
Structural Analysis of the Menangle Virus P Protein Reveals a Soft Boundary between Ordered and Disordered Regions

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Production and Purification
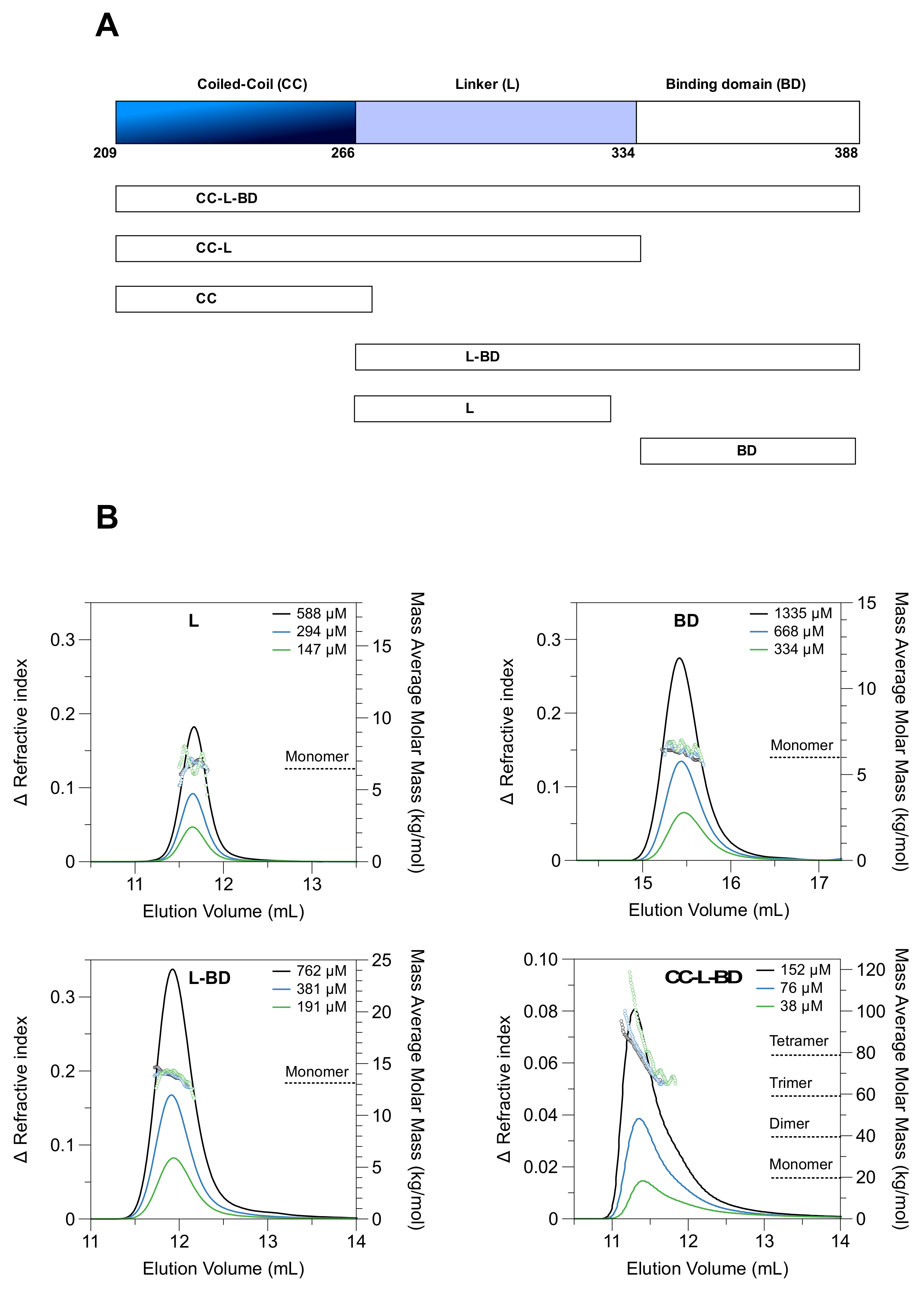
2.2. SEC-MALLS
2.3. Large Zone SEC
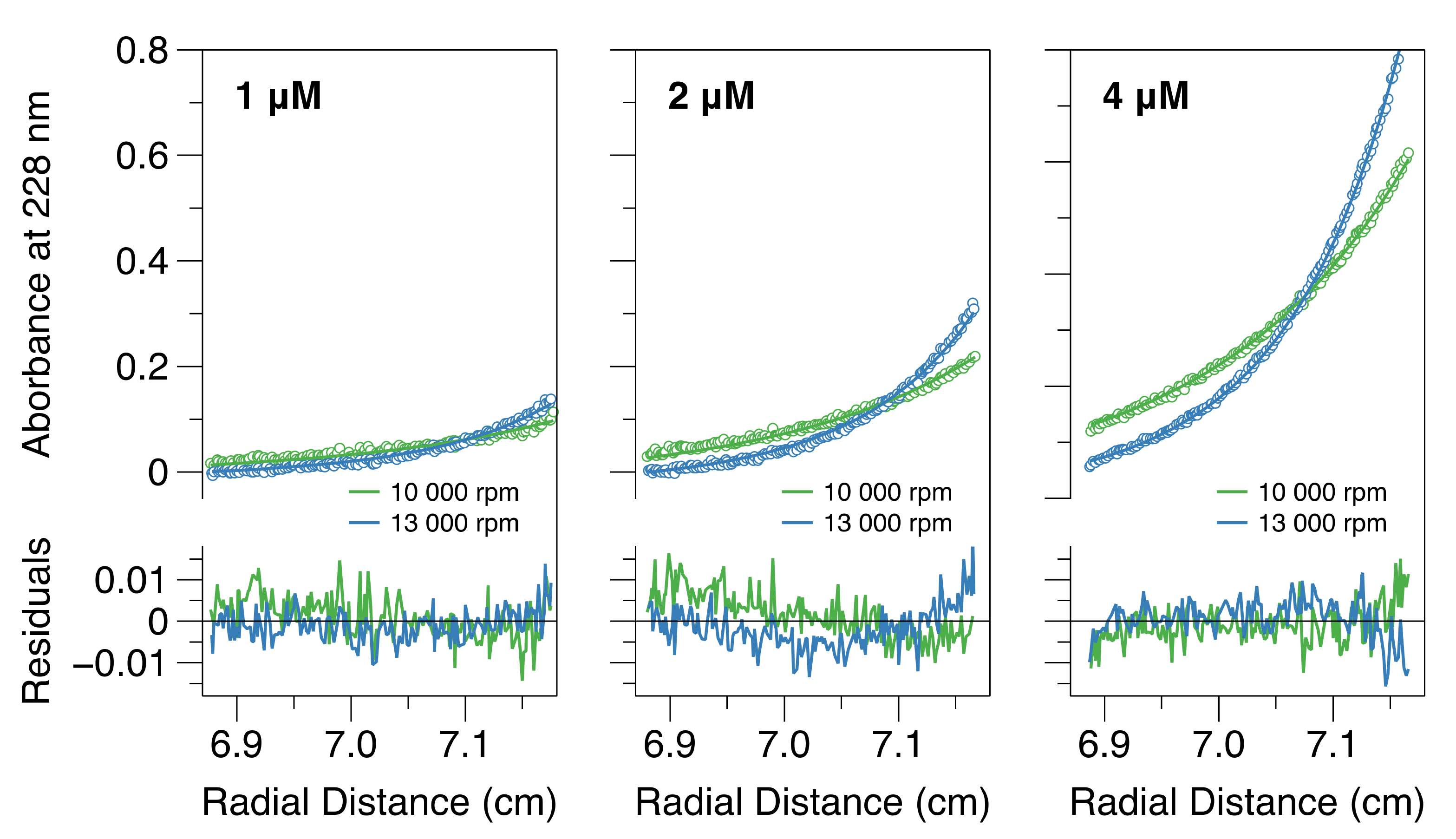
2.4. Analytical Ultracentrifugation
2.5. Small Angle X-ray Scattering (SAXS)
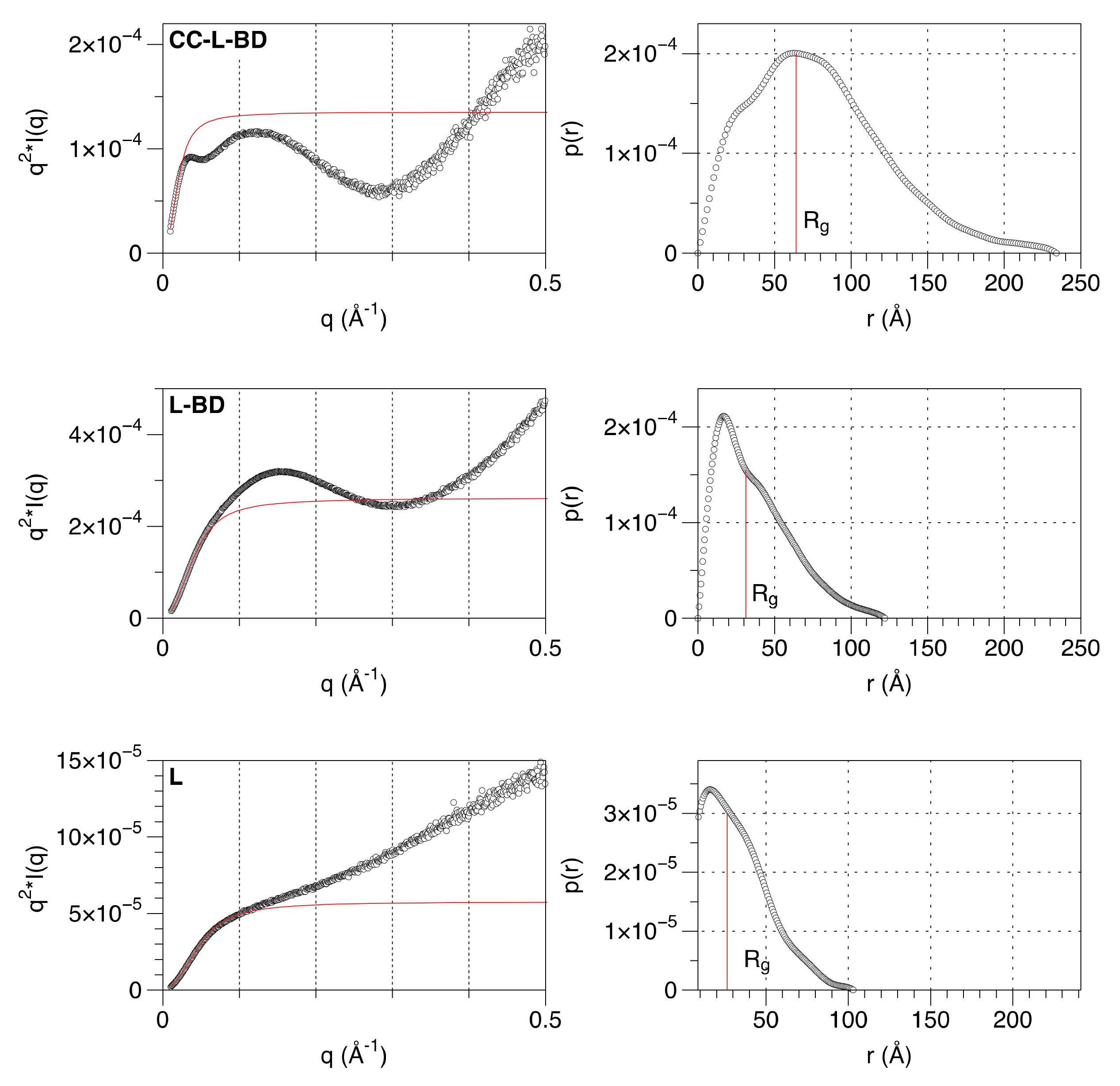
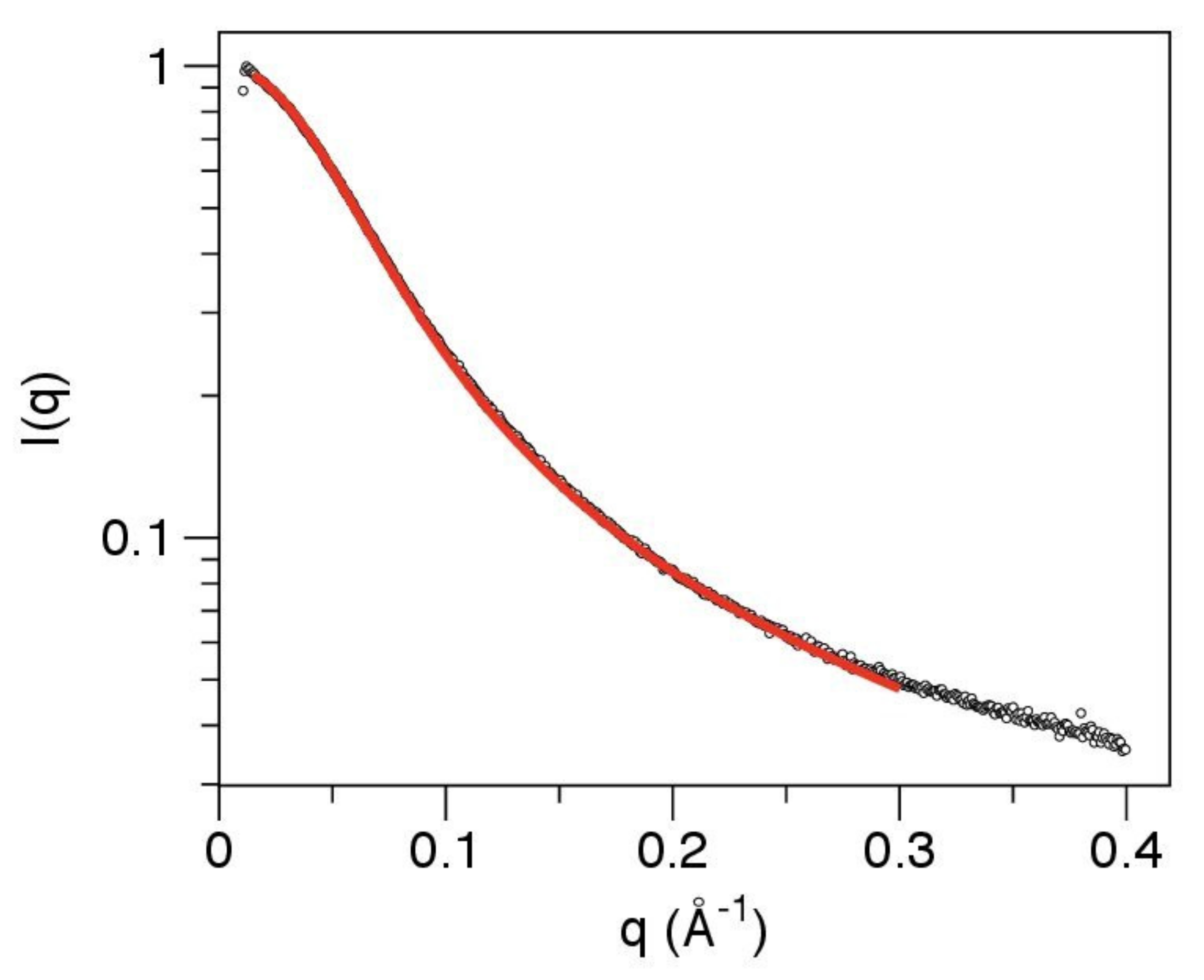
2.5.1. Data Collection and Model Free Analysis
2.5.2. Fit of a Polymer Physics Model to the Data
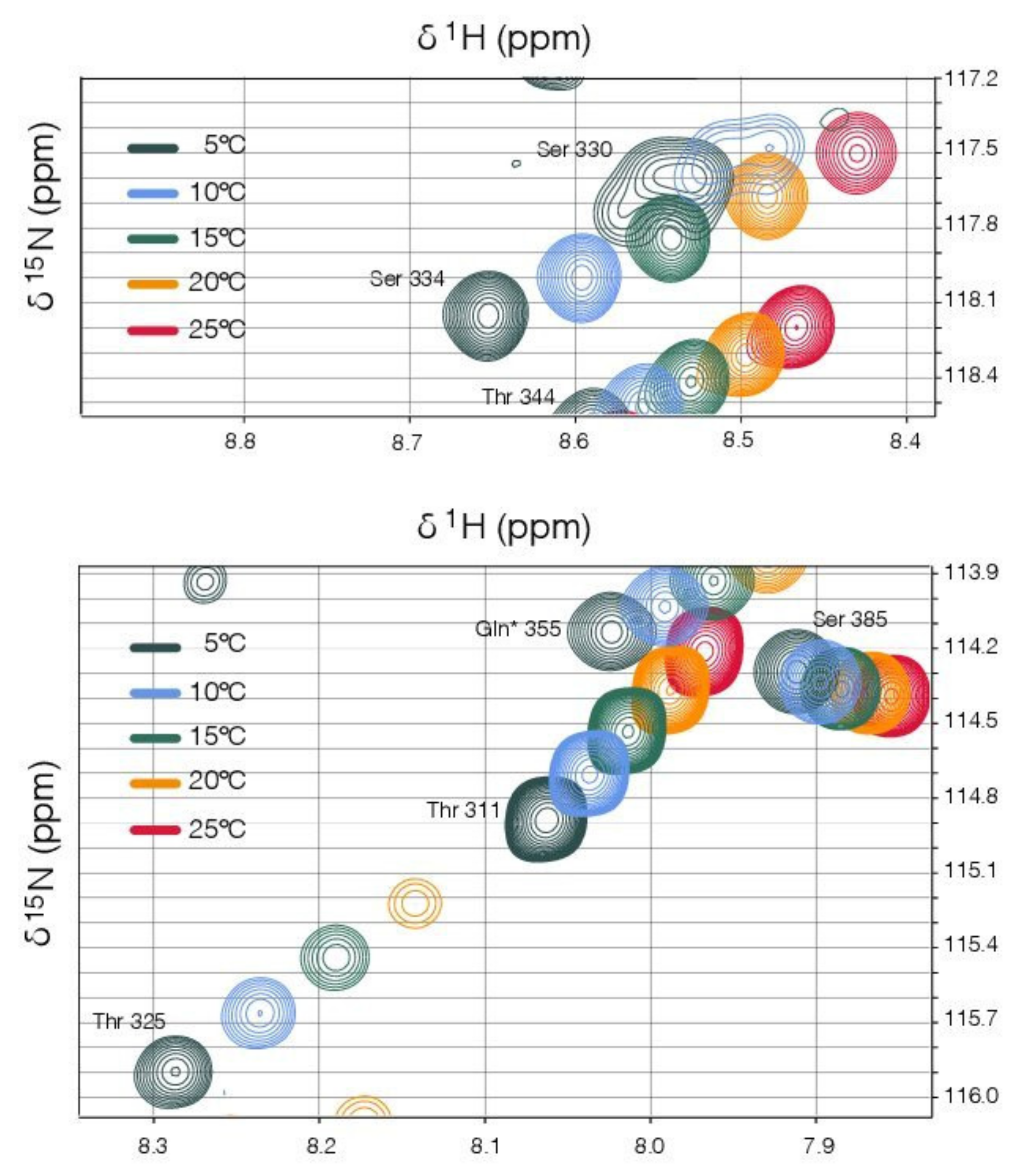
2.6. NMR Spectroscopy
2.7. X-ray Crystallography
3. Results
3.1. The Coiled-Coil Drives Tetramer Formation
3.2. A Dimer–Tetramer Equilibrium Exists at Sub-Micromolar Protein Concentrations
3.3. The Linker Sequence Can Be Modeled as A Worm-like Chain and Exhibits Only Weak Local Conformational Preferences
3.4. The Structural Transition between the Disordered Linker and the Ordered Binding Domain Is Not Abrupt
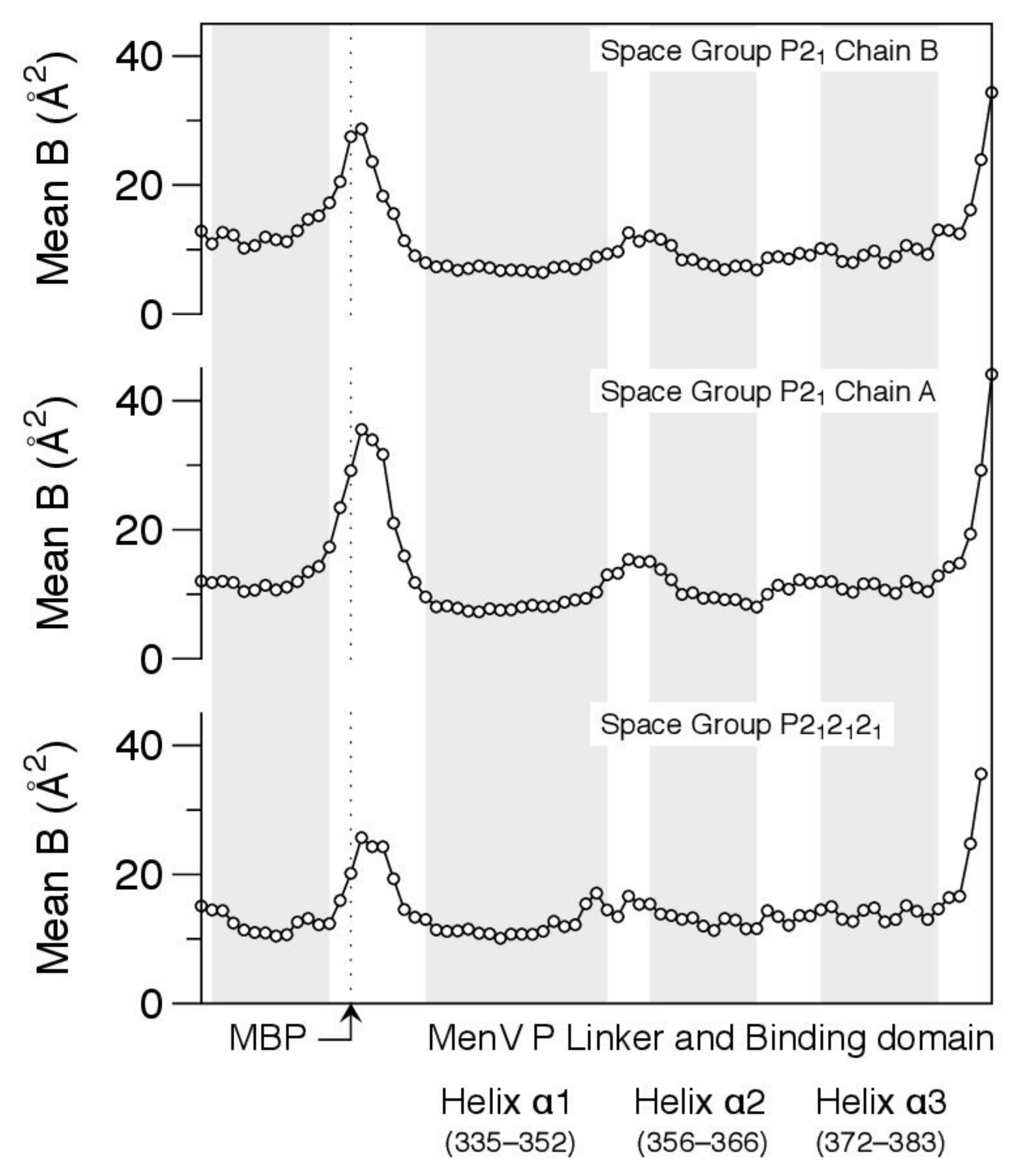
3.5. Crystallographic Model for the Interfacial Region Is Consistent with the NMR Observations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Drexler, J.F.; Corman, V.M.; Müller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, I.; Wang, L.-F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef]
- Hauser, N.; Gushiken, A.; Narayanan, S.; Kottilil, S.; Chua, J. Evolution of Nipah Virus Infection: Past, Present, and Future Considerations. Trop. Med. Infect. Dis. 2021, 6, 24. [Google Scholar] [CrossRef]
- Clayton, B.A. Nipah virus: Transmission of a zoonotic paramyxovirus. Curr. Opin. Virol. 2017, 22, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Ong, K.C.; Wong, K.T. Henipavirus Encephalitis: Recent Developments and Advances. Brain Pathol. 2015, 25, 605–613. [Google Scholar] [CrossRef]
- Bowden, T.R.; Westenbergb, M.; Wanga, L.-F.; Eaton, B.T.; Boyle, D.B. Molecular Characterization of Menangle Virus, a Novel Paramyxovirus which Infects Pigs, Fruit Bats, and Humans. Virology 2001, 283, 358–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barr, J.A.; Smith, C.; Marsh, G.A.; Field, H.; Wang, L.-F. Evidence of bat origin for Menangle virus, a zoonotic paramyxovirus first isolated from diseased pigs. J. Gen. Virol. 2012, 93, 2590–2594. [Google Scholar] [CrossRef]
- Philbey, A.W.; Kirkland, P.D.; Ross, A.D.; Davis, R.J.; Gleeson, A.B.; Love, R.J.; Daniels, P.W.; Gould, A.R.; Hyatt, A.D. An Apparently New Virus (Family Paramyxoviridae) Infectious for Pigs, Humans, and Fruit Bats. Emerg. Infect. Dis. 1998, 4, 269–271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alayyoubi, M.; Leser, G.P.; Kors, C.A.; Lamb, R.A. Structure of the paramyxovirus parainfluenza virus 5 nucleoprotein–RNA complex. Proc. Natl. Acad. Sci. USA 2015, 112, E1792–E1799. [Google Scholar] [CrossRef] [Green Version]
