
Specificity of the HIV-1 Protease on Substrates Representing the Cleavage Site in the Proximal Zinc-Finger of HIV-1 Nucleocapsid Protein



Abstract
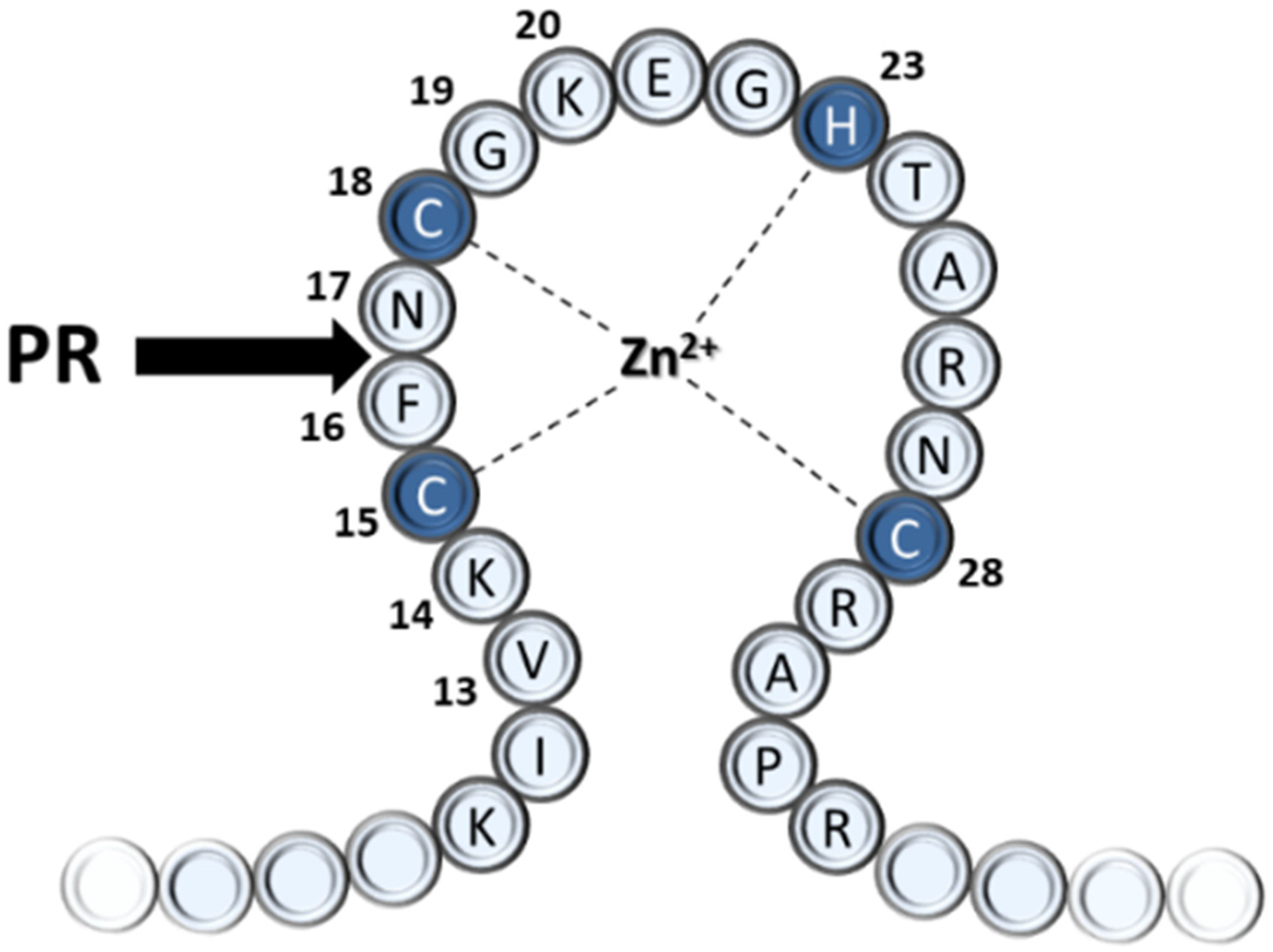
:1. Introduction
2. Materials and Methods
2.1. Oligopeptide Synthesis and Characterization
2.2. Enzyme Assay with Synthetic Peptide Substrates
2.3. Molecular Modeling
2.4. Design of Recombinant Fluorescent Protein Substrates
2.5. Expression, Purification and Cleavage of Protein Substrates
3. Results
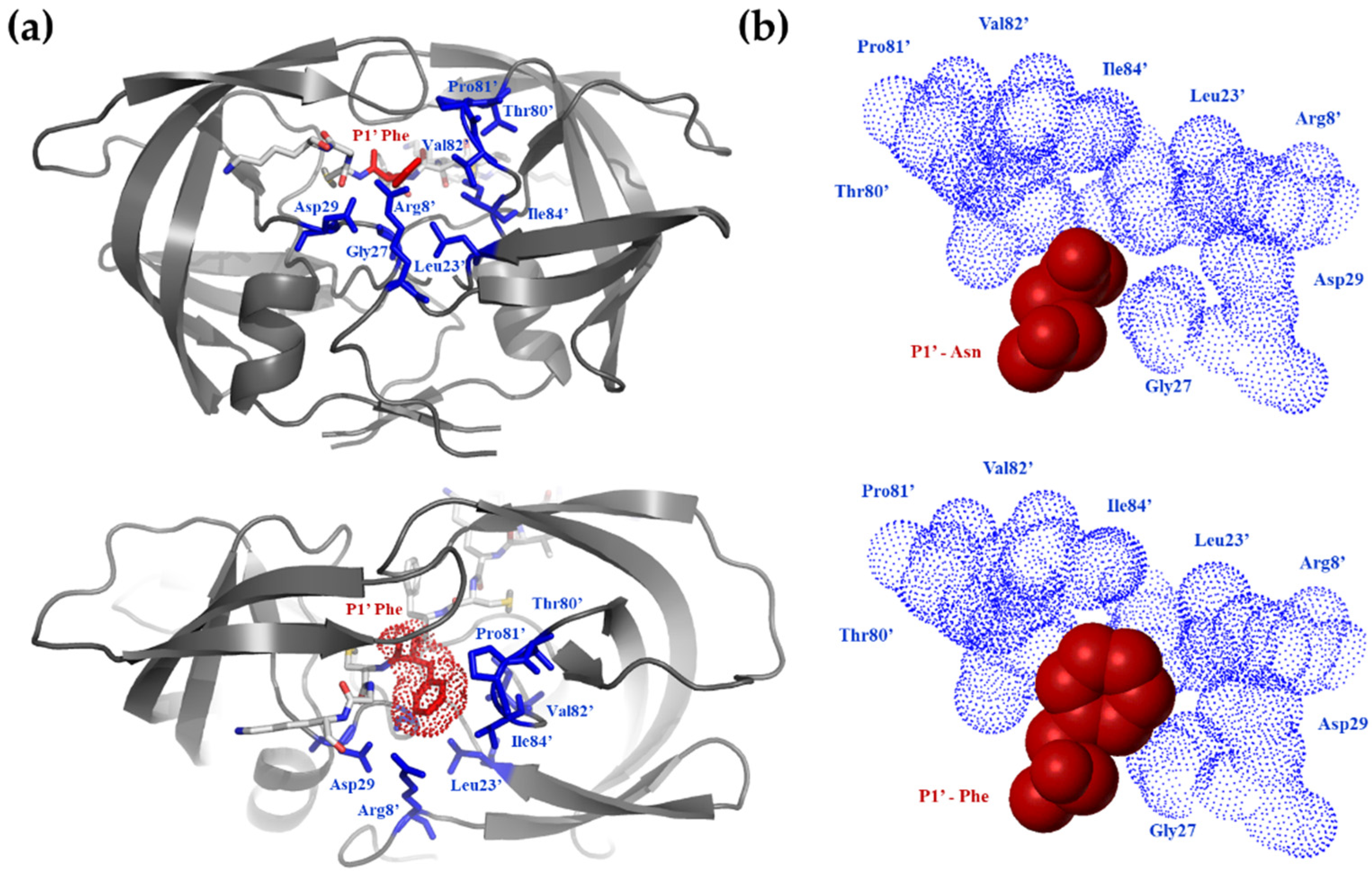
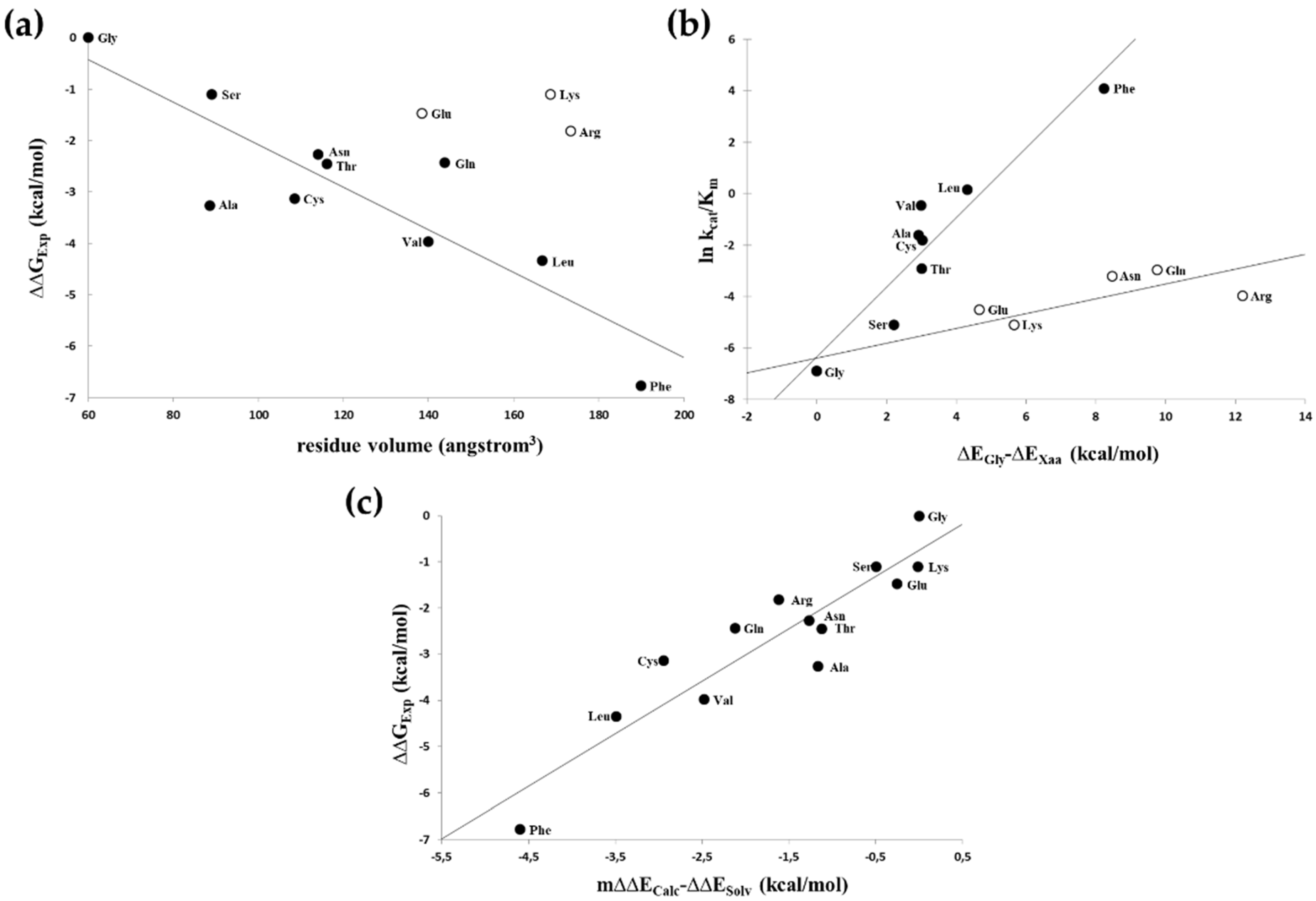
3.1. Probing the S1′ Binding Site of HIV-1 Protease Using Substituted Peptides Representing the Proximal Cleavage Site of HIV-1 NC
3.2. Design of Nucleocapsid Cleavage Site “Revertants”
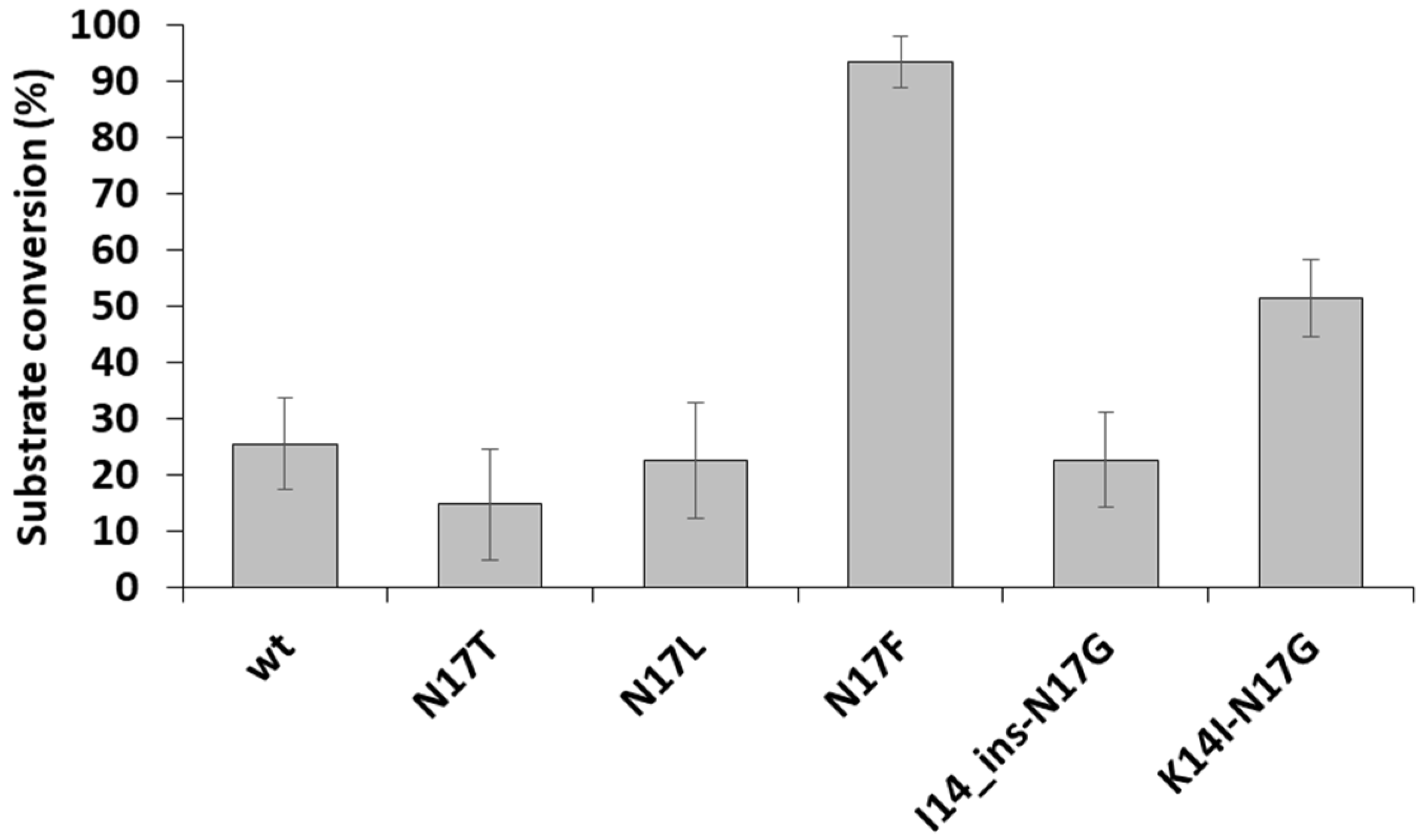
3.3. Cleavage Reactions Using Recombinant Protein Substrates
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tözsér, J.; Oroszlan, S. Proteolytic events of HIV-1 replication as targets for therapeutic intervention. Curr. Pharm. Des. 2003, 9, 1803–1815. [Google Scholar] [CrossRef]
- Tozser, J. Stages of HIV Replication and Targets for Therapeutic Intervention. Curr. Top. Med. Chem. 2003, 3, 1447–1457. [Google Scholar] [CrossRef] [PubMed]
- Rein, A.; Henderson, L.E.; Levin, J.G. Nucleic-acidchaperone activity of retroviral nucleocapsid proteins: Significance for viral replication. Trends Biochem. Sci. 1998, 23, 297–301. [Google Scholar] [CrossRef]
- Roberts, M.M.; Oroszlan, S. The preparation and biochemical characterization of intact capsids of equine infectious anemia virus. Biochem. Biophys. Res. Commun. 1989, 160, 486–494. [Google Scholar] [CrossRef]
- Roberts, M.M.; Copeland, T.D.; Oroszlan, S. In situ processing of a retroviral nucleocapsid protein by the viral proteinase. Protein Eng. 1991, 4, 695–700. [Google Scholar] [CrossRef]
