
Hepatitis D Virus Entry Inhibitors Based on Repurposing Intestinal Bile Acid Reabsorption Inhibitors

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. NTCP-Expressing Cell Lines
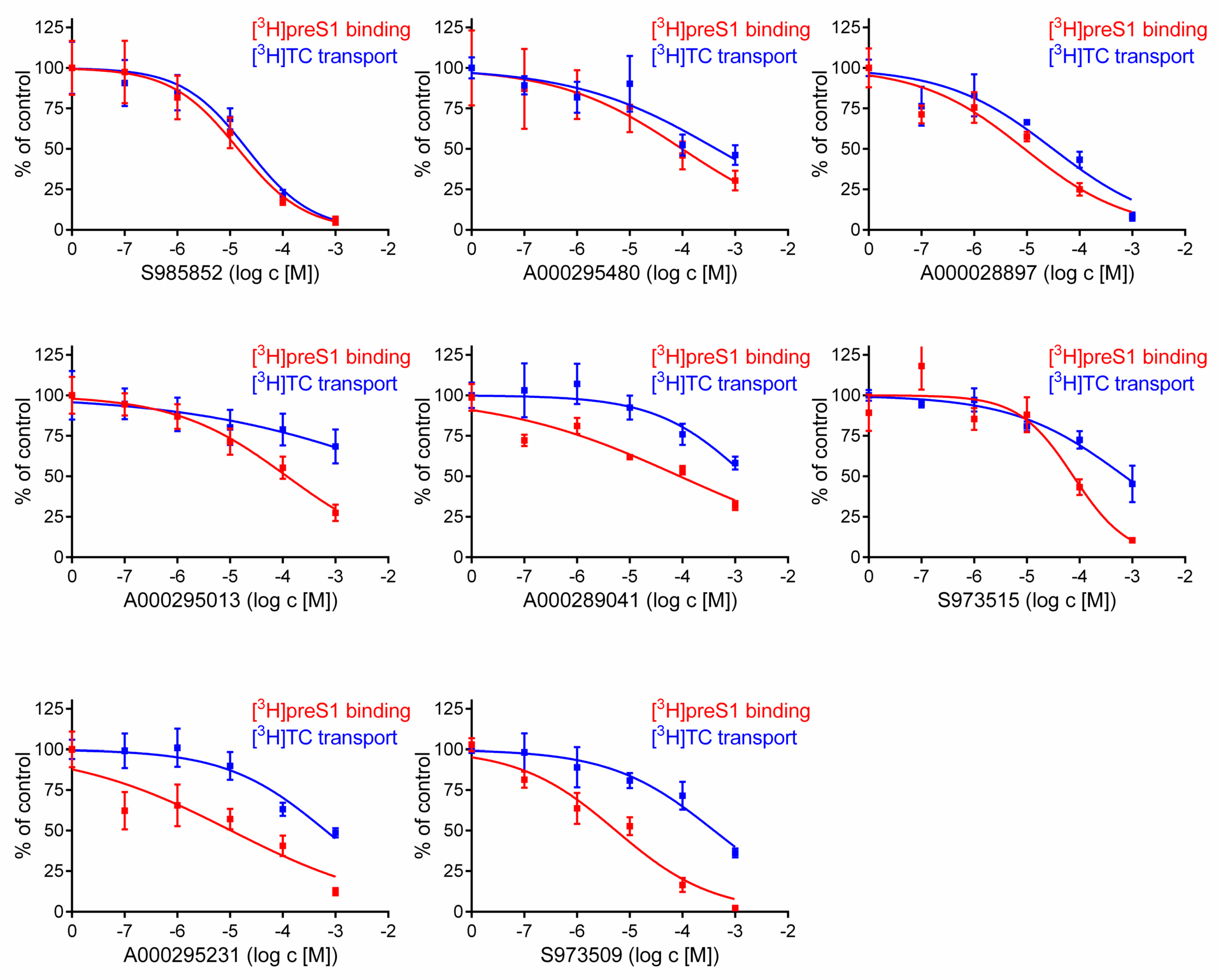
2.2. Inhibitory Concentrations (IC50) for [3H]preS1 Binding and [3H]Taurocholic Acid Transport
2.3. HDV Infection Experiments
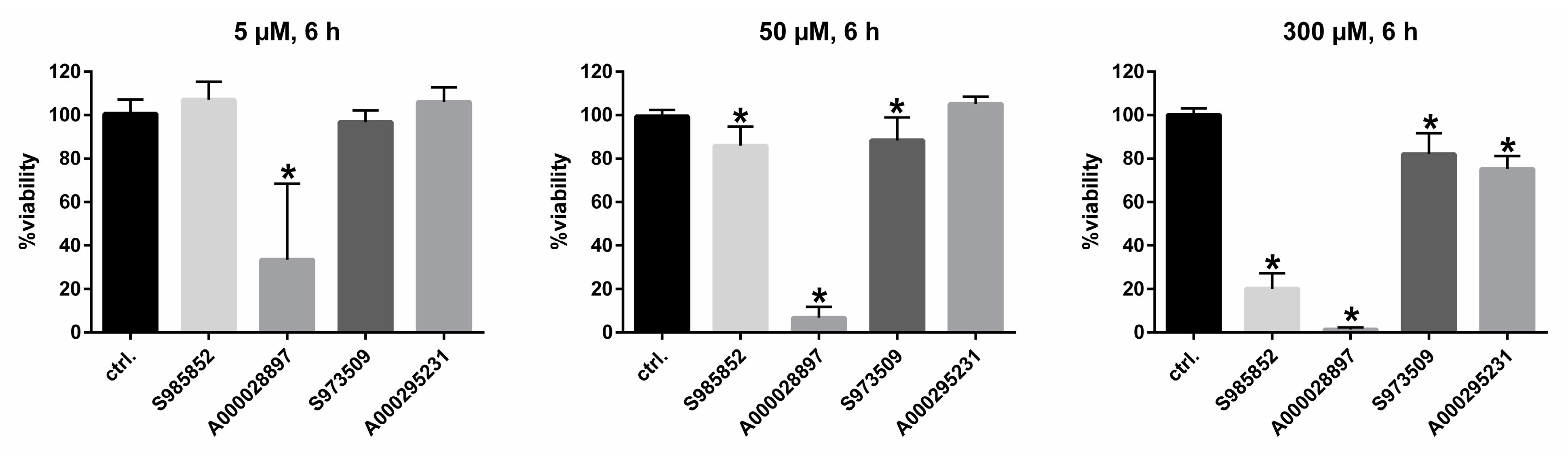
2.4. Cytotoxicity Assay
2.5. Structure Modeling
2.6. Statistics
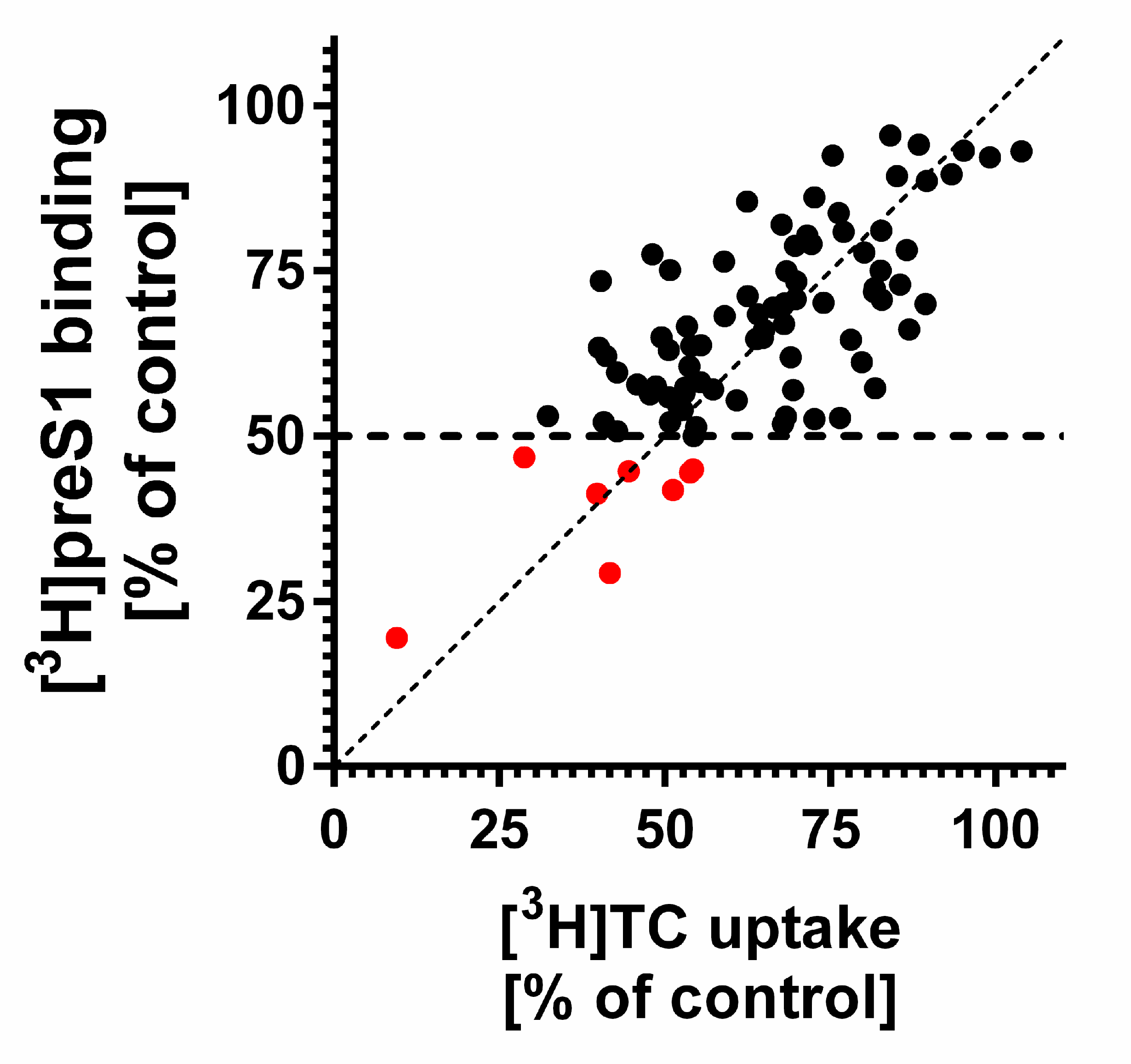