- Gutsche, I.; Desfosses, A.; Effantin, G.; Ling, W.L.; Haupt, M.; Ruigrok, R.W.H.; Sachse, C.; Schoehn, G. Structural virology. Near-atomic cryo-EM structure of the helical measles virus nucleocapsid. Science 2015, 348, 704–707. [Google Scholar] [CrossRef]
- Song, X.; Shan, H.; Zhu, Y.; Hu, S.; Xue, L.; Chen, Y.; Ding, W.; Niu, T.; Gu, J.; Ouyang, S.; et al. Self-capping of nucleoprotein filaments protects the Newcastle disease virus genome. eLife 2019, 8. [Google Scholar] [CrossRef]
- Ker, D.-S.; Jenkins, H.T.; Greive, S.J.; Antson, A.A. CryoEM structure of the Nipah virus nucleocapsid assembly. PLoS Pathog. 2021, 17, e1009740. [Google Scholar] [CrossRef] [PubMed]
- Zinzula, L.; Beck, F.; Klumpe, S.; Bohn, S.; Pfeifer, G.; Bollschweiler, D.; Nagy, I.; Plitzko, J.M.; Baumeister, W. Cryo-EM structure of the cetacean morbillivirus nucleoprotein-RNA complex. J. Struct. Biol. 2021, 213, 107750. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Shan, H.; Liu, M.; Li, T.; Luo, R.; Yang, L.; Qi, L.; Chu, X.; Su, X.; Wang, R.; et al. Structure and assembly of double-headed Sendai virus nucleocapsids. Commun. Biol. 2021, 4, 494. [Google Scholar] [CrossRef] [PubMed]
- Fearns, R.; Plemper, R.K. Polymerases of paramyxoviruses and pneumoviruses. Virus Res. 2017, 234, 87–102. [Google Scholar] [CrossRef] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef]
- Liang, B. Structures of the Mononegavirales Polymerases. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Velthuis, A.J.W.T.; Grimes, J.M.; Fodor, E. Structural insights into RNA polymerases of negative-sense RNA viruses. Nat. Rev. Microbiol. 2021, 19, 303–318. [Google Scholar] [CrossRef]
- Noton, S.L.; Fearns, R. Initiation and regulation of paramyxovirus transcription and replication. Virology 2015, 479–480, 545–554. [Google Scholar] [CrossRef] [Green Version]
- Tarbouriech, N.; Curran, J.; Ruigrok, R.W.; Burmeister, W.P. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat. Struct. Biol. 2000, 7, 777–781. [Google Scholar] [CrossRef]
- Communie, G.; Crepin, T.; Maurin, D.; Jensen, M.R.; Blackledge, M.; Ruigrok, R.W.H. Structure of the Tetramerization Domain of Measles Virus Phosphoprotein. J. Virol. 2013, 87, 7166–7169. [Google Scholar] [CrossRef] [Green Version]
- Cox, R.; Green, T.J.; Purushotham, S.; Deivanayagam, C.; Bedwell, G.J.; Prevelige, P.E.; Luo, M. Structural and Functional Characterization of the Mumps Virus Phosphoprotein. J. Virol. 2013, 87, 7558–7568. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.-M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef]
- Pickar, A.; Elson, A.; Yang, Y.; Xu, P.; Luo, M.; He, B. Oligomerization of Mumps Virus Phosphoprotein. J. Virol. 2015, 89, 11002–11010. [Google Scholar] [CrossRef] [Green Version]
- Douglas, J.; Drummond, A.J.; Kingston, R.L. Evolutionary history of cotranscriptional editing in the paramyxoviral phosphoprotein gene. Virus Evol. 2021, 7, veab028. [Google Scholar] [CrossRef] [PubMed]