- Welker, R.; Hohenberg, H.; Tessmer, U.; Huckhagel, C.; Krausslich, H.G. Biochemical and structural analysis of isolated mature cores of human immunodeficiency virus type 1. J. Virol. 2000, 74, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J.; Friedman, D.; Weber, I.T.; Blaha, I.; Oroszlan, S. Studies on the substrate specificity of the proteinase of equine infectious anemia virus using oligopeptide substrates. Biochemistry 1993, 32, 3347–3353. [Google Scholar] [CrossRef] [PubMed]
- Wondrak, E.M.; Sakaguchi, K.; Rice, W.G.; Kun, E.; Kimmel, A.R.; Louis, J.M. Removal of zinc is required for processing of the mature nucleocapsid protein of human immunodeficiency virus, type 1, by the viral protease. J. Biol. Chem. 1994, 269, 21948–21950. [Google Scholar] [CrossRef]
- Tözsér, J.; Shulenin, S.; Louis, J.M.; Copeland, T.D.; Oroszlan, S. In Vitro Processing of HIV-1 Nucleocapsid Protein by the Viral Proteinase: Effects of Amino Acid Substitutions at the Scissile Bond in the Proximal Zinc Finger Sequence. Biochemistry 2004, 43, 4304–4312. [Google Scholar] [CrossRef] [PubMed]
- Risco, C.; Menéndez-Arias, L.; Copeland, T.D.; Pinto da Silva, P.; Oroszlan, S. Intracellular transport of the murine leukemia virus during acute infection of NIH 3T3 cells: Nuclear import of nucleocapsid protein and integrase. J. Cell Sci. 1995, 108, 3039–3050. [Google Scholar] [CrossRef]
- Baboonian, C.; Dalgleish, A.; Bountiff, L.; Gross, J.; Oroszlan, S.; Rickett, G.; Smith, B.C.; Troke, P.; Merson, J. HIV-1 proteinase is required for synthesis of proviral DNA. Biochem. Biophys. Res. Commun. 1991, 179, 17–24. [Google Scholar] [CrossRef]
- Venaud, S.; Yahi, N.; Fehrentz, J.; Guettari, N.; Nisato, D.; Hirsch, I.; Chermann, J. Inhibition of HIV by an anti-HIV protease synthetic peptide blocks an early step of viral replication. Res. Virol. 1992, 143, 311–319. [Google Scholar] [CrossRef]
