
Phage-Displayed Peptides for Targeting Tyrosine Kinase Membrane Receptors in Cancer Therapy

 , , , , , and
, , , , , and
Abstract
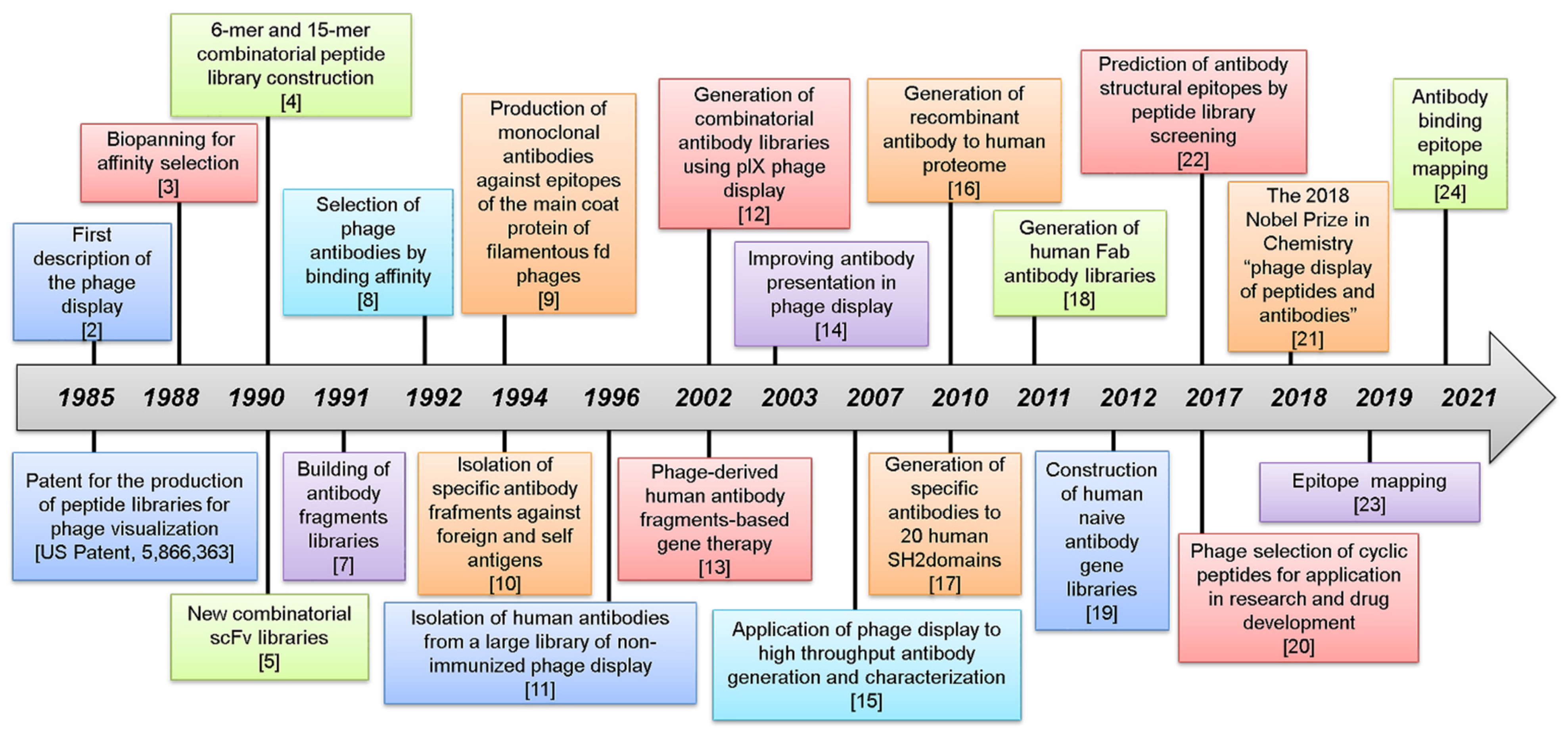
:1. The Evolution of Phage Display
2. Phages Used for Peptide Display
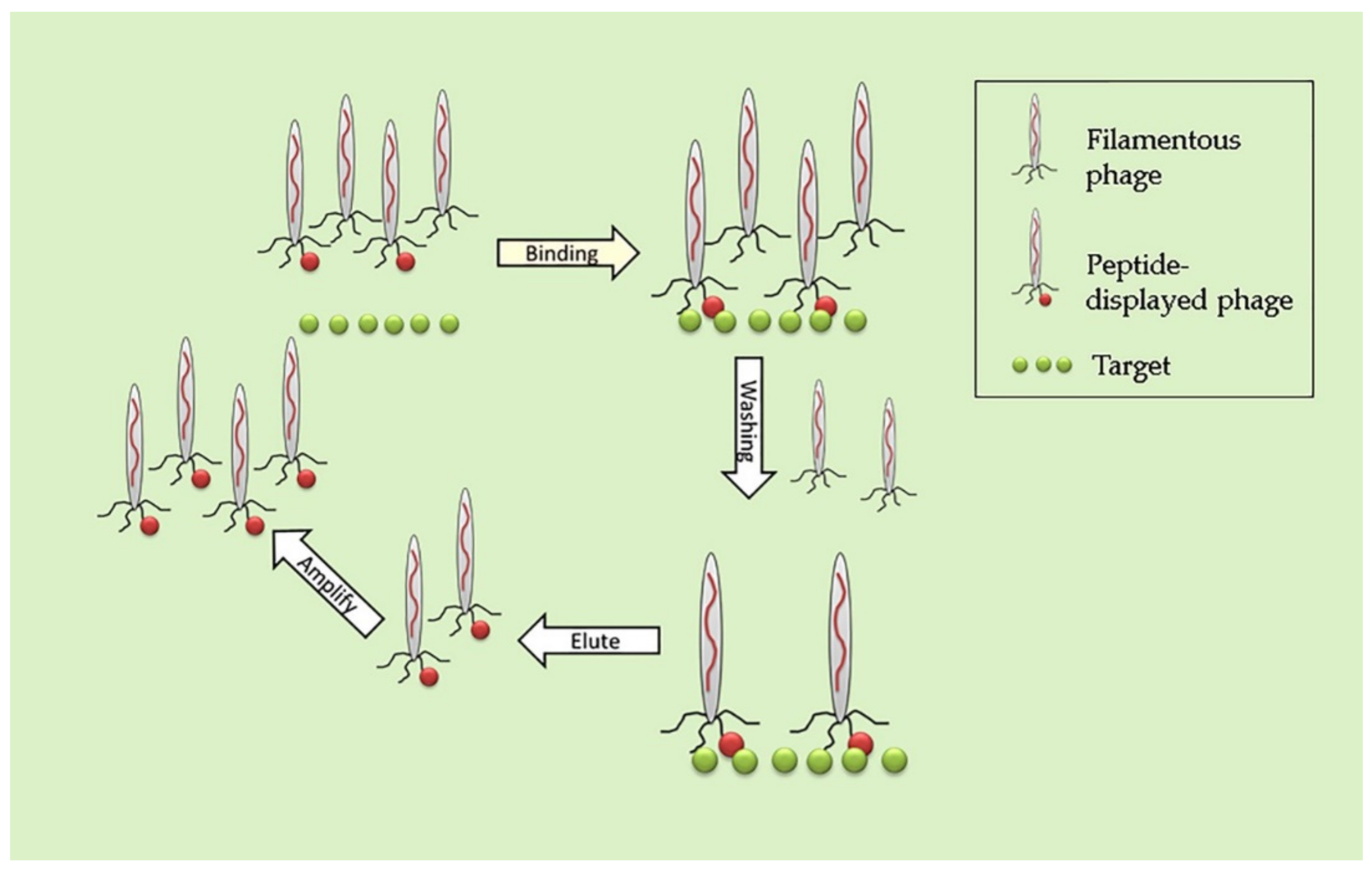
3. Biopanning
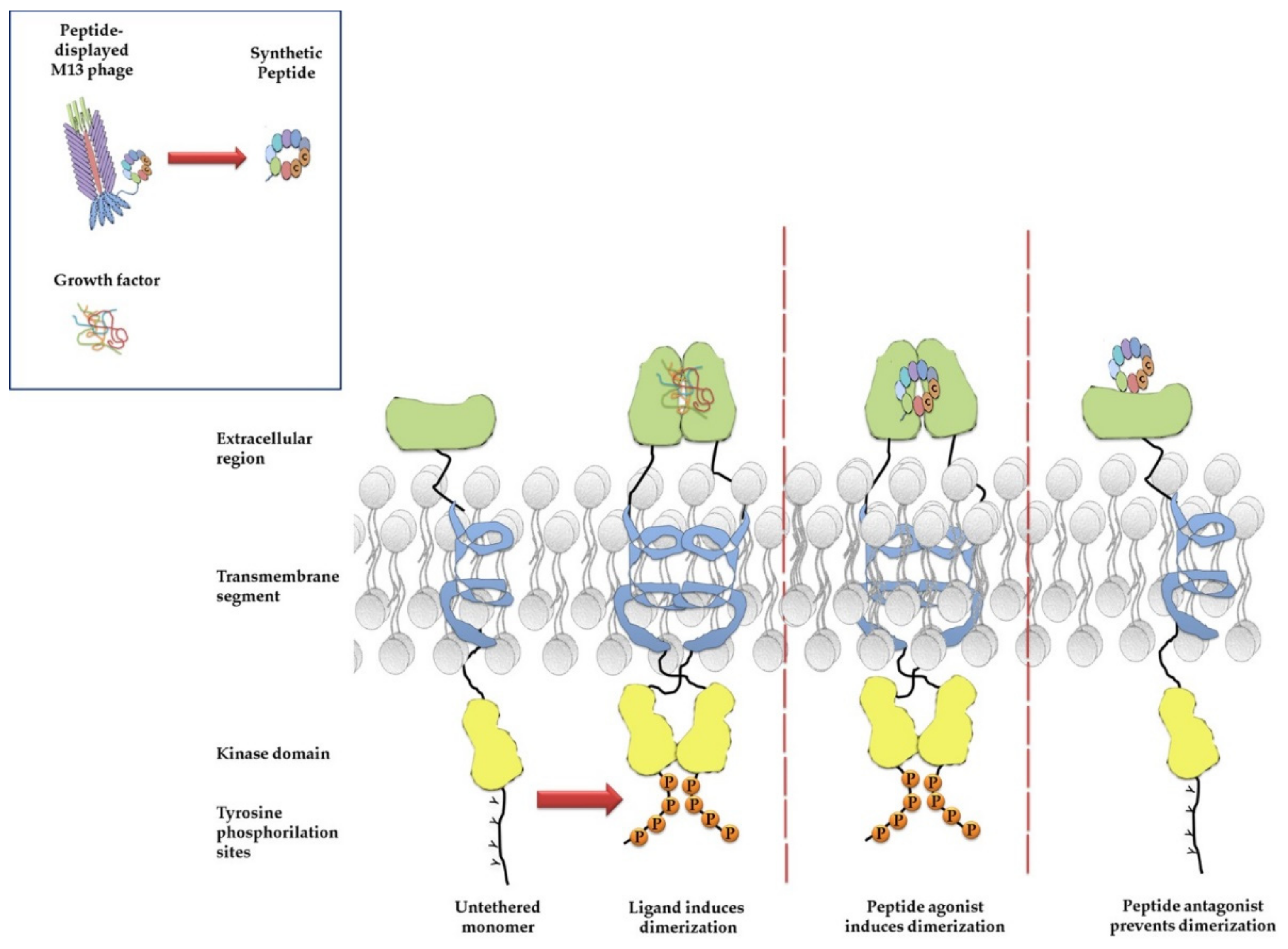
4. Phage Display for Selection of Peptide Ligands of Membrane Receptors
5. Phage Display for Analysis of Protein–Protein Interaction Domains
6. Optimization of Peptides for Targeting
7. Membrane Receptors in Cancer Cells
8. The Epidermal Growth Factor Receptor Family
9. Vascular Endothelial Growth Factor Receptor
10. Fibroblast Growth Factor Receptor
11. Platelet-Derived Growth Factor Receptor
12. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hertveldt, K.; Belien, T.; Volckaert, G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol. Biol. 2009, 502, 321–339. [Google Scholar]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Devlin, J.J.; Panganiban, L.C.; Devlin, P.E. Random peptide libraries: A source of specific protein binding molecules. Science 1990, 249, 404–406. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Clackson, T.; Hoogenboom, H.R.; Griffiths, A.D.; Winter, G. Making antibody fragments using phage display libraries. Nature 1991, 352, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, R.E.; Russell, S.J.; Winter, G. Selection of phage antibodies by binding affinity. Mimicking affinity maturation. J. Mol. Biol. 1992, 226, 889–896. [Google Scholar] [CrossRef]
- Micheel, B.; Heymann, S.; Scharte, G.; Bottger, V.; Vogel, F.; Dubel, S.; Breitling, F.; Little, M.; Behrsing, O. Production of monoclonal antibodies against epitopes of the main coat protein of filamentous fd phages. J. Immunol. Methods 1994, 171, 103–109. [Google Scholar] [CrossRef]
- Winter, G.; Griffiths, A.D.; Hawkins, R.E.; Hoogenboom, H.R. Making antibodies by phage display technology. Annu. Rev. Immunol. 1994, 12, 433–455. [Google Scholar] [CrossRef]
- Vaughan, T.J.; Williams, A.J.; Pritchard, K.; Osbourn, J.K.; Pope, A.R.; Earnshaw, J.C.; McCafferty, J.; Hodits, R.A.; Wilton, J.; Johnson, K.S. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat. Biotechnol. 1996, 14, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Mao, S.; Kaufmann, G.; Wirsching, P.; Lerner, R.A.; Janda, K.D. A method for the generation of combinatorial antibody libraries using pIX phage display. Proc. Natl. Acad. Sci. USA 2002, 99, 12612–12616. [Google Scholar] [CrossRef] [Green Version]
- Sanz, L.; Kristensen, P.; Blanco, B.; Facteau, S.; Russell, S.J.; Winter, G.; Alvarez-Vallina, L. Single-chain antibody-based gene therapy: Inhibition of tumor growth by in situ production of phage-derived human antibody fragments blocking functionally active sites of cell-associated matrices. Gene Ther. 2002, 9, 1049–1053. [Google Scholar] [CrossRef] [Green Version]
- Broders, O.; Breitling, F.; Dubel, S. Hyperphage. Improving antibody presentation in phage display. Methods Mol. Biol. 2003, 205, 295–302. [Google Scholar]
- Schofield, D.J.; Pope, A.R.; Clementel, V.; Buckell, J.; Chapple, S.; Clarke, K.F.; Conquer, J.S.; Crofts, A.M.; Crowther, S.R.; Dyson, M.R.; et al. Application of phage display to high throughput antibody generation and characterization. Genome Biol. 2007, 8, R254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubel, S.; Stoevesandt, O.; Taussig, M.J.; Hust, M. Generating recombinant antibodies to the complete human proteome. Trends Biotechnol. 2010, 28, 333–339. [Google Scholar] [CrossRef]
- Pershad, K.; Pavlovic, J.D.; Graslund, S.; Nilsson, P.; Colwill, K.; Karatt-Vellatt, A.; Schofield, D.J.; Dyson, M.R.; Pawson, T.; Kay, B.K.; et al. Generating a panel of highly specific antibodies to 20 human SH2 domains by phage display. Protein Eng. Des. Sel. 2010, 23, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Andris-Widhopf, J.; Steinberger, P.; Fuller, R.; Rader, C.; Barbas, C.F., 3rd. Generation of human Fab antibody libraries: PCR amplification and assembly of light- and heavy-chain coding sequences. Cold Spring Harb. Protoc. 2011, 2011. [Google Scholar] [CrossRef] [Green Version]
- Hust, M.; Frenzel, A.; Meyer, T.; Schirrmann, T.; Dubel, S. Construction of human naive antibody gene libraries. Methods Mol. Biol. 2012, 907, 85–107. [Google Scholar]
- Deyle, K.; Kong, X.D.; Heinis, C. Phage Selection of Cyclic Peptides for Application in Research and Drug Development. Acc. Chem. Res. 2017, 50, 1866–1874. [Google Scholar] [CrossRef]
- Barderas, R.; Benito-Pena, E. The 2018 Nobel Prize in Chemistry: Phage display of peptides and antibodies. Anal. Bioanal. Chem. 2019, 411, 2475–2479. [Google Scholar] [CrossRef]
- Ibsen, K.N.; Daugherty, P.S. Prediction of antibody structural epitopes via random peptide library screening and next generation sequencing. J. Immunol. Methods 2017, 451, 28–36. [Google Scholar] [CrossRef]
