | + [120] | |||||
| Epithelial | + [105] | + [102,117] | + [45] | + [119] | ||||||
| Macrophage | ||||||||||
| Neuron | + [112] | |||||||||
| Smooth muscle | ||||||||||
| NiV-B | Endothelial | + [119] | ||||||||
| Epithelial | + [105] | + [119] | ||||||||
| Macrophage | ||||||||||
| Neuron | ||||||||||
| Smooth muscle |
| HeV | NiV-M | NiV-B | |
|---|---|---|---|
| Monkey | Respiratory disorders [115] | Respiratory and neurological disorders [100,104,113] | Respiratory disorders [98,99,104] |
| Pig | Respiratory and neurological disorders [128,129,130,131] | No clinical sign [132] | |
| Dog | Mild non-specific symptoms [109] | ||
| Cat | Respiratory disorders [96] | Respiratory disorders, no neurological disorders [102,117] | |
| Ferret | Neurological disorders [133] | Respiratory and neurological disorders [119,134] | Respiratory and neurological disorder [119,134] |
| Guinea pig | Little to no respiratory disorder, neurological disorders [101,135] | Respiratory and neurological disorders [45,136] | |
| Hamster | Respiratory and neurological disorders [114,124,137,138] | Respiratory and neurological disorders [76,95,107,116,127,137,138,139,140,141,142,143] | Respiratory and neurological disorders [76,138] |
| Mouse | Neurological disorders [144] | No clinical sign [116,145] | No clinical sign [145] |
| Horse | Respiratory disorders [146] (neurological disorders also noted from field cases [147]) | ||
| Fruit bat | No clinical sign [93,135] | No clinical sign [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gamble, A.; Yeo, Y.Y.; Butler, A.A.; Tang, H.; Snedden, C.E.; Mason, C.T.; Buchholz, D.W.; Bingham, J.; Aguilar, H.C.; Lloyd-Smith, J.O. Drivers and Distribution of Henipavirus-Induced Syncytia: What Do We Know? Viruses 2021, 13, 1755. https://doi.org/10.3390/v13091755
Gamble A, Yeo YY, Butler AA, Tang H, Snedden CE, Mason CT, Buchholz DW, Bingham J, Aguilar HC, Lloyd-Smith JO. Drivers and Distribution of Henipavirus-Induced Syncytia: What Do We Know? Viruses. 2021; 13(9):1755. https://doi.org/10.3390/v13091755
Chicago/Turabian StyleGamble, Amandine, Yao Yu Yeo, Aubrey A. Butler, Hubert Tang, Celine E. Snedden, Christian T. Mason, David W. Buchholz, John Bingham, Hector C. Aguilar, and James O. Lloyd-Smith. 2021. "Drivers and Distribution of Henipavirus-Induced Syncytia: What Do We Know?" Viruses 13, no. 9: 1755. https://doi.org/10.3390/v13091755