- Nagy, K.; Young, M.; Baboonian, C.; Merson, J.; Whittle, P.; Oroszlan, S. Antiviral activity of human immunodeficiency virus type 1 protease inhibitors in a single cycle of infection: Evidence for a role of protease in the early phase. J. Virol. 1994, 68, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Stefanidou, M.; Herrera, C.; Armanasco, N.; Shattock, R.J. Saquinavir inhibits early events associated with establishment of HIV-1 infection: Potential role for protease inhibitors in prevention. Antimicrob. Agents Chemother. 2012, 56, 4381–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharmeen, L.; McQuade, T.; Heldsinger, A.; Gogliotti, R.; Domagala, J.; Gracheck, S. Inhibition of the early phase of HIV replication by an isothiazolone, PD 161374. Antivir. Res. 2001, 49, 101–114. [Google Scholar] [CrossRef]
- Jacobsen, H.; Ahlborn, L.L.; Gugel, R.; Mous, J. Progression of early steps of human immunodeficiency virus type 1 replication in the presence of an inhibitor of viral protease. J. Virol. 1992, 66, 5087–5091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, A.H.; Manchester, M.; Smith, T.; Yang, Y.L.; Swanstrom, R. Conditional human immunodeficiency virus type 1 protease mutants show no role for the viral protease early in virus replication. J. Virol. 1996, 70, 5840–5844. [Google Scholar] [CrossRef] [Green Version]
- Uchida, H.; Maeda, Y.; Mitsuya, H. HIV-1 protease does not play a critical role in the early stages of HIV-1 infection. Antivir. Res. 1997, 36, 107–113. [Google Scholar] [CrossRef]
- Foley, B.; Korber, B.T.M.; Leitner, T.K.; Apetrei, C.; Hahn, B.; Mizrachi, I.; Mullins, J.; Rambaut, A.; Wolinsky, S. HIV Sequence Compendium 2018; U.S. Department of Energy—Office of Scientific and Technical Information: Oak Ridge, TN, USA, 2018. [CrossRef] [Green Version]