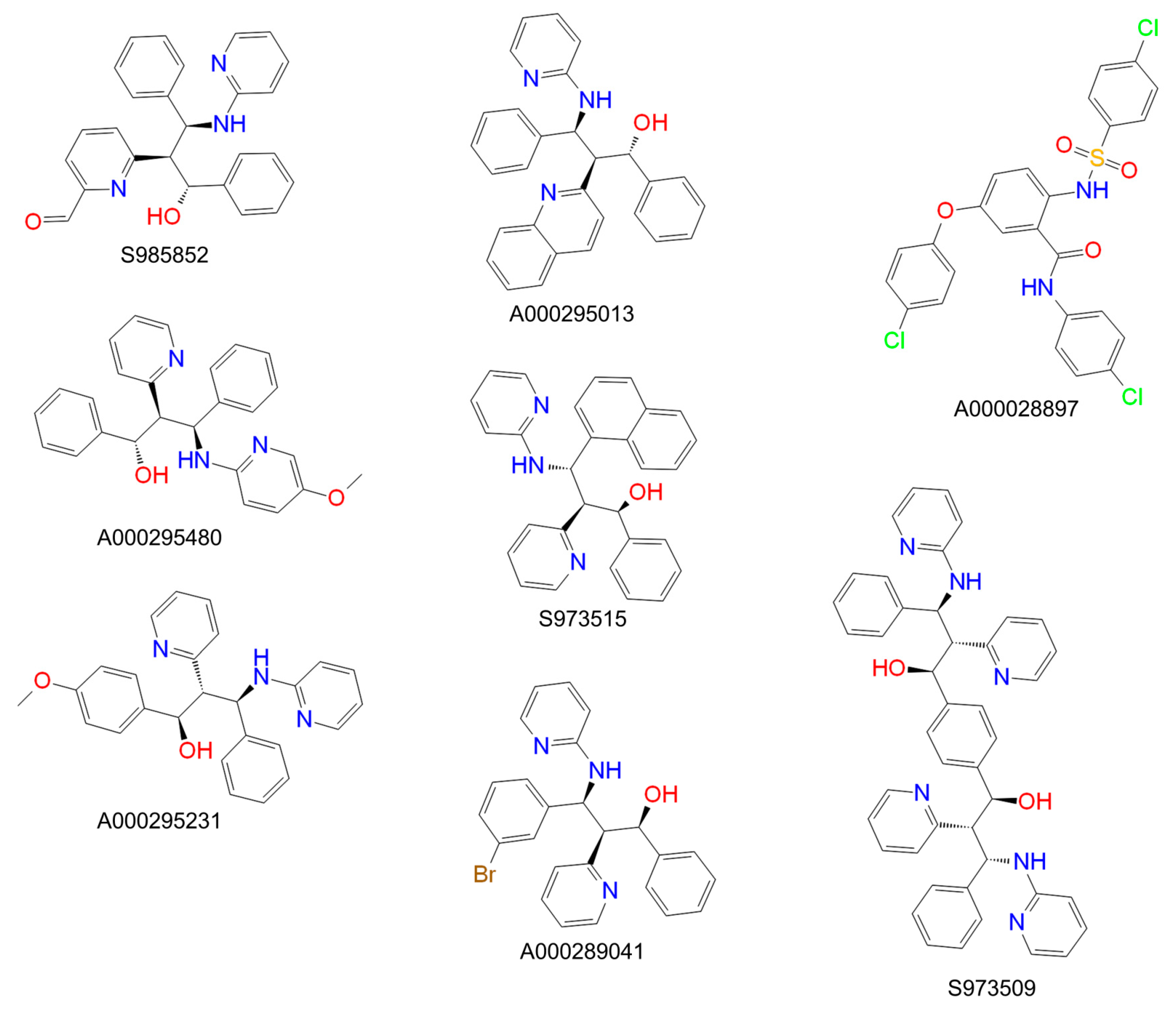
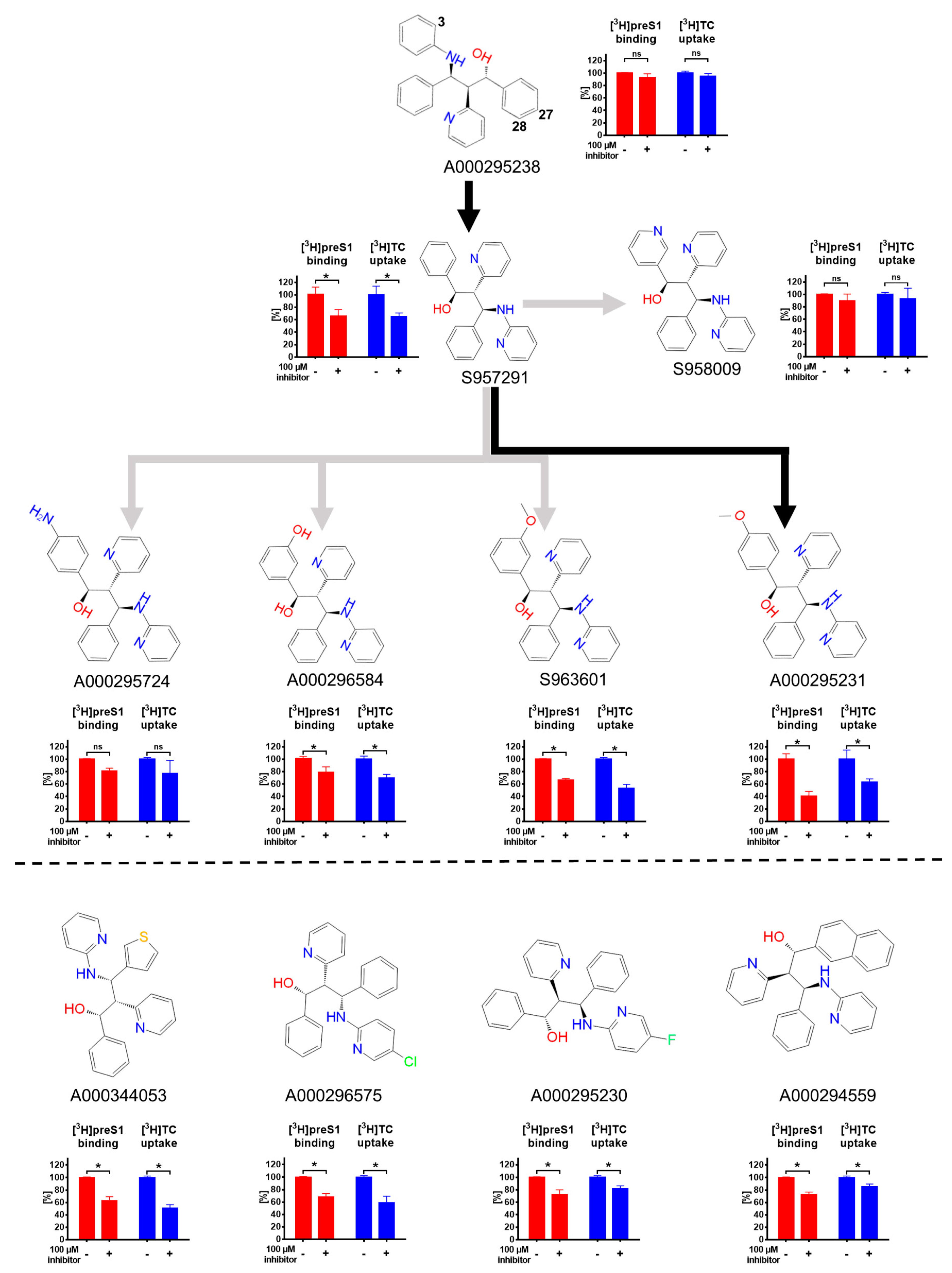
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Global Hepatitis Report 2017; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver. Dis. 2013, 33, 103–112. [Google Scholar] [CrossRef]
- Glebe, D.; Urban, S.; Knoop, E.V.; Cag, N.; Krass, P.; Grün, S.; Bulavaite, A.; Sasnauskas, K.; Gerlich, W.H. Mapping of the hepatitis B virus attachment site by use of infection-inhibiting preS1 