- Fuhner, V.; Heine, P.A.; Zilkens, K.J.C.; Meier, D.; Roth, K.D.R.; Moreira, G.; Hust, M.; Russo, G. Epitope Mapping via Phage Display from Single-Gene Libraries. Methods Mol. Biol. 2019, 1904, 353–375. [Google Scholar] [PubMed]
- Qi, H.; Ma, M.; Hu, C.; Xu, Z.W.; Wu, F.L.; Wang, N.; Lai, D.Y.; Li, Y.; Zhang, H.; Jiang, H.W.; et al. Antibody binding epitope Mapping (AbMap) of hundred antibodies in a single run. Mol. Cell Proteom. 2021, 20, 100059. [Google Scholar] [CrossRef] [PubMed]
- Malik, P.; Terry, T.D.; Gowda, L.R.; Langara, A.; Petukhov, S.A.; Symmons, M.F.; Welsh, L.C.; Marvin, D.A.; Perham, R.N. Role of capsid structure and membrane protein processing in determining the size and copy number of peptides displayed on the major coat protein of filamentous bacteriophage. J. Mol. Biol. 1996, 260, 9–21. [Google Scholar] [CrossRef]
- Azzazy, H.M.; Highsmith, W.E., Jr. Phage display technology: Clinical applications and recent innovations. Clin. Biochem. 2002, 35, 425–445. [Google Scholar] [CrossRef]
- Lowman, H.B. Phage Display for Protein Binding. In Encyclopedia of Biological Chemistry, 2nd ed.; Lennarz, W.J., Lane, M.D., Eds.; Academic Press: New York, NY, USA, 2013; pp. 431–436. ISBN 9780123786319. [Google Scholar]
- Glisic, S.; Veljkovic, V. Chapter 9—Design of targeting peptides for nanodrugs for treatment of infectious diseases and cancer. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, NY, USA, 2018; pp. 343–381. ISBN 9780128136898. [Google Scholar]
- Malys, N.; Chang, D.Y.; Baumann, R.G.; Xie, D.; Black, L.W. A bipartite bacteriophage T4 SOC and HOC randomized peptide display library: Detection and analysis of phage T4 terminase (gp17) and late sigma factor (gp55) interaction. J. Mol. Biol. 2002, 319, 289–304. [Google Scholar] [CrossRef]
- Deng, X.; Wang, L.; You, X.; Dai, P.; Zeng, Y. Advances in the T7 phage display system (Review). Mol. Med. Rep. 2018, 17, 714–720. [Google Scholar] [CrossRef] [Green Version]
- Nicastro, J.; Sheldon, K.; Slavcev, R.A. Bacteriophage lambda display systems: Developments and applications. Appl. Microbiol. Biotechnol. 2014, 98, 2853–2866. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef]
- Barbas, C.F., III; Burton, D.R.; Scott, J.K.; Silverman, G.J. Phage Display a Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Takakusagi, Y.; Takakusagi, K.; Sugawara, F.; Sakaguchi, K. Use of phage display technology for the determination of the targets for small-molecule therapeutics. Expert Opin. Drug Dis. 2010, 5, 361–389. [Google Scholar] [CrossRef]
- Rosenberg, A.; Griffin, K.; Studier, F.W.; McCormick, M.; Berg, J.; Novy, R.; Mierendorf, R. T7Select® Phage Display System: A powerful new protein display system based on bacteriophage T7. Innovations 1996, 6, 1–6. [Google Scholar]
- Castagnoli, L.; Zucconi, A.; Quondam, M.; Rossi, M.; Vaccaro, P.; Panni, S.; Paoluzi, S.; Santonico, E.; Dente, L.; Cesareni, G. Alternative bacteriophage display systems. Comb. Chem. High Throughput Screen. 2001, 4, 121–133. [Google Scholar] [CrossRef]
- McKenzie, K.M.; Videlock, E.J.; Splittgerber, U.; Austin, D.J. Simultaneous identification of multiple protein targets by using complementary-DNA phage display and a natural-product-mimetic probe. Angew. Chem. Int. Ed. 2004, 43, 4052–4055. [Google Scholar] [CrossRef]
- Takakusagi, Y.; Ohta, K.; Kuramochi, K.; Morohashi, K.; Kobayashi, S.; Sakaguchi, K.; Sugawara, F. Synthesis of a biotinylated camptothecin derivative and determination of the binding sequence by T7 phage display technology. Bioorg. Med. Chem. Lett. 2005, 15, 4846–4849. [Google Scholar] [CrossRef]
- Griffiths, A.; Miller, J.; Suzuki, D.; Lewontin, R.; Gelbart, W. An Introduction to Genetic Analysis, 7th ed.; W.H. Freeman & Co. Ltd: New York, NY, USA, 2000. [Google Scholar]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [Green Version]
- Bratkovic, T. Progress in phage display: Evolution of the technique and its applications. Cell. Mol. Life Sci 2010, 67, 749–767. [Google Scholar] [CrossRef]
- Hamzeh-Mivehroud, M.; Alizadeh, A.A.; Morris, M.B.; Church, W.B.; Dastmalchi, S. Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov. Today 2013, 18, 1144–1157. [Google Scholar] [CrossRef] [PubMed]
- Omidfar, K.; Daneshpour, M. Advances in phage display technology for drug discovery. Expert Opin. Drug Dis. 2015, 10, 651–669. [Google Scholar] [CrossRef]
- Kim, S.; Kim, D.; Jung, H.H.; Lee, I.H.; Kim, J.I.; Suh, J.Y.; Jon, S. Bio-Inspired Design and Potential Biomedical Applications of a Novel Class of High-Affinity Peptides. Angew. Chem. Int. Ed. 2012, 51, 1890–1894. [Google Scholar] [CrossRef]