- Mark-Danieli, M.; Laham, N.; Kenan-Eichler, M.; Castiel, A.; Melamed, D.; Landau, M.; Bouvier, N.M.; Evans, M.J.; Bacharach, E. Single point mutations in the zinc finger motifs of the human immunodeficiency virus type 1 nucleocapsid alter RNA binding specificities of the gag protein and enhance packaging and infectivity. J. Virol. 2005, 79, 7756–7767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kafaie, J.; Song, R.; Abrahamyan, L.; Mouland, A.J.; Laughrea, M. Mapping of nucleocapsid residues important for HIV-1 genomic RNA dimerization and packaging. Virology 2008, 375, 592–610. [Google Scholar] [CrossRef]
- Narayanan, N.; Gorelick, R.J.; DeStefano, J.J. Structure/function mapping of amino acids in the N-terminal zinc finger of the human immunodeficiency virus type 1 nucleocapsid protein: Residues responsible for nucleic acid helix destabilizing activity. Biochemistry 2006, 45, 12617–12628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, J.A.; Shulenin, S.; Coren, L.V.; Bosche, W.J.; Gagliardi, T.D.; Gorelick, R.J.; Oroszlan, S. Characterization of human immunodeficiency virus type 1 (HIV-1) containing mutations in the nucleocapsid protein at a putative HIV-1 protease cleavage site. Virology 2006, 354, 261–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorfman, T.; Luban, J.; Goff, S.P.; Haseltine, W.A.; Göttlinger, H.G. Mapping of functionally important residues of a cysteine-histidine box in the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol. 1993, 67, 6159–6169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tözsér, J.; Shulenin, S.; Young, M.R.; Briggs, C.J.; Oroszlan, S. Replication-dependent fitness recovery of HIV-1 harboring mutations of Asn17 of the nucleocapsid protein. J. Gen. Virol. 2006, 87, 961–965. [Google Scholar] [CrossRef] [Green Version]
- Grobelny, D.; Wondrak, E.M.; Galardy, R.E.; Oroszlan, S. Selective phosphinate transition-state analogue inhibitors of the protease of human immunodeficiency virus. Biochem. Biophys. Res. Commun. 1990, 169, 1111–1116. [Google Scholar] [CrossRef]