lipopeptides and tupaia hepatocytes. Gastroenterology 2005, 129, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, S.A.; Wedemeyer, H.; Harrison, P.M. Hepatitis delta virus. Lancet 2011, 378, 73–85. [Google Scholar] [CrossRef]
- Sureau, C.; Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.G.; Villeret, F.; Testoni, B.; Zoulim, F. Can we cure hepatitis B virus with novel direct-acting antivirals? Liver Int. 2020, 40 (Suppl. 1), 27–34. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Guidelines for the Prevention, Care and Treatment of Persons with Chronic Hepatitis B Infection; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1. [Google Scholar] [CrossRef] [PubMed]
- König, A.; Döring, B.; Mohr, C.; Geipel, A.; Geyer, J.; Glebe, D. Kinetics of the bile acid transporter and hepatitis B virus receptor Na+/taurocholate cotransporting polypeptide (NTCP) in hepatocytes. J. Hepatol. 2014, 61, 867–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamas Longarela, O.; Schmidt, T.T.; Schöneweis, K.; Romeo, R.; Wedemeyer, H.; Urban, S.; Schulze, A. Proteoglycans act as cellular hepatitis delta virus attachment receptors. PLoS ONE 2013, 8, e58340. [Google Scholar] [CrossRef] [Green Version]
- Fukano, K.; Tsukuda, S.; Watashi, K.; Wakita, T. Concept of Viral Inhibitors via NTCP. Semin. Liver Dis. 2019, 39, 78–85. [Google Scholar] [CrossRef]
- MYR Pharmaceuticals. 2020. Available online: http://myr-pharma.com/ (accessed on 24 February 2021).