- Zhao, P.; Grabinski, T.; Gao, C.; Skinner, R.S.; Giambernardi, T.; Su, Y.; Hudson, E.; Resau, J.; Gross, M.; Woude, G.F.V.; et al. Identification of a met-binding peptide from a phage display library. Clin. Cancer Res. 2007, 13, 6049–6055. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.M.; Zhang, C.; Liu, G.B.; Liu, H.; Zhou, C.P.; Lu, Y.X.; Zhou, C.; Yuan, L.; Li, X.N. A novel mouse CD133 binding-peptide screened by phage display inhibits cancer cell motility in vitro. Clin. Exp. Metastasis 2012, 29, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Laakkonen, P.; Porkka, K.; Hoffman, J.A.; Ruoslahti, E. A tumor-homing peptide with a targeting specificity related to lymphatic vessels. Nat. Med. 2002, 8, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Lin, C.T.; Wu, H.C. Hepatocellular carcinoma cell-specific peptide ligand for targeted drug delivery. Mol. Cancer Ther. 2008, 7, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Koivunen, E.; Wang, B.; Ruoslahti, E. Phage libraries displaying cyclic peptides with different ring sizes: Ligand specificities of the RGD-directed integrins. Biotechnology 1995, 13, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Pasqualini, R.; Ruoslahti, E. Organ targeting in vivo using phage display peptide libraries. Nature 1996, 380, 364–366. [Google Scholar] [CrossRef]
- Ruoslahti, E. Targeting tumor vasculature with homing peptides from phage display. Semin. Cancer Biol. 2000, 10, 435–442. [Google Scholar] [CrossRef]
- Arap, W.; Kolonin, M.G.; Trepel, M.; Lahdenranta, J.; Cardo-Vila, M.; Giordano, R.J.; Mintz, P.J.; Ardelt, P.U.; Yao, V.J.; Vidal, C.I.; et al. Steps toward mapping the human vasculature by phage display. Nat. Med. 2002, 8, 121–127. [Google Scholar] [CrossRef]
- Mueller, J.; Gaertner, F.C.; Blechert, B.; Janssen, K.P.; Essler, M. Targeting of tumor blood vessels: A phage-displayed tumor-homing peptide specifically binds to matrix metalloproteinase-2-processed collagen IV and blocks angiogenesis in vivo. Mol. Cancer Res. 2009, 7, 1078–1085. [Google Scholar] [CrossRef] [Green Version]
- Karkkainen, S.; Hiipakka, M.; Wang, J.H.; Kleino, I.; Vaha-Jaakkola, M.; Renkema, G.H.; Liss, M.; Wagner, R.; Saksela, K. Identification of preferred protein interactions by phage-display of the human Src homology-3 proteome. EMBO Rep. 2006, 7, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Voss, M.; Lettau, M.; Janssen, O. Identification of SH3 domain interaction partners of human FasL (CD178) by phage display screening. BMC Immunol. 2009, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Luck, K.; Trave, G. Phage display can select over-hydrophobic sequences that may impair prediction of natural domain-peptide interactions. Bioinformatics 2011, 27, 899–902. [Google Scholar] [CrossRef] [Green Version]
- Mimmi, S.; Vecchio, E.; Iaccino, E.; Rossi, M.; Lupia, A.; Albano, F.; Chiurazzi, F.; Fiume, G.; Pisano, A.; Ceglia, S.; et al. Evidence of shared epitopic reactivity among independent B-cell clones in chronic lymphocytic leukemia patients. Leukemia 2016, 30, 2419–2422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogenboom, H.R. Overview of antibody phage-display technology and its applications. Methods Mol. Biol. 2002, 178, 1–37. [Google Scholar] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The Current State of Peptide Drug Discovery: Back to the Future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Katz, B.A. Structural and mechanistic determinants of affinity and specificity of ligands discovered or engineered by phage display. Annu. Rev. Biophys. Biomol. 1997, 26, 27–45. [Google Scholar] [CrossRef] [Green Version]
- Dan, N.; Samanta, K.; Almoazen, H. An Update on Pharmaceutical Strategies for Oral Delivery of Therapeutic Peptides and Proteins in Adults and Pediatrics. Children 2020, 7, 307. [Google Scholar] [CrossRef]
- Ladner, R.C.; Sato, A.K.; Gorzelany, J.; de Souza, M. Phage display-derived peptides as a therapeutic alternatives to antibodies. Drug Discov. Today 2004, 9, 525–529. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Quinto, I.; Iaccino, E. Phage Display: An Overview in Context to Drug Discovery. Trends Pharmacol. Sci. 2019, 40, 87–91. [Google Scholar] [CrossRef]
- Choonara, B.F.; Choonara, Y.E.; Kumar, P.; Bijukumar, D.; du Toit, L.C.; Pillay, V. A review of advanced oral drug delivery technologies facilitating the protection and absorption of protein and peptide molecules. Biotechnol. Adv. 2014, 32, 1269–1282. [Google Scholar] [CrossRef]
- Tripathi, P.P.; Arami, H.; Banga, I.; Gupta, J.; Gandhi, S. Cell penetrating peptides in preclinical and clinical cancer diagnosis and therapy. Oncotarget 2018, 9, 37252–37267. [Google Scholar] [CrossRef] [Green Version]
- Boohaker, R.J.; Lee, M.W.; Vishnubhotla, P.; Perez, J.M.; Khaled, A.R. The use of therapeutic peptides to target and to kill cancer cells. Curr. Med. Chem. 2012, 19, 3794–3804. [Google Scholar] [CrossRef]