- Mahalingam, B.; Boross, P.; Wang, Y.F.; Louis, J.M.; Fischer, C.C.; Tozser, J.; Harrison, R.W.; Weber, I.T. Combining mutations in HIV-1 protease to understand mechanisms of resistance. Proteins 2002, 48, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S.J.; Kollman, P.A.; Nguyen, D.T.; Case, D.A. An all atom force-field for simulations of proteins and nucleic acids. J. Comp. Chem. 1986, 7, 230–252. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding; W.H. Freeman and Company: New York, NY, USA, 2002; pp. 332–336. [Google Scholar]
- Fauchere, J.L.; Plisca, V. Hydrophobic parameters of amino acid side chains from the partitioning of N-acetyl-amino acid amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Zamyatin, A.A. Protein volume in solution. Prog. Biophys. Mol. Biol. 1972, 24, 107–123. [Google Scholar] [CrossRef]
- Bozóki, B.; Gazda, L.; Tóth, F.; Miczi, M.; Mótyán, J.A.; Tőzsér, J. A recombinant fusion protein-based, fluorescent protease assay for high throughput-compatible substrate screening. Anal. Biochem. 2018, 540–541, 52–63. [Google Scholar] [CrossRef]
- Gazda, L.D.; Joóné Matúz, K.; Nagy, T.; Mótyán, J.A.; Tőzsér, J. Biochemical characterization of Ty1 retrotransposon protease. PLoS ONE 2020, 15, e0227062. [Google Scholar] [CrossRef]
- Mótyán, J.A.; Miczi, M.; Bozóki, B.; Tőzsér, J. Data supporting Ni-NTA magnetic bead-based fluorescent protease assay using recombinant fusion protein substrates. Data Brief 2018, 18, 203–208. [Google Scholar] [CrossRef]
- Bozóki, B.; Mótyán, J.A.; Miczi, M.; Gazda, L.D.; Tőzsér, J. Use of Recombinant Fusion Proteins in a Fluorescent Protease Assay Platform and Their In-gel Renaturation. J. Vis. Exp. 2019, 143, e58824. [Google Scholar] [CrossRef] [Green Version]
- Billich, A.; Winkler, G. Analysis of subsite preferences of HIV-1 proteinase using MA/CA junction peptides substituted at the P3-P1’ positions. Arch. Biochem. Biophys. 1991, 290, 186–190. [Google Scholar] [CrossRef]