- Geyer, J.; Wilke, T.; Petzinger, E. The solute carrier family SLC10: More than a family of bile acid transporters regarding function and phylogenetic relationships. Arch. Pharmacol. 2006, 372, 413–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirstgen, M.; Lowjaga, K.A.A.T.; Müller, S.F.; Goldmann, N.; Lehmann, F.; Alakurtti, S.; Yli-Kauhaluoma, J.; Glebe, D.; Geyer, J. Selective hepatitis B and D virus entry inhibitors from the group of pentacyclic lupane-type betulin-derived triterpenoids. Sci. Rep. 2020, 10, 21772. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Pan, G. An important intestinal transporter that regulates the enterohepatic circulation of bile acids and cholesterol homeostasis: The apical sodium-dependent bile acid transporter (SLC10A2/ASBT). Clin. Res. Hepatol. Gastroenterol. 2017, 41, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Kramer, W.; Glombik, H. Bile acid reabsorption inhibitors (BARI): Novel hypolipidemic drugs. Curr. Med. Chem. 2006, 13, 997–1016. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Oelkers, P. Bile acid transporters. Curr. Opin. Lipidol. 1995, 6, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Geyer, J.; Döring, B.; Meerkamp, K.; Ugele, B.; Bakhiya, N.; Fernandes, C.F.; Godoy, J.R.; Glatt, H.; Petzinger, E. Cloning and functional characterization of human sodium-dependent organic anion transporter (SLC10A6). J. Biol. Chem. 2007, 282, 19728–19741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasche, A.; Lehmann, F.; König, A.; Goldmann, N.; Corman, V.M.; Moreira-Soto, A.; Geipel, A.; van Riel, D.; Vakulenko, Y.A.; Sander, A.-L.; et al. Highly diversified shrew hepatitis B viruses corroborate ancient origins and divergent infection patterns of mammalian hepadnaviruses. Proc. Natl. Acad. Sci. USA 2019, 116, 17007–17012. [Google Scholar] [CrossRef] [Green Version]
- de Carvalho Dominguez Souza, B.F.; König, A.; Rasche, A.; de Oliveira Carneiro, I.; Stephan, N.; Corman, V.M.; Roppert, P.L.; Goldmann, N.; Kepper, R.; Müller, S.F.; et al. A novel hepatitis B virus species discovered in capuchin monkeys sheds new light on the evolution of primate hepadnaviruses. J. Hepatol. 2018, 68, 1114–1122. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.F.; König, A.; Döring, B.; Glebe, D.; Geyer, J. Characterisation of the hepatitis B virus cross-species transmission pattern via Na+/taurocholate co-transportingü polypeptides from 11 New World and Old World primate species. PLoS ONE 2018, 13, e0199200. [Google Scholar] [CrossRef]