- Scodeller, P.; Asciutto, E.K. Targeting Tumors Using Peptides. Molecules 2020, 25, 808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangini, M.; Iaccino, E.; Mosca, M.G.; Mimmi, S.; D’Angelo, R.; Quinto, I.; Scala, G.; Mariggio, S. Peptide-guided targeting of GPR55 for anti-cancer therapy. Oncotarget 2017, 8, 5179–5195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauquelin, G.; Charlton, S.J. Exploring avidity: Understanding the potential gains in functional affinity and target residence time of bivalent and heterobivalent ligands. Br. J. Pharmacol. 2013, 168, 1771–1785. [Google Scholar] [CrossRef] [Green Version]
- Bolhassani, A. Improvements in chemical carriers of proteins and peptides. Cell Biol. Int. 2019, 43, 437–452. [Google Scholar] [CrossRef]
- Morales, P.; Jimenez, M.A. Design and structural characterisation of monomeric water-soluble alpha-helix and beta-hairpin peptides: State-of-the-art. Arch. Biochem. Biophys. 2019, 661, 149–167. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.; Burn, A.; Tolbert, T.J. Increasing solubility of proteins and peptides by site-specific modification with betaine. Bioconjug. Chem. 2008, 19, 1113–1118. [Google Scholar] [CrossRef]
- Mousavizadeh, A.; Jabbari, A.; Akrami, M.; Bardania, H. Cell targeting peptides as smart ligands for targeting of therapeutic or diagnostic agents: A systematic review. Colloid Surf. B 2017, 158, 507–517. [Google Scholar] [CrossRef]
- Shukla, R.S.; Qin, B.; Cheng, K. Peptides used in the delivery of small noncoding RNA. Mol. Pharm. 2014, 11, 3395–3408. [Google Scholar] [CrossRef]
- Jeong, W.J.; Bu, J.; Kubiatowicz, L.J.; Chen, S.S.; Kim, Y.; Hong, S. Peptide-nanoparticle conjugates: A next generation of diagnostic and therapeutic platforms? Nano Converg. 2018, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Li, J.; Xu, Y.; Zhan, Y.; Li, Y.; Song, T.T.; Zheng, J.; Yang, H. Application of Phage-Displayed Peptides in Tumor Imaging Diagnosis and Targeting Therapy. Int. J. Pept. Res. Ther. 2021, 27, 587–595. [Google Scholar] [CrossRef]
- Palmieri, C.; Falcone, C.; Iaccino, E.; Tuccillo, F.M.; Gaspari, M.; Trimboli, F.; De Laurentiis, A.; Luberto, L.; Pontoriero, M.; Pisano, A.; et al. In vivo targeting and growth inhibition of the A20 murine B-cell lymphoma by an idiotype-specific peptide binder. Blood 2010, 116, 226–238. [Google Scholar] [CrossRef] [Green Version]
- Iaccino, E.; Mimmi, S.; Dattilo, V.; Marino, F.; Candeloro, P.; Di Loria, A.; Marimpietri, D.; Pisano, A.; Albano, F.; Vecchio, E.; et al. Monitoring multiple myeloma by idiotype-specific peptide binders of tumor-derived exosomes. Mol. Cancer 2017, 16, 159. [Google Scholar] [CrossRef]
- Mimmi, S.; Maisano, D.; Nistico, N.; Vecchio, E.; Chiurazzi, F.; Ferrara, K.; Iannalfo, M.; D’Ambrosio, A.; Fiume, G.; Iaccino, E.; et al. Detection of chronic lymphocytic leukemia subpopulations in peripheral blood by phage ligands of tumor immunoglobulin B cell receptors. Leukemia 2021, 35, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Deller, M.C.; Jones, E.Y. Cell surface receptors. Curr. Opin. Struct. Biol. 2000, 10, 213–219. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Guo, M.; Hay, B.A. Cell proliferation and apoptosis. Curr. Opin. Cell Biol. 1999, 11, 745–752. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, I.N. Mechanisms of Activation of Receptor Tyrosine Kinases: Monomers or Dimers. Cells 2014, 3, 304–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.F.; Lovly, C.M. Mechanisms of receptor tyrosine kinase activation in cancer. Mol. Cancer 2018, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Rusten, T.E.; Stenmark, H. Aberrant receptor signaling and trafficking as mechanisms in oncogenesis. Crit. Rev. Oncog. 2007, 13, 39–74. [Google Scholar]
- Santos, R.; Ursu, O.; Gaulton, A.; Bento, A.P.; Donadi, R.S.; Bologa, C.G.; Karlsson, A.; Al-Lazikani, B.; Hersey, A.; Oprea, T.I.; et al. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017, 16, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Ardito, F.; Giuliani, M.; Perrone, D.; Troiano, G.; Lo Muzio, L. The crucial role of protein phosphorylation in cell signaling and its use as targeted therapy. Int. J. Mol. Med. 2017, 40, 271–280. [Google Scholar] [CrossRef] [Green Version]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Loktev, A.; Haberkorn, U.; Mier, W. Multicyclic Peptides as Scaffolds for the Development of Tumor Targeting Agents. Curr. Med. Chem. 2017, 24, 2141–2155. [Google Scholar] [CrossRef] [PubMed]
- Elzoghby, A.O.; Elgohary, M.M.; Kamel, N.M. Implications of Protein- and Peptide-Based Nanoparticles as Potential Vehicles for Anticancer Drugs. Adv. Protein Chem. Struct. Biol. 2015, 98, 169–221. [Google Scholar]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar]