- Tözsér, J.; Zahuczky, G.; Bagossi, P.; Louis, J.M.; Copeland, T.D.; Oroszlan, S.; Harrison, R.W.; Weber, I.T. Comparison of the substrate specificity of the human T-cell leukemia virus and human immunodeficiency virus proteinases. Eur. J. Biochem. 2000, 267, 6287–6295. [Google Scholar] [CrossRef] [Green Version]
- Tözsér, J.; Bagossi, P.; Weber, I.T.; Louis, J.M.; Copeland, T.D.; Oroszlan, S. Studies on the Symmetry and Sequence Context Dependence of the HIV-1 Proteinase Specificity. J. Biol. Chem. 1997, 272, 16807–16814. [Google Scholar] [CrossRef] [Green Version]
- Bagossi, P.; Sperka, T.; Fehér, A.; Kádas, J.; Zahuczky, G.; Miklóssy, G.; Boross, P.; Tözsér, J. Amino Acid Preferences for a Critical Substrate Binding Subsite of Retroviral Proteases in Type 1 Cleavage Sites. J. Virol. 2005, 79, 4213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eizert, H.; Bander, P.; Bagossi, P.; Sperka, T.; Miklóssy, G.; Boross, P.; Weber, I.T.; Tözsér, J. Amino Acid Preferences of Retroviral Proteases for Amino-Terminal Positions in a Type 1 Cleavage Site. J. Virol. 2008, 82, 10111–10117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tözsér, J.; Weber, I.T.; Gustchina, A.; Bláha, I.; Copeland, T.D.; Louis, J.M.; Oroszlan, S. Kinetic and modeling studies of S3-S3’ subsites of HIV proteinases. Biochemistry 1992, 31, 4794–4800. [Google Scholar] [CrossRef]
- Tőzsér, J.; Gustchina, A.; Weber, I.T.; Bláha, I.; Wondrak, E.M.; Oroszlan, S. Studies on the role of the S4 substrate binding site of HIV proteinases. FEBS Lett. 1991, 279, 356–360. [Google Scholar] [CrossRef] [Green Version]
- Thomas, J.A.; Gorelick, R.J. Nucleocapsid protein function in early infection processes. Virus Res. 2008, 134, 39–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Cleavage Site Sequence/Oligonucleotide Primer Sequence |
|---|---|
