
Processing Hundreds of SARS-CoV-2 Samples with an In-House PCR-Based Method without Robotics

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Material and Lysis Buffers
2.2. Primers and Probes
2.3. Viral RNA Extraction
2.4. One Step RT-qPCR
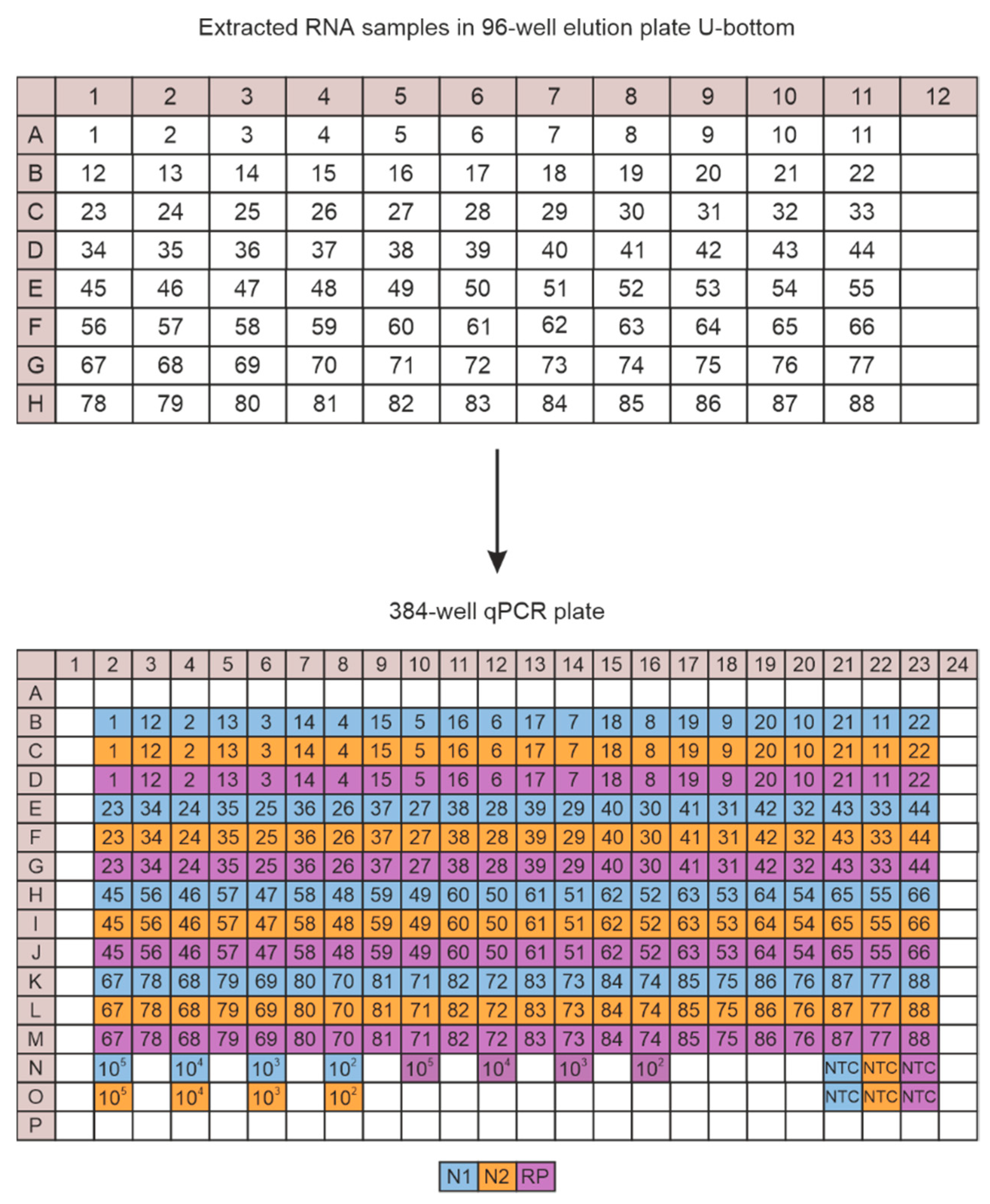
2.5. CFX Plate Loading Strategy
2.6. Data Analysis
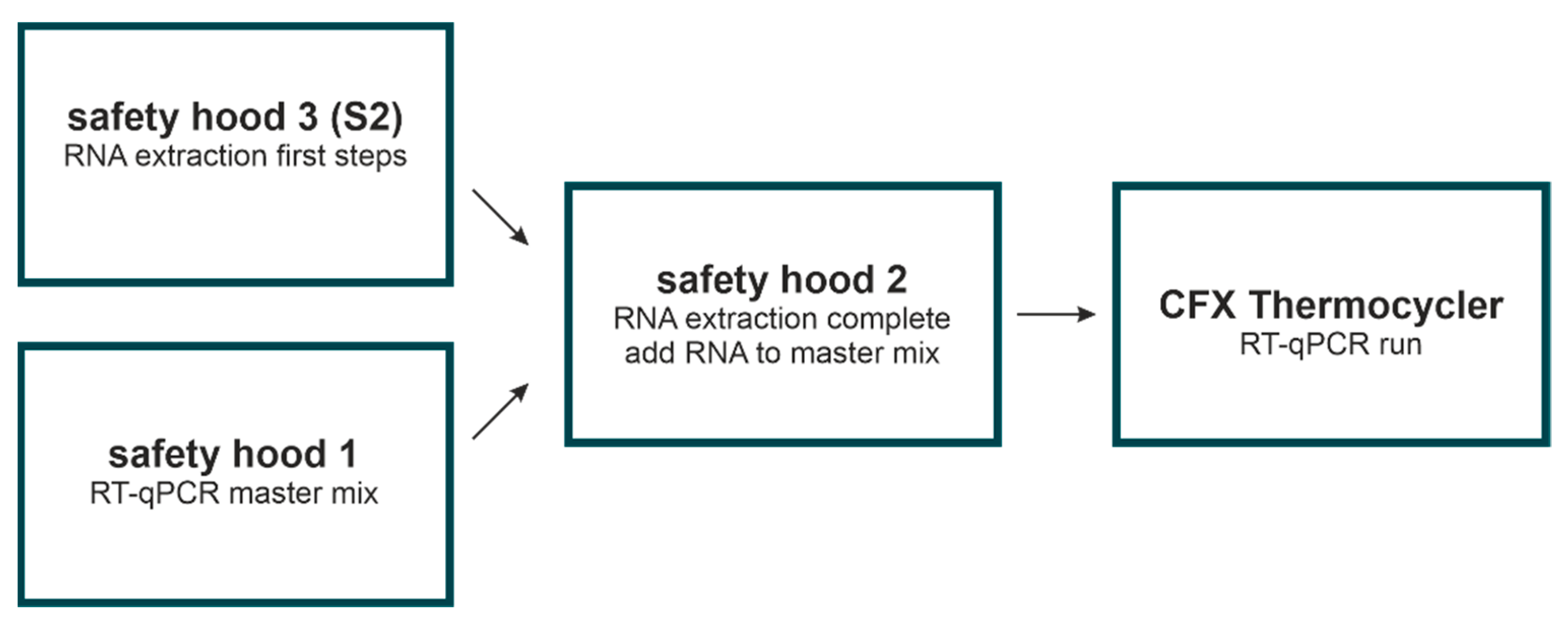