- Sabbah, D.A.; Hajjo, R.; Sweidan, K. Review on Epidermal Growth Factor Receptor (EGFR) Structure, Signaling Pathways, Interactions, and Recent Updates of EGFR Inhibitors. Curr. Top. Med. Chem. 2020, 20, 815–834. [Google Scholar] [CrossRef]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Engelman, J.A.; Cantley, L.C. A sweet new role for EGFR in cancer. Cancer Cell 2008, 13, 375–376. [Google Scholar] [CrossRef] [Green Version]
- Rajaram, P.; Chandra, P.; Ticku, S.; Pallavi, B.K.; Rudresh, K.B.; Mansabdar, P. Epidermal growth factor receptor: Role in human cancer. Indian J. Dent. Res. 2017, 28, 687–694. [Google Scholar]
- Mazzarella, L.; Guida, A.; Curigliano, G. Cetuximab for treating non-small cell lung cancer. Expert Opin. Biol. Ther. 2018, 18, 483–493. [Google Scholar] [CrossRef]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of Resistance to Epidermal Growth Factor Receptor-Tyrosine Kinase Inhibitors and a Potential Treatment Strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef] [Green Version]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7, 73618–73637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, K.; Hung, M.C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Yavari, B.; Mahjub, R.; Saidijam, M.; Raigani, M.; Soleimani, M. The Potential Use of Peptides in Cancer Treatment. Curr. Protein Pept. Sci. 2018, 19, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Xue, E.Y.; Wong, R.C.H.; Wong, C.T.T.; Fong, W.P.; Ng, D.K.P. Synthesis and biological evaluation of an epidermal growth factor receptor-targeted peptide-conjugated phthalocyanine-based photosensitiser. RSC Adv. 2019, 9, 20652–20662. [Google Scholar] [CrossRef] [Green Version]
- Caprini, A.; Silva, D.; Zanoni, I.; Cunha, C.; Volonte, C.; Vescovi, A.; Gelain, F. A novel bioactive peptide: Assessing its activity over murine neural stem cells and its potential for neural tissue engineering. New Biotechnol. 2013, 30, 552–562. [Google Scholar] [CrossRef]
- Kelly, K.A.; Carson, J.; McCarthy, J.R.; Weissleder, R. Novel Peptide Sequence (“IQ-tag”) with High Affinity for NIR Fluorochromes Allows Protein and Cell Specific Labeling for In Vivo Imaging. PLoS ONE 2007, 2, e665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, R.; Jin, C.C.; Quan, J.; Nie, H.L.; Zhu, L.M. A Novel Self-Assembling Peptide with UV-Responsive Properties. Biopolymers 2014, 101, 272–278. [Google Scholar] [CrossRef]
- Li, Z.H.; Zhao, R.J.; Wu, X.H.; Sun, Y.; Yao, M.; Li, J.J.; Xu, Y.H.; Gu, J.R. Identification and characterization of a novel peptide ligand of epidermal growth factor receptor for targeted delivery of therapeutics. FASEB J. 2005, 19, 1978–1985. [Google Scholar] [CrossRef]
- Li, X.L.; Hu, K.Z.; Liu, W.F.; Wei, Y.F.; Sha, R.H.; Long, Y.X.; Han, Y.J.; Sun, P.H.; Wu, H.B.; Li, G.P.; et al. Synthesis and evaluation of [F-18]FP-Lys-GE11 as a new radiolabeled peptide probe for epidermal growth factor receptor (EGFR) imaging. Nucl. Med. Biol. 2020, 90–91, 84–92. [Google Scholar] [CrossRef]
- Hamzeh-Mivehroud, M.; Mahmoudpour, A.; Dastmalchi, S. Identification of New Peptide Ligands for Epidermal Growth Factor Receptor Using Phage Display and Computationally Modeling their Mode of Binding. Chem. Biol. Drug Des. 2012, 79, 246–259. [Google Scholar] [CrossRef]
- Ki, M.K.; Kang, K.J.; Shimi, H. Phage Display Selection of EGFR-specific Antibodies by Capture-sandwich Panning. Biotechnol. Bioprocess Eng. 2010, 15, 152–156. [Google Scholar] [CrossRef]
- Wang, A.D.; Cui, M.; Qu, H.; Di, J.B.; Wang, Z.Z.; Xing, J.D.; Wu, F.; Wu, W.; Wang, X.C.; Shen, L.; et al. Induction of anti-EGFR immune response with mimotopes identified from a phage display peptide library by panitumumab. Oncotarget 2016, 7, 75293–75306. [Google Scholar] [CrossRef] [Green Version]
- Perrier, A.; Gligorov, J.; Lefevre, G.; Boissan, M. The extracellular domain of Her2 in serum as a biomarker of breast cancer. Lab. Investig. 2018, 98, 696–707. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Human Epidermal Growth Factor Receptor 2 (HER2) in Cancers: Overexpression and Therapeutic Implications. Mol. Biol. Int. 2014, 2014, 852748. [Google Scholar] [CrossRef]
- Ocana, A.; Amir, E.; Pandiella, A. HER2 heterogeneity and resistance to anti-HER2 antibody-drug conjugates. Breast Cancer Res. 2020, 22, 15. [Google Scholar] [CrossRef]
- Rye, I.H.; Trinh, A.; Saetersdal, A.B.; Nebdal, D.; Lingjaerde, O.C.; Almendro, V.; Polyak, K.; Borresen-Dale, A.L.; Helland, A.; Markowetz, F.; et al. Intratumor heterogeneity defines treatment-resistant HER2+ breast tumors. Mol. Oncol. 2018, 12, 1838–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diderich, P.; Heinis, C. Phage selection of bicyclic peptides binding Her2. Tetrahedron 2014, 70, 7733–7739. [Google Scholar] [CrossRef]
- Houimel, M.; Schneider, P.; Terskikh, A.; Mach, J.P. Selection of peptides and synthesis of pentameric peptabody molecules reacting specifically with ERBB-2 receptor. Int. J. Cancer 2001, 92, 748–755. [Google Scholar] [CrossRef]