| Wild-type | KIVKCFNCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGAAATGCTTTAACTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCAGTTAAAGCATTTCACAATTTTTTTAAT-3′ |
| N17F | KIVKCFFCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGAAATGCTTTTTTTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCAAAAAAAGCATTTCACAATTTTTTTAAT-3′ |
| N17L | KIVKCFLCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGAAATGCTTTCTGTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCACAGAAAGCATTTCACAATTTTTTTAAT-3′ |
| N17T | KIVKCFTCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGAAATGCTTTACCTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCAGGTAAAGCATTTCACAATTTTTTTAAT-3′ |
| K14I-N17G | KIVICFGCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGATTTGCTTTGGCTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCAGCCAAAGCAAATCACAATTTTTTTAAT-3′ |
| I14_ins-N17G | KIVKICFGCGKEGHTARNCRAPR |
| FWD | 5′-TAAAAAAATTGTGAAAATTTGCTTTGGCTGCGGCAAAGAAGGCCATACCGCGCGTAACTGCCGTGCGCCGCGTG-3′ |
| REV | 5′-GATCCACGCGGCGCACGGCAGTTACGCGCGGTATGGCCTTCTTTGCCGCAGCCAAAGCAAATTTTCACAATTTTTTTAAT-3′ |
| sequencing primer | 5′-GATGAAGCCCTGAAAGACGCGCAG-3′ |
| Name | Sequence | Cleavage Site | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) | kcat/Km Fold-Change | |
|---|---|---|---|---|---|---|---|
| 1. | wild-type | KIVKCFNCGK | CF↓NC | 0.43 1 | 0.017 1 | 0.040 1 | - |
| 2. | N17F | KIVKCFFCGK | CF↓FC | 0.02 1 | 0.900 1 | 59.6 1 | 1490.00 |
| 3. | N17L | KIVKCFLCGK | CF↓LC | 0.17 1 | 0.196 1 | 1.153 1 | 28.83 |
| 4. | N17V | KIVKCFVCGK | CF↓VC | 0.45 | 0.284 | 0.632 | 15.80 |
| 5. | N17A | KIVKCFACGK 2 | CF↓AC | 0.32 1 | 0.064 1 | 0.200 1 | 5.00 |
| 6. | N17C | KIVKCFCCGK 2 | CF↓CC | 1.52 | 0.247 | 0.162 1 | 4.05 |
| 7. | N17T | KIVKCFTCGK | CF↓TC | 0.31 | 0.017 | 0.054 | 1.35 |
| 8. | N17Q | KIVKCFQCGK | CF↓QC | 0.67 | 0.035 | 0.052 | 1.30 |
| 9. | N17S | KIVKCFSCGK 2 | CF↓SC | 0.63 | 0.004 | 0.006 1 | 0.15 |