2.7. Room Separation and Decontamination
3. Results
3.1. Principle of Workflow
3.2. Two Different RT-qPCR Kits Can Be Used
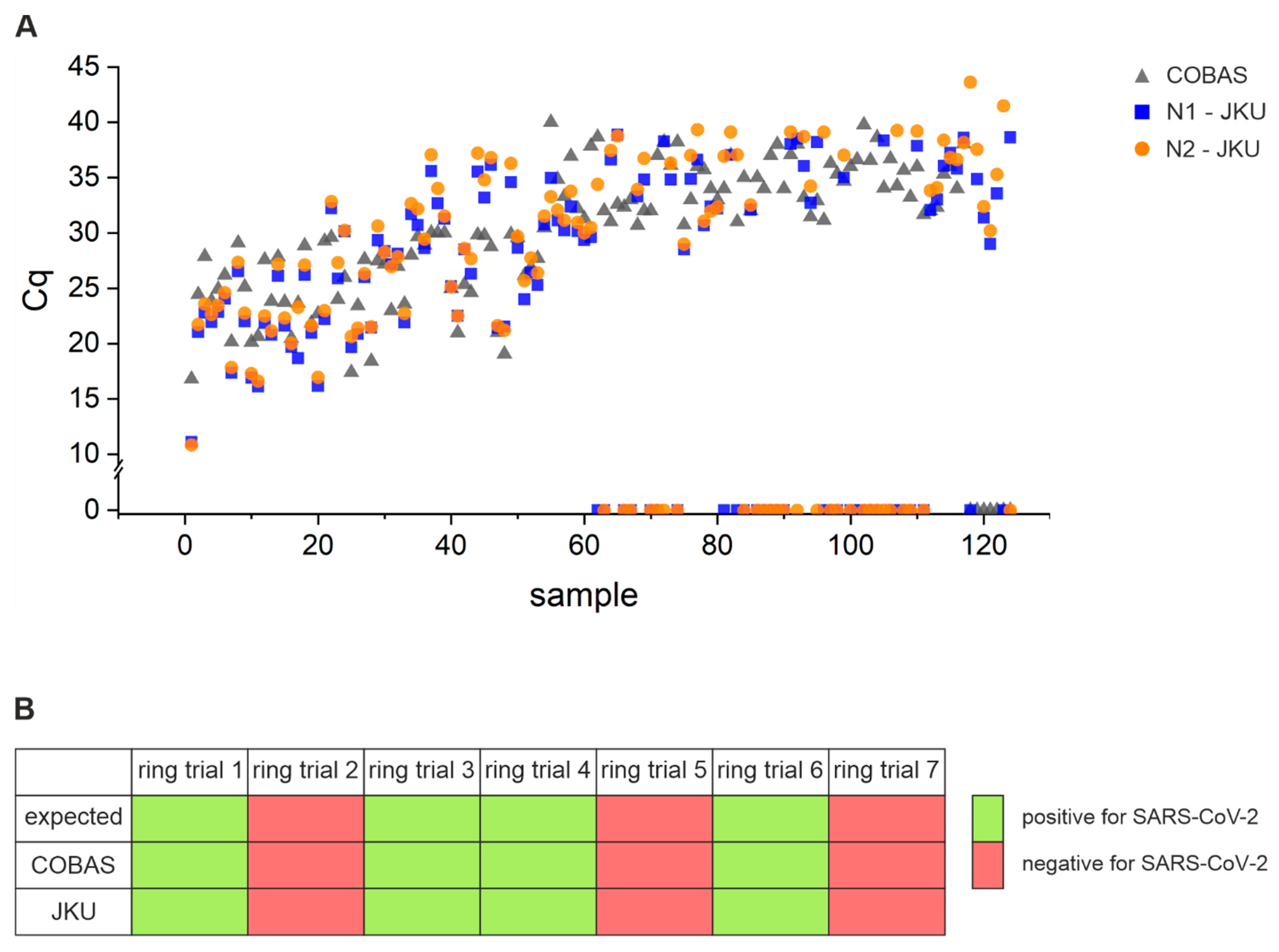
3.3. Assay Sensitivity: Comparison of Performance of Open System Assay with COBAS 6800
3.4. Technical Sensitivity and Reproducibility of Our Open System Assay
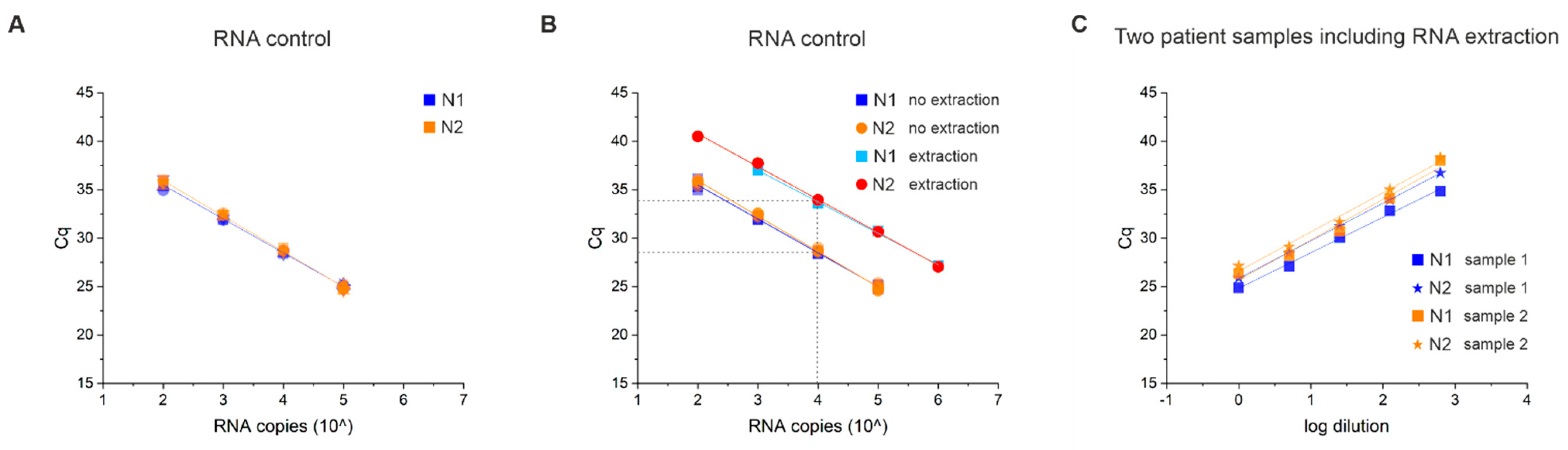