- Du Pont, V.; Jiang, Y.; Plemper, R.K. Bipartite interface of the measles virus phosphoprotein X domain with the large polymerase protein regulates viral polymerase dynamics. PLoS Pathog. 2019, 15, e1007995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal Domain of the Measles Virus Nucleoprotein Is Intrinsically Disordered and Folds upon Binding to the C-terminal Moiety of the Phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingston, R.L.; Hamel, D.J.; Gay, L.S.; Dahlquist, F.W.; Matthews, B.W. Structural basis for the attachment of a paramyxoviral polymerase to its template. Proc. Natl. Acad. Sci. USA 2004, 101, 8301–8306. [Google Scholar] [CrossRef] [Green Version]
- Houben, K.; Marion, D.; Tarbouriech, N.; Ruigrok, R.W.H.; Blanchard, L. Interaction of the C-Terminal Domains of Sendai Virus N and P Proteins: Comparison of Polymerase-Nucleocapsid Interactions within the Paramyxovirus Family. J. Virol. 2007, 81, 6807–6816. [Google Scholar] [CrossRef] [Green Version]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.H.; Jamin, M.; Jensen, M.R.; et al. Atomic Resolution Description of the Interaction between the Nucleoprotein and Phosphoprotein of Hendra Virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef]
- Sourimant, J.; Thakkar, V.D.; Cox, R.M.; Plemper, R.K. Viral evolution identifies a regulatory interface between paramyxovirus polymerase complex and nucleocapsid that controls replication dynamics. Sci. Adv. 2020, 6, eaaz1590. [Google Scholar] [CrossRef] [Green Version]
- Johansson, K.; Bourhis, J.-M.; Campanacci, V.; Cambillau, C.; Canard, B.; Longhi, S. Crystal Structure of the Measles Virus Phosphoprotein Domain Responsible for the Induced Folding of the C-terminal Domain of the Nucleoprotein. J. Biol. Chem. 2003, 278, 44567–44573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanchard, L.; Tarbouriech, N.; Blackledge, M.; Timmins, P.; Burmeister, W.P.; Ruigrok, R.W.; Marion, D. Structure and dynamics of the nucleocapsid-binding domain of the Sendai virus phosphoprotein in solution. Virology 2004, 319, 201–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingston, R.L.; Gay, L.S.; Baase, W.S.; Matthews, B.W. Structure of the Nucleocapsid-Binding Domain from the Mumps Virus Polymerase; an Example of Protein Folding Induced by Crystallization. J. Mol. Biol. 2008, 379, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Yegambaram, K.; Bulloch, E.; Kingston, R.L. Protein domain definition should allow for conditional disorder. Protein Sci. 2013, 22, 1502–1518. [Google Scholar] [CrossRef] [Green Version]
- Hausrath, A.C.; Kingston, R.L. Conditionally disordered proteins: Bringing the environment back into the fold. Cell. Mol. Life Sci. 2017, 74, 3149–3162. [Google Scholar] [CrossRef] [PubMed]
- Pace, C.N.; Vajdos, F.; Fee, L.; Grimsley, G.; Gray, T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995, 4, 2411–2423. [Google Scholar] [CrossRef] [Green Version]