- Watashi, K.; Sluder, A.; Daito, T.; Matsunaga, S.; Ryo, A.; Nagamori, S.; Iwamoto, M.; Nakajima, S.; Tsukuda, S.; Borroto-Esoda, K.; et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 2014, 59, 1726–1737. [Google Scholar] [CrossRef]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Linder, T.; Esser-Nobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 2014, 60, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Shimura, S.; Watashi, K.; Fukano, K.; Peel, M.; Sluder, A.; Kawai, F.; Iwamoto, M.; Tsukuda, S.; Takeuchi, J.S.; Miyake, T. Cyclosporin derivatives inhibit hepatitis B virus entry without interfering with NTCP transporter activity. J. Hepatol. 2017, 66, 685–692. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ruan, H.; Li, Y.; Sun, G.; Liu, X.; He, W.; Mao, F.; He, M.; Yan, L.; Zhong, G.; et al. Potent and Specific Inhibition of NTCP-Mediated HBV/HDV Infection and Substrate Transporting by a Novel, Oral-Available Cyclosporine A Analogue. J. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Wang, X.J.; Hu, W.; Zhang, T.-Y.; Mao, Y.-Y.; Liu, N.-N.; Wang, S.-Q. Irbesartan, an FDA approved drug for hypertension and diabetic nephropathy, is a potent inhibitor for hepatitis B virus entry by disturbing Na(+)-dependent taurocholate cotransporting polypeptide activity. Antiviral Res. 2015, 120, 140–146. [Google Scholar] [CrossRef]
- Blanchet, M.; Sureau, C.; Labonté, P. Use of FDA approved therapeutics with hNTCP metabolic inhibitory properties to impair the HDV lifecycle. Antiviral Res. 2014, 106, 111–115. [Google Scholar] [CrossRef]
- Huang, H.C.; Tao, M.-H.; Hung, T.-M.; Chen, J.-C.; Lin, Z.-J.; Huang, C. (-)-Epigallocatechin-3-gallate inhibits entry of hepatitis B virus into hepatocytes. Antiviral Res. 2014, 111, 100–111. [Google Scholar] [CrossRef]
- Kaneko, M.; Watashi, K.; Kamisuki, S.; Matsunaga, H.; Iwamoto, M.; Kawai, F.; Ohashi, H.; Tsukuda, S.; Shimura, S.; Suzuki, R.; et al. A Novel Tricyclic Polyketide, Vanitaracin A, Specifically Inhibits the Entry of Hepatitis B and D Viruses by Targeting Sodium Taurocholate Cotransporting Polypeptide. J. Virol. 2015, 89, 11945–11953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukuda, S.; Watashi, K.; Iwamoto, M.; Suzuki, R.; Aizaki, H.; Okada, M.; Sugiyama, M.; Kojima, S.; Tanaka, Y.; Mizokami, M.; et al. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J. Biol. Chem. 2015, 290, 5673–5684. [Google Scholar] [CrossRef] [Green Version]
- Tsukuda, S.; Watashi, K.; Hojima, T.; Isogawa, M.; Iwamoto, M.; Omagari, K.; Suzuki, R.; Aizaki, H.; Kojima, S.; Sugiyama, M.; et al. A new class of hepatitis B and D virus entry inhibitors, proanthocyanidin and its analogs, that directly act on the viral large surface proteins. Hepatology 2017, 65, 1104–1116. [Google Scholar] [CrossRef] [Green Version]