- Karasseva, N.G.; Glinsky, V.V.; Chen, N.X.; Komatireddy, R.; Quinn, T.P. Identification and characterization of peptides that bind human ErbB-2 selected from a bacteriophage display library. J. Protein Chem. 2002, 21, 287–296. [Google Scholar] [CrossRef]
- Park, J.; Park, S.; Kim, S.; Lee, I.H.; Saw, P.E.; Lee, K.; Kim, Y.C.; Kim, Y.J.; Farokhzad, O.C.; Jeong, Y.Y.; et al. HER2-specific aptide conjugated magneto-nanoclusters for potential breast cancer imaging and therapy. J. Mater. Chem. B 2013, 1, 4576–4583. [Google Scholar] [CrossRef]
- Ducharme, M.; Lapi, S.E. Peptide Based Imaging Agents for HER2 Imaging in Oncology. Mol. Imaging 2020, 19. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15, 339–351. [Google Scholar] [CrossRef]
- Koch, S.; Tugues, S.; Li, X.J.; Gualandi, L.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Biochem. J. 2011, 437, 169–183. [Google Scholar] [CrossRef] [Green Version]
- Albuquerque, R.J.C.; Hayashi, T.; Cho, W.G.; Kleinman, M.E.; Dridi, S.; Takeda, A.; Baffi, J.Z.; Yamada, K.; Kaneko, H.; Green, M.G.; et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat. Med. 2009, 15, 1023–1030. [Google Scholar] [CrossRef]
- Stevens, M.; Oltean, S. Modulation of Receptor Tyrosine Kinase Activity through Alternative Splicing of Ligands and Receptors in the VEGF-A/VEGFR Axis. Cells 2019, 8, 288. [Google Scholar] [CrossRef] [Green Version]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurwitz, H. Integrating the anti-VEGF-A humanized monoclonal antibody bevacizumab with chemotherapy in advanced colorectal cancer. Clin. Colorectal Cancer 2004, 4 (Suppl. S2), S62–S68. [Google Scholar] [CrossRef]
- Zhang, J.; Li, H.; Wang, X.; Qi, H.; Miao, X.; Zhang, T.; Chen, G.; Wang, M. Phage-derived fully human antibody scFv fragment directed against human vascular endothelial growth factor receptor 2 blocked its interaction with VEGF. Biotechnol. Prog. 2012, 28, 981–989. [Google Scholar] [CrossRef]
- Lamdan, H.; Gavilondo, J.V.; Munoz, Y.; Pupo, A.; Huerta, V.; Musacchio, A.; Perez, L.; Ayala, M.; Rojas, G.; Balint, R.F.; et al. Affinity maturation and fine functional mapping of an antibody fragment against a novel neutralizing epitope on human vascular endothelial growth factor. Mol. Biosyst. 2013, 9, 2097–2106. [Google Scholar] [CrossRef] [PubMed]
- Kordi, S.; Rahmati-Yamchi, M.; Asghari Vostakolaei, M.; Etemadie, A.; Barzegari, A.; Abdolalizadeh, J. Isolation of a Novel Anti-KDR3 Single-chain Variable Fragment Antibody from a Phage Display Library. Iran. J. Allergy Asthma Immunol. 2019, 18, 289–299. [Google Scholar] [CrossRef]
- Giordano, R.J.; Cardo-Vila, M.; Salameh, A.; Anobom, C.D.; Zeitlin, B.D.; Hawke, D.H.; Valente, A.P.; Almeida, F.C.; Nor, J.E.; Sidman, R.L.; et al. From combinatorial peptide selection to drug prototype (I): Targeting the vascular endothelial growth factor receptor pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 5112–5117. [Google Scholar] [CrossRef] [Green Version]
- Binetruy-Tournaire, R.; Demangel, C.; Malavaud, B.; Vassy, R.; Rouyre, S.; Kraemer, M.; Plouet, J.; Derbin, C.; Perret, G.; Mazie, J.C. Identification of a peptide blocking vascular endothelial growth factor (VEGF)-mediated angiogenesis. EMBO J. 2000, 19, 1525–1533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.; Chen, H.; Hao, D.; Zhang, X.; Wang, F. The functions and applications of A7R in anti-angiogenic therapy, imaging and drug delivery systems. Asian J. Pharm. Sci. 2019, 14, 595–608. [Google Scholar] [CrossRef]
- Giordano, R.J.; Cardo-Vila, M.; Lahdenranta, J.; Pasqualini, R.; Arap, W. Biopanning and rapid analysis of selective interactive ligands. Nat. Med. 2001, 7, 1249–1253. [Google Scholar] [CrossRef]
- Hetian, L.; Ping, A.; Shumei, S.; Xiaoying, L.; Luowen, H.; Jian, W.; Lin, M.; Meisheng, L.; Junshan, Y.; Chengchao, S. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J. Biol. Chem. 2002, 277, 43137–43142. [Google Scholar] [CrossRef] [Green Version]
- An, P.; Lei, H.; Zhang, J.; Song, S.; He, L.; Jin, G.; Liu, X.; Wu, J.; Meng, L.; Liu, M.; et al. Suppression of tumor growth and metastasis by a VEGFR-1 antagonizing peptide identified from a phage display library. Int. J. Cancer 2004, 111, 165–173. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [Green Version]
- Mohammadi, M.; Olsen, S.K.; Ibrahimi, O.A. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev. 2005, 16, 107–137. [Google Scholar] [CrossRef]
- Dode, C.; Levilliers, J.; Dupont, J.M.; De Paepe, A.; Le Du, N.; Soussi-Yanicostas, N.; Coimbra, R.S.; Delmaghani, S.; Compain-Nouaille, S.; Baverel, F.; et al. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003, 33, 463–465. [Google Scholar] [CrossRef] [Green Version]