- Webby, M.N.; Sullivan, M.P.; Yegambaram, K.M.; Radjainia, M.; Keown, J.; Kingston, R.L. A method for analyzing the composition of viral nucleoprotein complexes, produced by heterologous expression in bacteria. Virology 2019, 527, 159–168. [Google Scholar] [CrossRef]
- Valdes, R.; Ackers, G.K. Study of protein subunit association equilibria by elution gel chromatography. Methods Enzymol. 1979, 61, 125–142. [Google Scholar] [CrossRef]
- Winzor, D.J. Analytical exclusion chromatography. J. Biochem. Biophys. Methods 2003, 56, 15–52. [Google Scholar] [CrossRef]
- Schuck, P. Size-Distribution Analysis of Macromolecules by Sedimentation Velocity Ultracentrifugation and Lamm Equation Modeling. Biophys. J. 2000, 78, 1606–1619. [Google Scholar] [CrossRef] [Green Version]
- Dam, J.; Schuck, P. Calculating Sedimentation Coefficient Distributions by Direct Modeling of Sedimentation Velocity Concentration Profiles. Methods Enzymol. 2004, 384, 185–212. [Google Scholar] [CrossRef]
- Laue, T.M.; Shah, B.D.; Ridgeway, T.M.; Pelletier, S.L. Computer-Aided Interpretation of Analytical Sedimentation Data for Proteins. In Analytical Ultracentrifugation in Biochemistry and Polymer Science; Harding, S.E., Rowe, A.J., Horton, J.C., Eds.; Royal Society of Chemistry: London, UK, 1992. [Google Scholar]
- Schuck, P. On the analysis of protein self-association by sedimentation velocity analytical ultracentrifugation. Anal. Biochem. 2003, 320, 104–124. [Google Scholar] [CrossRef]
- Vistica, J.; Dam, J.; Balbo, A.; Yikilmaz, E.; Mariuzza, R.A.; Rouault, T.A.; Schuck, P. Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 2004, 326, 234–256. [Google Scholar] [CrossRef] [Green Version]
- Taylor, I.A.; Eccleston, J.F.; Rittinger, K.; Fu, H. Sedimentation Equilibrium Studies. Methods Mol. Biol. 2004, 261, 119–136. [Google Scholar] [CrossRef]
- Schuck, P. Sedimentation Equilibrium Analytical Ultracentrifugation for Multicomponent Protein Interactions. In Protein Interactions; Schuck, P., Ed.; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2007; pp. 289–316. [Google Scholar]
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.; Svergun, D.I. New developments in theATSASprogram package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Konarev, P.; Volkov, V.V.; Sokolova, A.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Blanchet, C.E.; Svergun, D. Small-Angle X-Ray Scattering on Biological Macromolecules and Nanocomposites in Solution. Annu. Rev. Phys. Chem. 2013, 64, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.; Bloomfield, V.A. Light scattering from wormlike chains with excluded volume effects. Biopolymers 1968, 6, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Brulet, A.; Boue, F.; Cotton, J.P. About the Experimental Determination of the Persistence Length of Wormlike Chains of Polystyrene. J. Phys. II Fr. 1996, 6, 885–891. [Google Scholar] [CrossRef]
- Pötschke, D. Analysis of the conformation of worm-like chains by small-angle scattering: Monte-Carlo simulations in com-parison to analytical theory. Macromol. Theory Simul. 2000, 9, 345–353. [Google Scholar] [CrossRef]