3.5. RNA Extraction Efficiency and Limit of Detection (LOD)
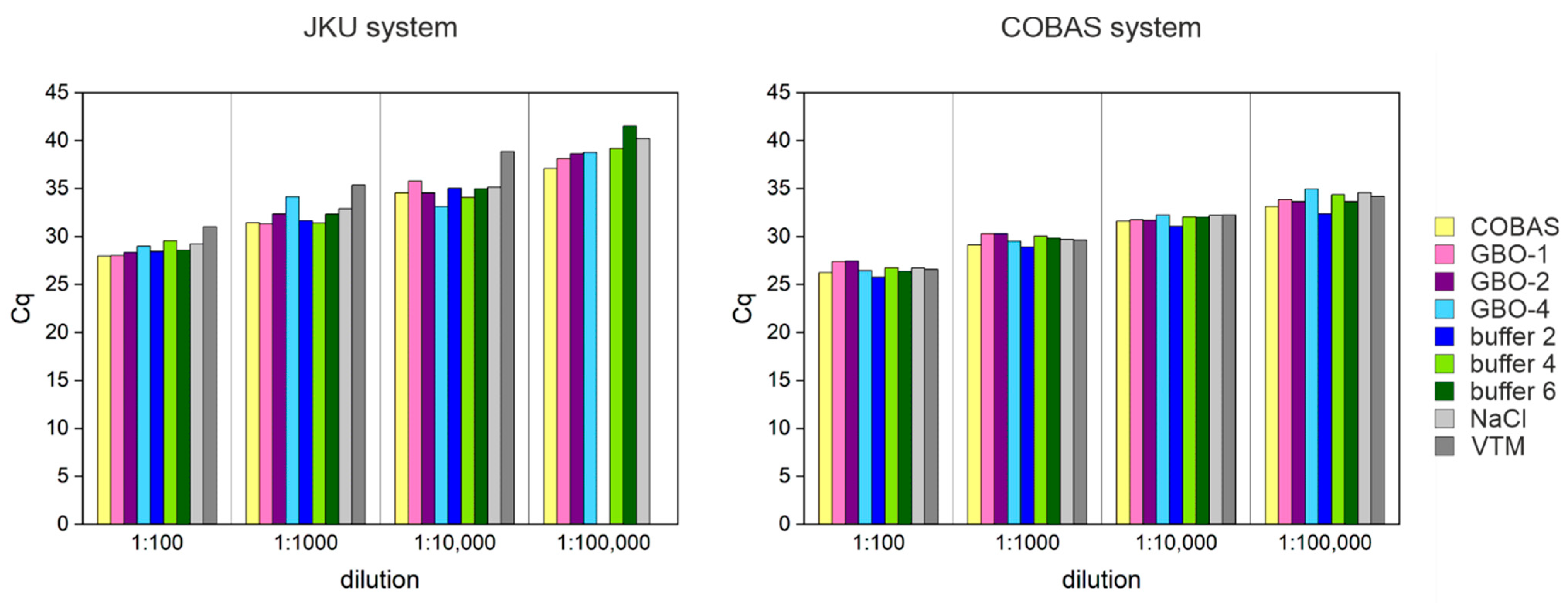
3.6. Lysis Buffer Can Affect the Test Performance
3.7. Sample Stability
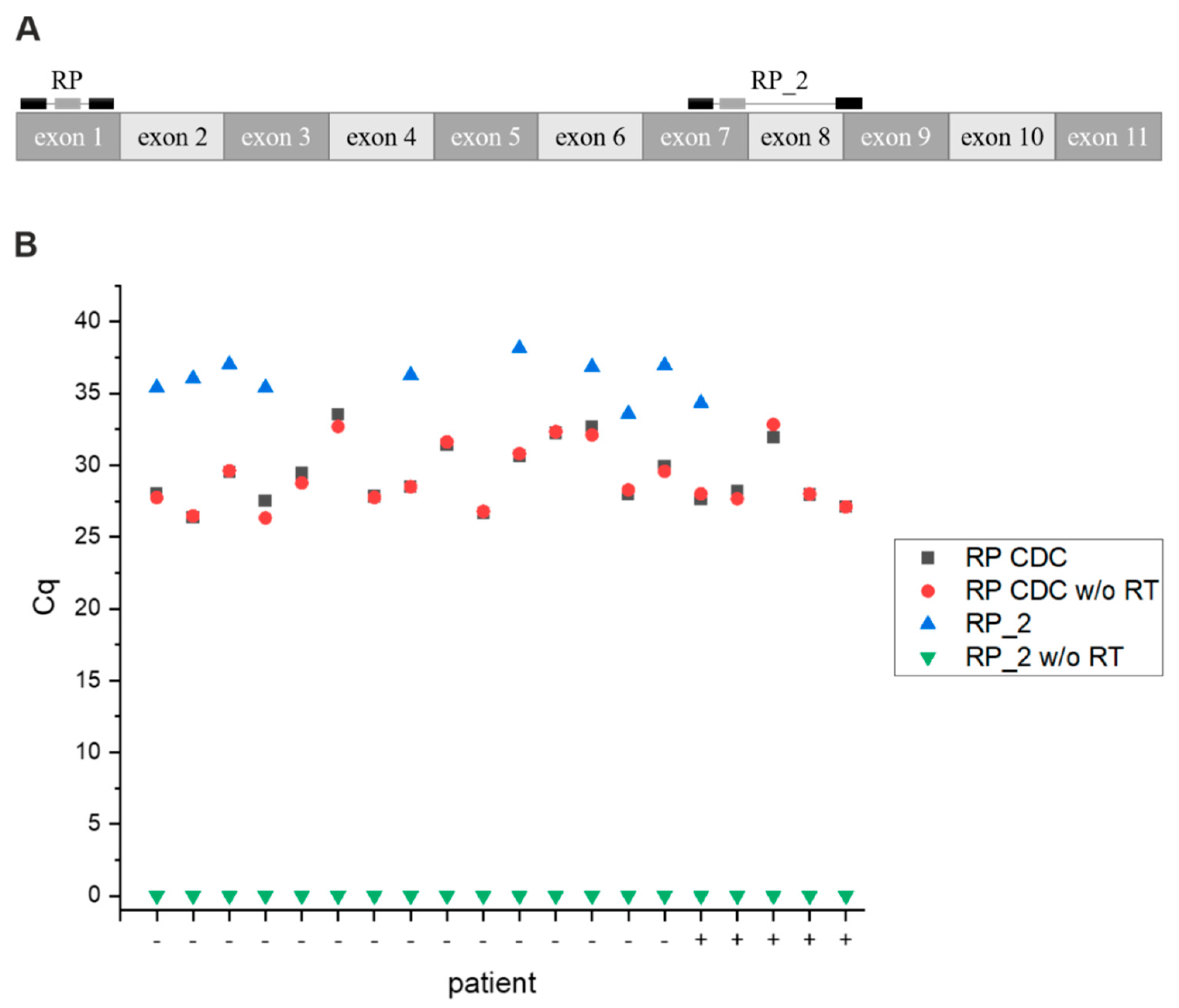
3.8. Primer Test
4. Discussion
4.1. Sample Handling
4.2. Reproducibility at Low Viral Loads
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.W.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef] [PubMed]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Crozier, A.; Rajan, S.; Buchan, I.; McKee, M. Put to the test: Use of rapid testing technologies for covid-19. BMJ 2021, 372, n208. [Google Scholar] [CrossRef] [PubMed]
- Guglielmi, G. Rapid coronavirus tests: A guide for the perplexed. Nature 2021, 590, 202–205. [Google Scholar] [CrossRef] [PubMed]
- Kashir, J.; Yaqinuddin, A. Loop mediated isothermal amplification (LAMP) assays as a rapid diagnostic for COVID-19. Med. Hypotheses 2020, 141, 109786. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Wu, X.; Wan, Z.; Li, Y.; Jin, X.; Zhang, C. A Novel Reverse Transcription Loop-Mediated Isothermal Amplification Method for Rapid Detection of SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.B.T.; Brünink, S.; Drosten, C. Diagnostic Detection of 2019-nCoV by Real-Time RT-PCR. Protocol and Preliminary Evaluation as of Jan 17, 2020; World Health Organization, 2020; Available online: https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf?sfvrsn=a9ef618c_2 (accessed on 5 July 2021).