- Kan, S.H.; Elanko, N.; Johnson, D.; Cornejo-Roldan, L.; Cook, J.; Reich, E.W.; Tomkins, S.; Verloes, A.; Twigg, S.R.; Rannan-Eliya, S.; et al. Genomic screening of fibroblast growth-factor receptor 2 reveals a wide spectrum of mutations in patients with syndromic craniosynostosis. Am. J. Hum. Genet. 2002, 70, 472–486. [Google Scholar] [CrossRef] [Green Version]
- Webster, M.K.; Donoghue, D.J. FGFR activation in skeletal disorders: Too much of a good thing. Trends Genet. 1997, 13, 178–182. [Google Scholar] [CrossRef]
- Wang, J.; Stockton, D.W.; Ittmann, M. The fibroblast growth factor receptor-4 Arg388 allele is associated with prostate cancer initiation and progression. Clin. Cancer Res. 2004, 10 Pt 1, 6169–6178. [Google Scholar] [CrossRef] [Green Version]
- Massari, F.; Ciccarese, C.; Santoni, M.; Lopez-Beltran, A.; Scarpelli, M.; Montironi, R.; Cheng, L. Targeting fibroblast growth factor receptor (FGFR) pathway in renal cell carcinoma. Expert Rev. Anticancer Ther. 2015, 15, 1367–1369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.; Li, X.; You, B.; Shan, Y.; Cao, X.; You, Y. High Expression of FGFR4 Enhances Tumor Growth and Metastasis in Nasopharyngeal Carcinoma. J. Cancer 2015, 6, 1245–1254. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Vida, A.; Saggese, M.; Hughes, S.; Rudman, S.; Chowdhury, S.; Smith, N.R.; Lawrence, P.; Rooney, C.; Dougherty, B.; Landers, D.; et al. Complexity of FGFR signalling in metastatic urothelial cancer. J. Hematol. Oncol. 2015, 8, 119. [Google Scholar] [CrossRef] [Green Version]
- Criscitiello, C.; Esposito, A.; De Placido, S.; Curigliano, G. Targeting fibroblast growth factor receptor pathway in breast cancer. Curr. Opin. Oncol. 2015, 27, 452–456. [Google Scholar] [CrossRef]
- Wu, X.P.; Huang, H.X.; Wang, C.; Lin, S.Q.; Huang, Y.D.; Wang, Y.; Liang, G.; Yan, Q.X.; Xiao, J.; Wu, J.Z.; et al. Identification of a novel peptide that blocks basic fibroblast growth factor-mediated cell proliferation. Oncotarget 2013, 4, 1819–1828. [Google Scholar] [CrossRef]
- Fan, H.; Duan, Y.; Zhou, H.; Li, W.; Li, F.; Guo, L.; Roeske, R.W. Selection of peptide ligands binding to fibroblast growth factor receptor 1. IUBMB Life 2002, 54, 67–72. [Google Scholar] [CrossRef]
- Wang, W.; Chen, X.; Li, T.; Li, Y.; Wang, R.; He, D.; Luo, W.; Li, X.; Wu, X. Screening a phage display library for a novel FGF8b-binding peptide with anti-tumor effect on prostate cancer. Exp. Cell Res. 2013, 319, 1156–1164. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yan, Q.; Huang, Y.; Huang, H.; Su, Z.; Xiao, J.; Zeng, Y.; Wang, Y.; Nie, C.; Yang, Y.; et al. Isolation of a novel basic FGF-binding peptide with potent antiangiogenetic activity. J. Cell. Mol. Med. 2010, 14, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, S.Q.; Li, X.K.; Wu, X.P. Mechanism of inhibitory effect of P7 on 3T3 cell proliferation induced by basic fibroblast growth factor. Yao Xue Xue Bao 2010, 45, 314–317. [Google Scholar]
- Li, Q.; Gao, S.; Yu, Y.; Wang, W.; Chen, X.; Wang, R.; Li, T.; Wang, C.; Li, X.; Wu, X. A novel bFGF antagonist peptide inhibits breast cancer cell growth. Mol. Med. Rep. 2012, 6, 210–214. [Google Scholar] [PubMed] [Green Version]
- Chen, Q.; Yang, Z.; Chen, X.; Shu, L.; Qu, W. Peptide P7 inhibits the bFGF-stimulated proliferation and invasion of SKOV3 cells. Exp. Ther. Med. 2019, 17, 3003–3008. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Westermark, B. Platelet-derived growth factor: Three isoforms and two receptor types. Trends Genet. 1989, 5, 108–111. [Google Scholar] [CrossRef]
- Cao, Y.; Cao, R.; Hedlund, E.M. R Regulation of tumor angiogenesis and metastasis by FGF and PDGF signaling pathways. J. Mol. Med. (Berl.) 2008, 86, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.T. Signal transduction by the platelet-derived growth factor receptor. Science 1989, 243, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Ostman, A. PDGF receptors in tumor stroma: Biological effects and associations with prognosis and response to treatment. Adv. Drug Deliv. Rev. 2017, 121, 117–123. [Google Scholar] [CrossRef]
- Pietras, K.; Rubin, K.; Sjoblom, T.; Buchdunger, E.; Sjoquist, M.; Heldin, C.H.; Ostman, A. Inhibition of PDGF receptor signaling in tumor stroma enhances antitumor effect of chemotherapy. Cancer Res. 2002, 62, 5476–5484. [Google Scholar] [PubMed]
- Shen, J.Q.; Vil, M.D.; Prewett, M.; Damoci, C.; Zhang, H.F.; Li, H.L.; Jimenez, X.N.; Deevi, D.S.; Iacolina, M.; Kayas, A.; et al. Development of a Fully Human Anti-PDGFR beta Antibody That Suppresses Growth of Human Tumor Xenografts and Enhances Antitumor Activity of an Anti-VEGFR2 Antibody. Neoplasia 2009, 11, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klosowska-Wardega, A.; Hasumi, Y.; Burmakin, M.; Ahgren, A.; Stuhr, L.; Moen, I.; Reed, R.K.; Rubin, K.; Hellberg, C.; Heldin, C.H. Combined anti-angiogenic therapy targeting PDGF and VEGF receptors lowers the interstitial fluid pressure in a murine experimental carcinoma. PLoS ONE 2009, 4, e8149. [Google Scholar] [CrossRef] [Green Version]