- Pérez, J.; Vachette, P.; Russo, D.; Desmadril, M.; Durand, D. Heat-induced unfolding of neocarzinostatin, a small all-β protein investigated by small-angle X-ray scattering. J. Mol. Biol. 2001, 308, 721–743. [Google Scholar] [CrossRef] [PubMed]
- Boze, H.; Marlin, T.; Durand, D.; Pérez, J.; Vernhet, A.; Canon, F.; Sarni-Manchado, P.; Cheynier, V.; Cabane, B. Proline-Rich Salivary Proteins Have Extended Conformations. Biophys. J. 2010, 99, 656–665. [Google Scholar] [CrossRef] [Green Version]
- Receveur-Brechot, V.; Durand, D. How Random are Intrinsically Disordered Proteins? A Small Angle Scattering Perspective. Curr. Protein Pept. Sci. 2012, 13, 55–75. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, D.P.; Hernandez, B.; Durand, D.; Hourdel, V.; Sotomayor-Pérez, A.-C.; Vachette, P.; Ghomi, M.; Chamot-Rooke, J.; Ladant, D.; Brier, S.; et al. Structural models of intrinsically disordered and calcium-bound folded states of a protein adapted for secretion. Sci. Rep. 2015, 5, 14223. [Google Scholar] [CrossRef] [Green Version]
- Rawiso, M.; Duplessix, R.; Picot, C. Scattering function of polystyrene. Macromolecules 1987, 20, 630–648. [Google Scholar] [CrossRef]
- Häckel, M.; Hinz, H.J.; Hedwig, G.R. Partial molar volumes of proteins: Amino acid side-chain contributions derived from the partial molar volumes of some tripeptides over the temperature range 10-90 degrees C. Biophys. Chem. 1999, 82, 35–50. [Google Scholar] [CrossRef]
- Herr, N.; Webby, M.N.; Bulloch, E.; Schmitz, M.; Kingston, R.L. NMR chemical shift assignment of the C-terminal region of the Menangle virus phosphoprotein. Biomol. NMR Assign. 2019, 13, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Roche, J.; Ying, J.; Shen, Y.; Torchia, D.A.; Bax, A. ARTSY-J: Convenient and precise measurement of 3JHNHα couplings in medium-size proteins from TROSY-HSQC spectra. J. Magn. Reson. 2016, 268, 73–81. [Google Scholar] [CrossRef] [Green Version]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Niklasson, M.; Otten, R.; Ahlner, A.; Andresen, C.; Schlagnitweit, J.; Petzold, K.; Lundström, P. Comprehensive analysis of NMR data using advanced line shape fitting. J. Biomol. NMR 2017, 69, 93–99. [Google Scholar] [CrossRef] [Green Version]
- Willard, L.; Ranjan, A.; Zhang, H.; Monzavi, H.; Boyko, R.F.; Sykes, B.D.; Wishart, D.S. VADAR: A web server for quantitative evaluation of protein structure quality. Nucleic Acids Res. 2003, 31, 3316–3319. [Google Scholar] [CrossRef] [PubMed]
- Vogeli, B.; Ying, J.; Grishaev, A.; Bax, A. Limits on Variations in Protein Backbone Dynamics from Precise Measurements of Scalar Couplings. J. Am. Chem. Soc. 2007, 129, 9377–9385. [Google Scholar] [CrossRef] [PubMed]