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Buffer | Components | Source |
|---|---|---|
| COBAS | ≤40% (w/w) Guanidine hydrochloride | Roche |
| Tris-HCl buffer | ||
| GBO-1 | Guanidinium-based | Greiner Bio-One, Kremsmünster, AUT |
| GBO-2 | Guanidinium-based | |
| GBO-4 | PBS-based | |
| buffer 2 | 5.4 M GITC | Protocol by Vienna BioCenter |
| 56.8 mM Tris-HCl, pH 6.4 | ||
| 33.1 mM EDTA, pH 7.99 | ||
| 1.9% Triton X-100 | ||
| 54 mM DTT | ||
| buffer 4 | 0.15 M NaCl | Protocol by Jena (https://www.schubert-group.uni-jena.de/iomc2media/news-seite/corona+2020/preparation+of+buffers+for+viral+rna+extraction+for+detection+of+a+sars-cov-2-infection_schubert_jena+(1).pdf, accessed on 5 July 2021) |
| 0.01 M Tris-HCl, pH 7.4 | ||
| 0.25% Triton X-100 | ||
| buffer 6 | 4.5 M guanidine-hydrochloride NaCl | |
| 50 mM Tris-HCl, pH7.4 | ||
| NaCl | physiological saline solution (0.9% NaCl) | |
| PBS | phosphate-buffered saline solution | |
| VTM (viral transport medium) | 2% FBS | Falko Schüllner-Apotheke des A.ö. Landeskrankenhauses—Univ.-Kliniken Innsbruck based on the following protocol (https://www.cdc.gov/coronavirus/2019-ncov/downloads/Viral-Transport-Medium.pdf, accessed on 5 July 2021) |
| 100 µg/mL gentamicin | ||
| 0.5 µg/mL amphotericin B | ||
| in 1X HBSS with Ca2+ and Mg2+, no phenol red | ||
| Copan | Amies medium |
| Name | Target | Sequence | Purpose |
|---|---|---|---|
| 2019-nCoV_N1-F | N-gene | GACCCCAAAATCAGCGAAAT | N1 forward |
| 2019-nCoV_N1-R | N-gene | TCTGGTTACTGCCAGTTGAATCTG | N1 reverse |
| 2019-nCoV_N1-P | N-gene | FAM-ACCCCGCATTACGTTTGGTGGACC-BHQ1 | N1 probe |
| 2019-nCoV_N2-F | N-gene | TTACAAACATTGGCCGCAAA | N2 forward |
| 2019-nCoV_N2-R | N-gene | GCGCGACATTCCGAAGAA | N2 reverse |
| 2019-nCoV_N2-P | N-gene | FAM-ACAATTTGCCCCCAGCGCTTCAG-BHQ1 | N2 probe |
| RP-F | RNase P | AGATTTGGACCTGCGAGCG | RP forward |
| RP-R | RNase P | GAGCGGCTGTCTCCACAAGT | RP reverse |
| RP-P | RNase P | FAM–TTCTGACCTGAAGGCTCTGCGCG–BHQ1 | RP probe |
| RP_2-F | RNase P | GCCCTGCTATCAAAGACTCC | RP_2 forward |
| RP_2-R | RNase P | TGGCCCTCTTATTTCTAAAGGC | RP_2 reverse |
| RP_2-P | RNase P | Cy5-TCCAGTGCCCTCAATTTGATGCAA-3BHQ1 | RP_2 probe |
| E-Sarbeco-F | E-gene | ACAGGTACGTTAATAGTTAATAGCGT | E-Sarbeco forward |
| E-Sarbeco-R | E-gene | ATATTGCAGCAGTACGCACACA | E-Sarbeco reverse |
| E-Sarbeco-P | E-gene | HEX-ACACTAGCCATCCTTACTGCGCTTCG-3BHQ1 | E-Sarbeco probe |
| Virus-Specific 1 | Virus-Specific 2 | RP/RP_2 | Result |
|---|---|---|---|
| + | + | + | positive |
| + | + | - | positive |
| + | - | + | inconclusive |
| - | + | + | inconclusive |
| + | - | - | inconclusive |
| - | + | - | inconclusive |