| 10. | N17R | KIVKCFRCGK 2 | CF↓RC | 1.05 | 0.020 | 0.019 1 | 0.48 |
| 11. | N17K | KIVKCFKCGK | CF↓KC | 0.86 1 | 0.005 1 | 0.006 1 | 0.15 |
| 12. | N17E | KIVKCFECGK | CF↓EC | 1.21 | 0.013 | 0.011 | 0.28 |
| 13. | N17D | KIVKCFDCGK | not hydrolysed | - | |||
| 14. | N17G | KIVKCFGCGK | CF↓GC | 1.34 1 | 0.002 1 | 0.001 1 | 0.03 |
| Name | Sequence | Cleavage Site | Km (mM) | kcat (s−1) | kcat/Km 2 (mM−1 s−1) | kcat/Km 2 Fold-Change | kcat/Km 3 (mM−1 s−1) | |
|---|---|---|---|---|---|---|---|---|
| 1. | wild-type | KIVKCFNCGK 1 | CF↓NC | 0.43 | 0.017 | 0.040 | - | - |
| 14. | N17G | KIVKCFGCGK 1 | CF↓GC | 1.34 | 0.002 | 0.001 | 0.03 | - |
| 15. | K14T-N17G | KIVTCFGCGK | n.d. 4 | - | <0.010 | - | - | - |
| 16. | K14I-N17G | RKIVICFGCGKR | IC↓FG | 0.035 | 0.034 | 0.97 5 | 24.25 | 0.51 |
| 17. | I14_ins-N17G | KIVKICFGCGKR | IC↓FG | 0.070 | 0.025 | 0.35 | 8.75 | 0.48 |
| 18. | N17G-G19I | RKIVKCFGCIKR | n.d. 4 | - | <0.010 | - | - | - |
| 19. | K14I-N17G-G19I | RKIVICFGCIKR | IC↓FG | <0.010 | 0.015 | n.d. 4 | - | 0.48 |
| 20. | K14L-N17G | RKIVLCFGCGKR | VL↓CF/LC↓FG | - | n.d. 4 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mótyán, J.A.; Miczi, M.; Oroszlan, S.; Tőzsér, J. Specificity of the HIV-1 Protease on Substrates Representing the Cleavage Site in the Proximal Zinc-Finger of HIV-1 Nucleocapsid Protein. Viruses 2021, 13, 1092. https://doi.org/10.3390/v13061092
Mótyán JA, Miczi M, Oroszlan S, Tőzsér J. Specificity of the HIV-1 Protease on Substrates Representing the Cleavage Site in the Proximal Zinc-Finger of HIV-1 Nucleocapsid Protein. Viruses. 2021; 13(6):1092. https://doi.org/10.3390/v13061092
Chicago/Turabian StyleMótyán, János András, Márió Miczi, Stephen Oroszlan, and József Tőzsér. 2021. "Specificity of the HIV-1 Protease on Substrates Representing the Cleavage Site in the Proximal Zinc-Finger of HIV-1 Nucleocapsid Protein" Viruses 13, no. 6: 1092. https://doi.org/10.3390/v13061092