- Kingston, R.L.; Baker, H.M.; Baker, E.N. Search designs for protein crystallization based on orthogonal arrays. Acta Crystallogr. D 1994, 50, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.; Winn, M.D.; Storoni, L.C.; Read, R. Phasercrystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.; Cowtan, K.D. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D 2011, 67, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Stevens, F.J.; Schiffer, M. Computer simulation of protein self-association during small-zone gel filtration. Estimation of equilibrium constants. Biochem. J. 1981, 195, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Nenortas, E.; Beckett, D. Reduced-Scale Large-Zone Analytical Gel Filtration Chromatography for Measurement of Protein Association Equilibria. Anal. Biochem. 1994, 222, 366–373. [Google Scholar] [CrossRef]
- Bailey, G.D.; Hyun, J.K.; Mitra, A.; Kingston, R.L. Proton-Linked Dimerization of a Retroviral Capsid Protein Initiates Capsid Assembly. Structure 2009, 17, 737–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, P.; Radu, C.; Ward, E. Sedimentation equilibrium analysis of recombinant mouse FcRn with murine IgG1. Mol. Immunol. 1999, 36, 1117–1125. [Google Scholar] [CrossRef]
- Rambo, R.P.; Tainer, J. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers 2011, 95, 559–571. [Google Scholar] [CrossRef] [Green Version]
- Kohn, J.E.; Millett, I.S.; Jacob, J.; Zagrovic, B.; Dillon, T.M.; Cingel, N.; Dothager, R.S.; Seifert, S.; Thiyagarajan, P.; Sosnick, T.R.; et al. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12491–12496. [Google Scholar] [CrossRef] [Green Version]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle Neutron and X-ray Scattering; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Müller-Späth, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Rüegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef] [Green Version]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Garnier, J.; Gibrat, J.-F.; Robson, B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol. 1996, 266, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Tamiola, K.; Mulder, F.A.A. Using NMR chemical shifts to calculate the propensity for structural order and disorder in proteins. Biochem. Soc. Trans. 2012, 40, 1014–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishima, R.; Torchia, D.A. Estimating the time scale of chemical exchange of proteins from measurements of transverse relaxation rates in solution. J. Biomol. NMR 1999, 14, 369–372. [Google Scholar] [CrossRef]
- Millet, O.; Loria, J.P.; Kroenke, C.D.; Pons, M.; Palmer, A.G., III. The Static Magnetic Field Dependence of Chemical Exchange Linebroadening Defines the NMR Chemical Shift Time Scale. J. Am. Chem. Soc. 2000, 122, 2867–2877. [Google Scholar] [CrossRef]
- Baxter, N.J.; Williamson, M.P. Temperature dependence of 1H chemical shifts in proteins. J. Biomol. NMR 1997, 9, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.F.; Mueller, G.; Zhong, X.; Pedersen, L. A synergistic approach to protein crystallization: Combination of a fixed-arm carrier with surface entropy reduction. Protein Sci. 2010, 19, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. Identification of helix capping and β-turn motifs from NMR chemical shifts. J. Biomol. NMR 2012, 52, 211–232. [Google Scholar] [CrossRef]