| - | - | + | negative/not detected |
| - | - | - | invalid |
| Luna Kit | TaqMan Kit | ||||||
|---|---|---|---|---|---|---|---|
| Cq (COBAS) | Cq (N1) | Cq (N2) | Cq (COBAS) | Cq (N1) | Cq (N2) | ||
| Cq | 25.3 | 23.8 | 24.7 | 26.9 | 28.8 | 29.2 | Cq ≤ 30 |
| σ | 3.3 | 5.4 | 5.6 | 3.1 | 3.7 | 4.2 | |
| n | 35 | 35 | 35 | 9 | 9 | 9 | |
| Cq | 34.8 | 34.2 | 35.3 | 33.5 | 33.4 | 34.0 | Cq > 30 |
| σ | 2.5 | 2.8 | 3.2 | 2.1 | 2.7 | 3.0 | |
| n | 22 | 22 | 22 | 10 | 10 | 10 | |
| High Viral Load Cq ≤ 30 | Low Viral Load Cq > 30 | Negative | ||||
|---|---|---|---|---|---|---|
| COBAS | JKU | COBAS | JKU | COBAS | JKU | |
| positive | 53 | 53 | 64 | 36 | 0 | 0 |
| inconclusive | 0 | 0 | 0 | 13 | 0 | 0 |
| negative | 0 | 0 | 7 | 22 | 95 | 95 |
| Total | 53 | 71 | 95 | |||
| Replicate 1 | Replicate 2 | |||||
|---|---|---|---|---|---|---|
| Cq (COBAS) | Cq (N1) | Cq (N2) | Cq (N1) | Cq (N2) | ||
| Cq | 26.9 | 28.7 | 29.4 | 29.1 | 29.4 | Cq ≤ 30 |
| σ | 3.4 | 4.7 | 5.1 | 5.4 | 4.9 | |
| n | 18 | 18 | 18 | 18 | 18 | |
| Cq | 35.0 | 34.8 | 36.1 | 35.1 | 35.9 | Cq > 30 |
| σ | 2.4 | 2.9 | 3.7 | 2.9 | 3.2 | |
| n | 49 | 23 | 26 | 25 | 24 | |
| High Viral Load Cq ≤ 30 | Low Viral Load Cq > 30 | Negatives | |||||||
|---|---|---|---|---|---|---|---|---|---|
| COBAS | JKU | COBAS | JKU | COBAS | JKU | ||||
| Day 1 | Day 1 | Day 3 | Day 1 | Day 1 | Day 3 | Day 1 | Day 1 | Day 3 | |
| positive | 12 | 12 | 12 | 8 | 3 | 3 | 0 | 0 | 1 |
| inconclusive | 0 | 0 | 0 | 0 | 2 | 1 | 0 | 0 | 0 |
| negative | 0 | 0 | 0 | 0 | 3 | 4 | 15 | 15 | 14 |
| Cq | 24.0 | 25.8 | 26.9 | 33.2 | 32.7 | 33.8 | n.a. | n.a. | 37.1 |
| σ | 4.1 | 5.1 | 5.0 | 2.5 | 1.1 | 1.0 | n.a. | n.a. | n.a. |
| CI | 2.3 | 2.9 | 2.8 | 1.7 | 1.2 | 1.2 | n.a. | n.a. | n.a. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mair, T.; Ivankovic, M.; Paar, C.; Salzer, H.J.F.; Heissl, A.; Lamprecht, B.; Schreier-Lechner, E.; Tiemann-Boege, I. Processing Hundreds of SARS-CoV-2 Samples with an In-House PCR-Based Method without Robotics. Viruses 2021, 13, 1712. https://doi.org/10.3390/v13091712
Mair T, Ivankovic M, Paar C, Salzer HJF, Heissl A, Lamprecht B, Schreier-Lechner E, Tiemann-Boege I. Processing Hundreds of SARS-CoV-2 Samples with an In-House PCR-Based Method without Robotics. Viruses. 2021; 13(9):1712. https://doi.org/10.3390/v13091712
Chicago/Turabian StyleMair, Theresa, Maja Ivankovic, Christian Paar, Helmut J. F. Salzer, Angelika Heissl, Bernd Lamprecht, Elisabeth Schreier-Lechner, and Irene Tiemann-Boege. 2021. "Processing Hundreds of SARS-CoV-2 Samples with an In-House PCR-Based Method without Robotics" Viruses 13, no. 9: 1712. https://doi.org/10.3390/v13091712