- Starling, N.; Hawkes, E.A.; Chau, I.; Watkins, D.; Thomas, J.; Webb, J.; Brown, G.; Thomas, K.; Barbachano, Y.; Oates, J.; et al. A dose escalation study of gemcitabine plus oxaliplatin in combination with imatinib for gemcitabine-refractory advanced pancreatic adenocarcinoma. Ann. Oncol. 2012, 23, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Tsioumpekou, M.; Cunha, S.I.; Ma, H.; Ahgren, A.; Cedervall, J.; Olsson, A.K.; Heldin, C.H.; Lennartsson, J. Specific targeting of PDGFRbeta in the stroma inhibits growth and angiogenesis in tumors with high PDGF-BB expression. Theranostics 2020, 10, 1122–1135. [Google Scholar] [CrossRef]
- Roberts, W.G.; Whalen, P.M.; Soderstrom, E.; Moraski, G.; Lyssikatos, J.P.; Wang, H.F.; Cooper, B.; Baker, D.A.; Savage, D.; Dalvie, D.; et al. Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451. Cancer Res. 2005, 65, 957–966. [Google Scholar]
- Ramakrishnan, V.; Escobedo, M.A.; Fretto, L.J.; Seroogy, J.J.; Tomlinson, J.E.; Wolf, D.L. A Novel Monoclonal-Antibody Dependent on Domain-5 of the Platelet-Derived Growth Factor-Beta-Receptor Inhibits Ligand-Binding Receptor Activation. Growth Factors 1993, 8, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Askoxylakis, V.; Marr, A.; Altmann, A.; Markert, A.; Mier, W.; Debus, J.; Huber, P.E.; Haberkorn, U. Peptide-Based Targeting of the Platelet-Derived Growth Factor Receptor Beta. Mol. Imaging Biol. 2013, 15, 212–221. [Google Scholar] [CrossRef] [Green Version]
- Jitariu, A.A.; Raica, M.; Cimpean, A.M.; Suciu, S.C. The role of PDGF-B/PDGFR-BETA axis in the normal development and carcinogenesis of the breast. Crit. Rev. Oncol. Hematol. 2018, 131, 46–52. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Xiao, W.; Lam, K.S. Tumor-targeting peptides from combinatorial libraries. Adv. Drug Deliv. Rev. 2017, 110–111, 13–37. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, M.R.; Benoit, D.S.W. In Vivo Translation of Peptide-Targeted Drug Delivery Systems Discovered by Phage Display. Bioconjug. Chem. 2018, 29, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- McGuire, M.J.; Li, S.; Brown, K.C. Biopanning of phage displayed peptide libraries for the isolation of cell-specific ligands. Methods Mol. Biol. 2009, 504, 291–321. [Google Scholar] [PubMed] [Green Version]
- Molek, P.; Strukelj, B.; Bratkovic, T. Peptide phage display as a tool for drug discovery: Targeting membrane receptors. Molecules 2011, 16, 857–887. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Benzoni, P.; De Luca, A.; Crescini, E.; Dell’era, P. Phage displayed peptides/antibodies recognizing growth factors and their tyrosine kinase receptors as tools for anti-cancer therapeutics. Int. J. Mol. Sci. 2012, 13, 5254–5277. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Bacteriophage | Length | Size | Genome | Proteins | Displayed Copies/ Virions | Viral Life Cycle | ||
|---|---|---|---|---|---|---|---|---|
| M13 | 930 nm | 6.4 kb | ss DNA | Replication proteins | pII, pX, pV | 3–5 on pIII | 110 Kda | Lysogenic |
| Morphogenetic proteins | pI, pIV | 2700 on pVIII | 10 Kda | |||||
| Structural proteins | pIII, pVIII, pVI, pVII, pIX | |||||||
| T7 | Head 55 nm | 40 kb | ds DNA | Capsid proteins | gp10A, gp10B | Up to 1200 polypeptide | 132 Kda | Lytic |
| Inner core proteins | gp16, gp15, gp14 | |||||||
| Tail 19 nm | Connector proteins | gp8, gp6, gp7, gp11, gp12 | 1–415 peptide | |||||
| Tail protein | gp17 | |||||||
| T4 | 90 nm wide | 168 kb | ds DNA | Head protein | gp20, gp23, gp24 | 810 copies of polypeptides on SOC proteins | 710 Kda | Lytic |
| 200 nm long | Tail protein | gp15, gp13, gp14, gp18, gp19, gp34, gp35, gp36, gp9, gp10, gp11, gp12 | 155 copies of polypeptides on HOC proteins | |||||
| Lambda | Head 64 nm | 48.5 kb | ds DNA | Head protein | gpD, gpE, gpC, gpB, gpW | 405 on pD proteins | Lysogenic/ lytic | |
| Tail 150 nm | Tail protein | gpU, gpV, gpJ, gpH | 6 copies on pV proteins | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aloisio, A.; Nisticò, N.; Mimmi, S.; Maisano, D.; Vecchio, E.; Fiume, G.; Iaccino, E.; Quinto, I. Phage-Displayed Peptides for Targeting Tyrosine Kinase Membrane Receptors in Cancer Therapy. Viruses 2021, 13, 649. https://doi.org/10.3390/v13040649
Aloisio A, Nisticò N, Mimmi S, Maisano D, Vecchio E, Fiume G, Iaccino E, Quinto I. Phage-Displayed Peptides for Targeting Tyrosine Kinase Membrane Receptors in Cancer Therapy. Viruses. 2021; 13(4):649. https://doi.org/10.3390/v13040649
Chicago/Turabian StyleAloisio, Annamaria, Nancy Nisticò, Selena Mimmi, Domenico Maisano, Eleonora Vecchio, Giuseppe Fiume, Enrico Iaccino, and Ileana Quinto. 2021. "Phage-Displayed Peptides for Targeting Tyrosine Kinase Membrane Receptors in Cancer Therapy" Viruses 13, no. 4: 649. https://doi.org/10.3390/v13040649