- Salvador, P. Dependencies of J-Couplings upon Dihedral Angles on Proteins. Annu. Rep. NMR Spectrosc. 2014, 81, 185–227. [Google Scholar] [CrossRef] [Green Version]
- Guseva, S.; Milles, S.; Jensen, M.R.; Schoehn, G.; Ruigrok, R.W.; Blackledge, M. Structure, dynamics and phase separation of measles virus RNA replication machinery. Curr. Opin. Virol. 2020, 41, 59–67. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles Virus Forms Inclusion Bodies with Properties of Liquid Organelles. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ma, D.; George, C.X.; Nomburg, J.L.; Pfaller, C.K.; Cattaneo, R.; Samuel, C.E. Upon Infection, Cellular WD Repeat-Containing Protein 5 (WDR5) Localizes to Cytoplasmic Inclusion Bodies and Enhances Measles Virus Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fearns, R.; Young, D.F.; Randall, R. Evidence that the paramyxovirus simian virus 5 can establish quiescent infections by remaining inactive in cytoplasmic inclusion bodies. J. Gen. Virol. 1994, 75, 3525–3539. [Google Scholar] [CrossRef] [PubMed]
- Su, J.M.; Wilson, M.Z.; Samuel, C.E.; Ma, D. Formation and Function of Liquid-Like Viral Factories in Negative-Sense Single-Stranded RNA Virus Infections. Viruses 2021, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P.; Blanchard, L.; Timmins, P.; Marion, D.; Ruigrok, R.W.H.; Blackledge, M. A structural model for unfolded proteins from residual dipolar couplings and small-angle X-ray scattering. Proc. Natl. Acad. Sci. USA 2005, 102, 17002–17007. [Google Scholar] [CrossRef] [Green Version]
- Houben, K.; Blanchard, L.; Blackledge, M.; Marion, D. Intrinsic Dynamics of the Partly Unstructured PX Domain from the Sendai Virus RNA Polymerase Cofactor P. Biophys. J. 2007, 93, 2830–2844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guseva, S.; Milles, S.; Jensen, M.R.; Salvi, N.; Kleman, J.-P.; Maurin, D.; Ruigrok, R.W.H.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | |||
|---|---|---|---|
| “CC-L-BD” MenV P209-388 | “L-BD” MenV P267-388 | “L” MenV P267-328 | |
| Instrument | Australian Synchrotron SAXS/WAXS beamline, Pilatus 1M detector | Australian Synchrotron SAXS/WAXS beamline, Pilatus 1M detector | Australian Synchrotron SAXS/WAXS beamline, Pilatus 1M detector |
| Wavelength (Å) | 1.0332 | 1.0332 | 1.0332 |
| q range (Å−1) | 0.01–0.60 | 0.01–0.60 | 0.01–0.60 |
| Exposure time (s) a | 1 | 2 | 1 |
| Protein concentration range (g/L) | 0.1–4.8 | 0.35–11.1 | 0.45–3.62 |
| Buffer composition | 12.5 mM MOPS/KOH pH 7.0, 250 mM NaCl | 12.5 mM MOPS/KOH pH 7.0, 150 mM NaCl | 12.5 mM Tris/HCl pH 8.5, 150 mM NaCl |
| Temperature (°C) | 10 | 10 | 10 |
| SASDB accession codes | SASDLG9 | SASDLH9 | SASDLJ9 |
| Crystal Form I | Crystal Form II | |