- Donkers, J.M.; Zehnder, B.; van Westen, G.J.P.; Kwakkenbos, M.J.; Jzerman, A.P.I.; Oude Elferink, R.P.J.; Beuers, U.; Urban, S.; van de Graaf, S.F. Reduced hepatitis B and D viral entry using clinically applied drugs as novel inhibitors of the bile acid transporter NTCP. Sci. Rep. 2017, 7, 15307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, A.; Eidam, A.; Haag, M.; Hohmann, N.; Burhenne, J.; Schwab, M.; van de Graaf, S.F.; Meyer, M.R.; Maurer, H.H.; Meier, K.; et al. The NTCP-inhibitor Myrcludex B: Effects on Bile Acid Disposition and Tenofovir Pharmacokinetics. Clin. Pharmacol. Ther. 2018, 103, 341–348. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Mueller, R.; Dawson, E.S.; Meiler, J.; Rodriguez, A.L.; Chauder, B.A.; Bates, B.S.; Felts, A.S.; Lamb, J.P.; Menon, U.N.; Jadhav, S.B.; et al. Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu(5)): From an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem 2012, 7, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.; Auld, D.S.; Inglese, J. Apparent activity in high-throughput screening: Origins of compound-dependent assay interference. Curr. Opin. Chem. Biol. 2010, 14, 315–324. [Google Scholar] [CrossRef] [Green Version]
- Butkiewicz, M.; Lowe Jr., E.W.; Mueller, R.; Mendenhall, J.L.; Teixeira, P.L.; Weaver, C.D.; Meiler, J. Benchmarking ligand-based virtual High-Throughput Screening with the PubChem database. Molecules 2013, 18, 735–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
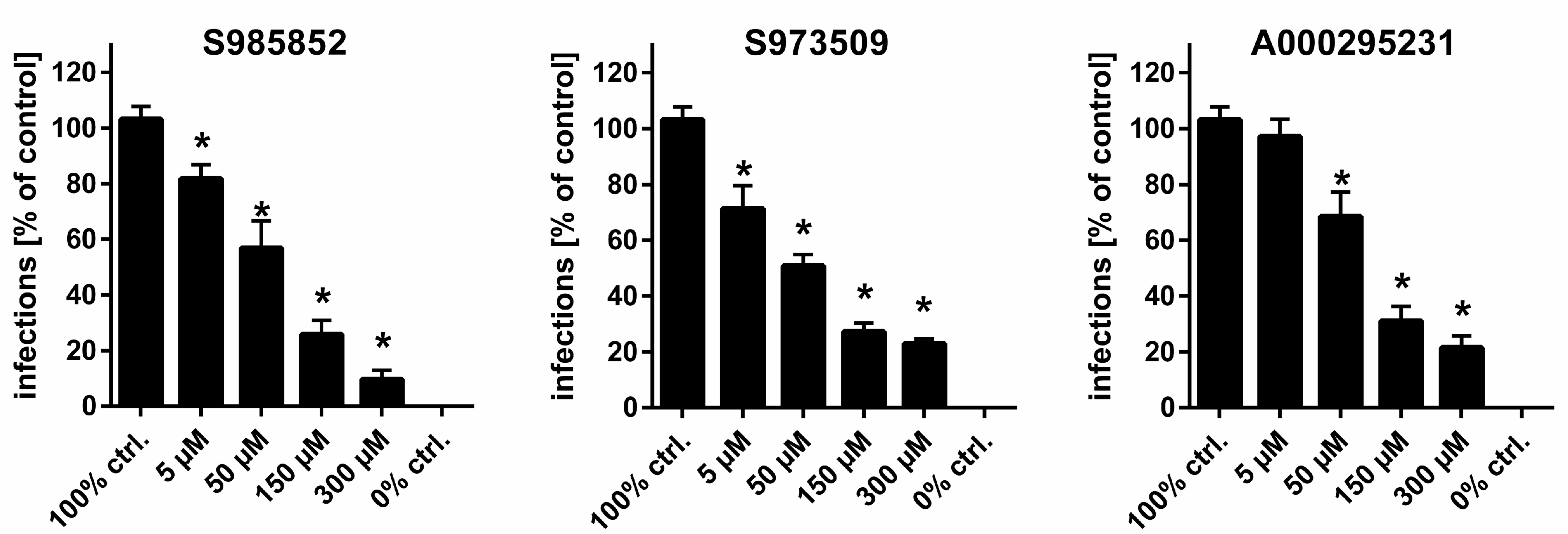
| Compound | IC50 ([3H]TC Uptake) [µM] a | IC50 ([3H]preS1 Binding) [µM] b | Selectivity Index c | In Vitro HDV Infection d |
|---|---|---|---|---|
| S985852 | 13 to 34 | 8 to 24 | 2 | IC50 ~ 15 µM |
| A000028897 | 16 to 62 | 3 to 24 | 3 | toxic |
| A000295480 | 145 to 1323 | 32 to 297 | 4 | ND |
| S973515 | 413 to 1238 | 39 to 148 | 9 | ND |
| A000289041 | >1000 | 35 to 224 | 18 | ND |
| S973509 | 202 to 775 | 3 to 10 | 53 | IC50 ~ 70 µM |
| A000295231 | 341 to 1221 | 4 to 29 | 65 | IC50 ~ 40 µM |
| A000295013 | >1000 | 73 to 194 | 313 | ND |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirstgen, M.; Lowjaga, K.A.A.T.; Müller, S.F.; Goldmann, N.; Lehmann, F.; Glebe, D.; Baringhaus, K.-H.; Geyer, J. Hepatitis D Virus Entry Inhibitors Based on Repurposing Intestinal Bile Acid Reabsorption Inhibitors. Viruses 2021, 13, 666. https://doi.org/10.3390/v13040666
Kirstgen M, Lowjaga KAAT, Müller SF, Goldmann N, Lehmann F, Glebe D, Baringhaus K-H, Geyer J. Hepatitis D Virus Entry Inhibitors Based on Repurposing Intestinal Bile Acid Reabsorption Inhibitors. Viruses. 2021; 13(4):666. https://doi.org/10.3390/v13040666
Chicago/Turabian StyleKirstgen, Michael, Kira Alessandra Alicia Theresa Lowjaga, Simon Franz Müller, Nora Goldmann, Felix Lehmann, Dieter Glebe, Karl-Heinz Baringhaus, and Joachim Geyer. 2021. "Hepatitis D Virus Entry Inhibitors Based on Repurposing Intestinal Bile Acid Reabsorption Inhibitors" Viruses 13, no. 4: 666. https://doi.org/10.3390/v13040666