|---|---|---|
| Crystallization Conditions | ||
| Protein concentration (μM) | 3300 | 3300 |
| Crystallization Method | Sitting Drop Vapor diffusion | Sitting Drop Vapor diffusion |
| Reservoir Solution | 1.65 M Ammonium Sulphate 0.20 M Malic acid/KOH pH 5.5 | 20% (w/v) PEG 5000 monomethyl ether 0.2 M PIPES/KOH pH 6.7 0.1 M Proline |
| Temperature (°C) | 18 | 18 |
| X-ray diffraction data | ||
| Cryoprotectant | 1.65 M Ammonium Sulphate 0.20 M Malic acid/KOH pH 5.5 1M Lithium Sulphate 5 mM Maltose | 20% (w/v) PEG 5000 monomethyl ether 0.2 M PIPES/KOH pH 6.7 0.1 M Proline 20% (v/v) Ethlyene glycol 5 mM Maltose |
| Space group | P21 | P212121 |
| Unit cell dimensions | a = 57.6, b = 70.1, c = 111.7 Å β = 96.5° | a = 67.6 b = 77.4, c = 79.1 Å |
| X-ray source | Australian Synchrotron Beamline MX1 | Rigaku MicroMax-007 HF Rotating Copper Anode |
| X-ray wavelength (Å) | 0.95370 | 1.54179 |
| Sample Temperature (K) | 110 | 110 |
| Data resolution limits (Å) a | 59.26–1.31 (1.33–1.31) | 31.24–1.55 (1.57–1.55) |
| Number of unique observations a | 149703 (307) | 59991 (2017) |
| Mean Redundancy a | 3.2 (1.5) | 6.8 (5.6) |
| Completeness (%) a | 70.7 (2.9) | 97.4 (66.5) |
| Rmeasure a | 0.061 (0.857) | 0.026 (0.204) |
| Rmerge a | 0.051 (0.607) | 0.026 (0.166) |
| CC1/2 a | (0.783) | (0.982) |
| Crystallographic models | ||
| Number of protein molecules in the asymmetric unit | 2 × 431 residues | 1 × 431 residues |
| Rwork/Rfree b | 14.9/19.6 | 16.7/19.0 |
| Total number of protein atoms | 6998 | 3569 |
| Number of water molecules | 1321 | 505 |
| Other ligands | Maltose, Sulphate | Maltose, Ethylene Glycol, PIPES, Proline |
| Disorder Model | Individual Anisotropic B-factors | Individual Isotropic B-factors |
| Mean total isotropic B-factor, all protein atoms (Å2): | 11.7 | 15.0 |
| Bulk Solvent model | Mask | Mask |
| RMSD from ideal geometry: Bond lengths (Å)/Bond angles (°) | 0.009/1.495 | 0.010/1.398 |
| Residues in Favoured/Allowed regions of Ramachandran plot (%) c | 99/1 | 99/1 |
| PDB ID | 7KD4 | 7KD5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webby, M.N.; Herr, N.; Bulloch, E.M.M.; Schmitz, M.; Keown, J.R.; Goldstone, D.C.; Kingston, R.L. Structural Analysis of the Menangle Virus P Protein Reveals a Soft Boundary between Ordered and Disordered Regions. Viruses 2021, 13, 1737. https://doi.org/10.3390/v13091737
Webby MN, Herr N, Bulloch EMM, Schmitz M, Keown JR, Goldstone DC, Kingston RL. Structural Analysis of the Menangle Virus P Protein Reveals a Soft Boundary between Ordered and Disordered Regions. Viruses. 2021; 13(9):1737. https://doi.org/10.3390/v13091737
Chicago/Turabian StyleWebby, Melissa N., Nicole Herr, Esther M. M. Bulloch, Michael Schmitz, Jeremy R. Keown, David C. Goldstone, and Richard L. Kingston. 2021. "Structural Analysis of the Menangle Virus P Protein Reveals a Soft Boundary between Ordered and Disordered Regions" Viruses 13, no. 9: 1737. https://doi.org/10.3390/v13091737