
Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Methodology
2.1. Ethical Statement
2.2. Study Samples
2.3. Immune Response Classification
2.4. Viral RNA Isolation and Quantification
2.5. Virus Genome Deep Sequencing
2.6. Bioinformatics Data Processing
2.7. Phylogenetic Analysis
2.8. Intrahost Viral Genetic Diversity Assessment
2.9. Natural Selection Assessment
2.10 Statistical Analysis
3. Results
3.1. Samples Characteristics
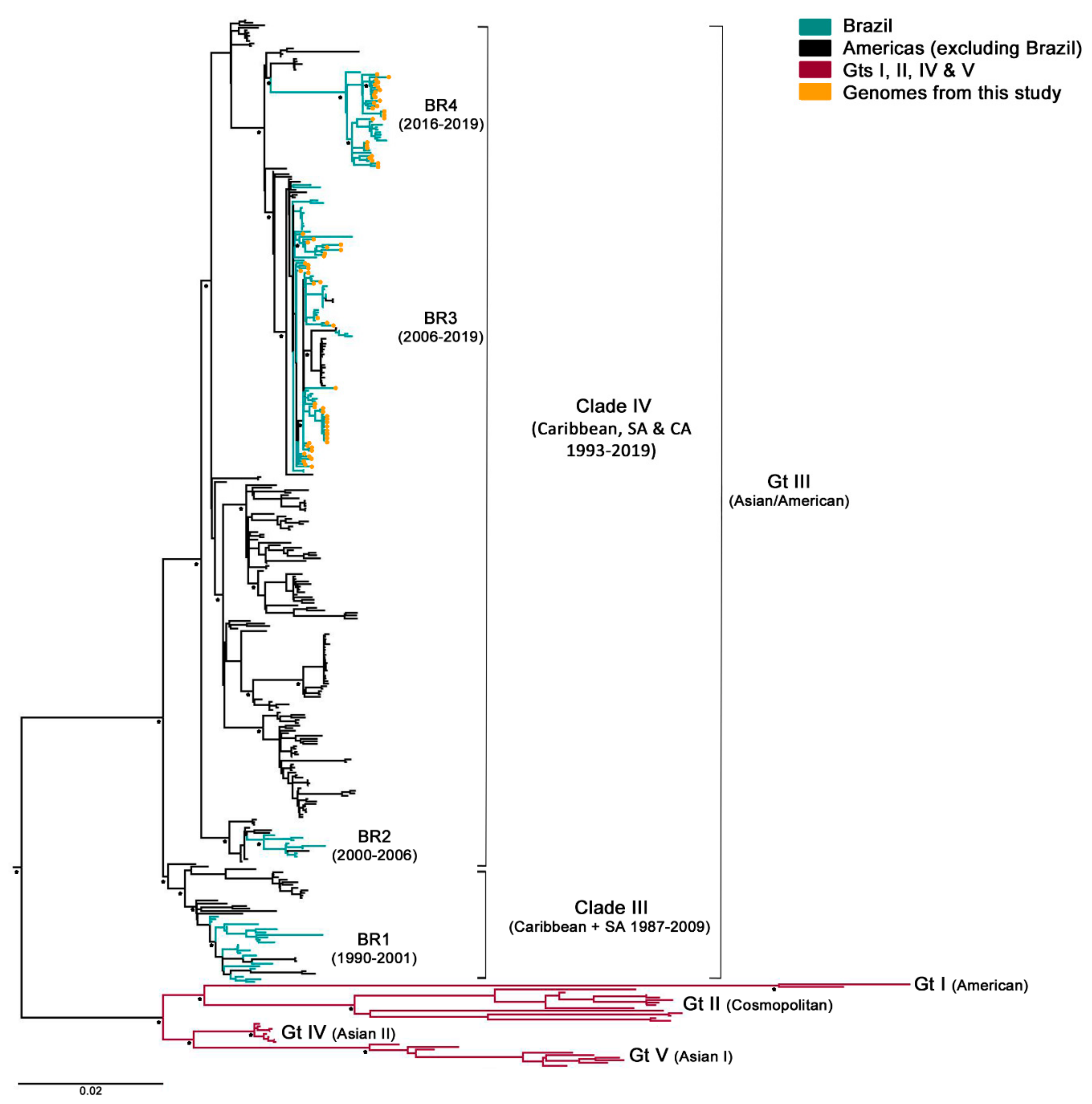
3.2. Phylogenetic Analysis
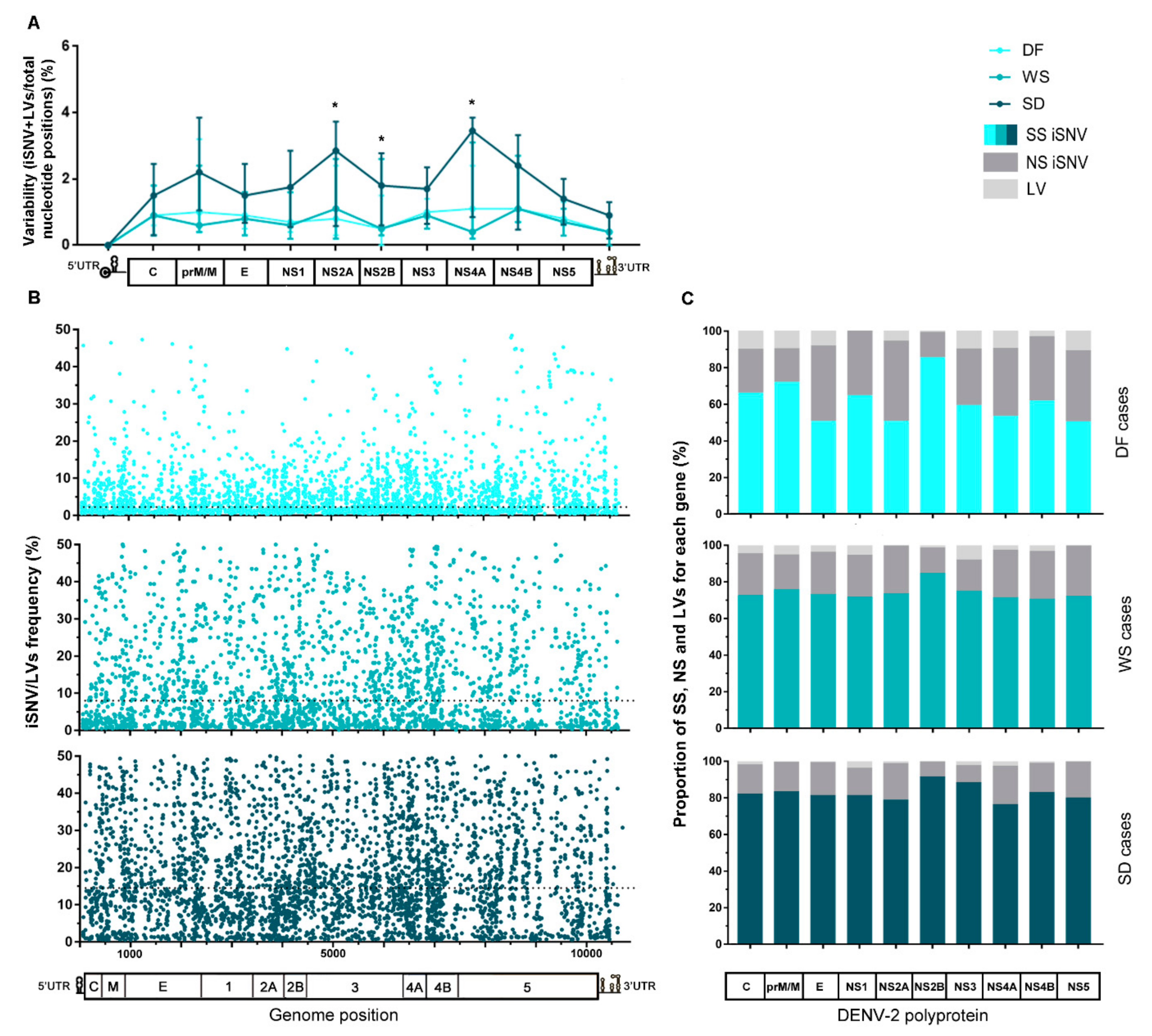
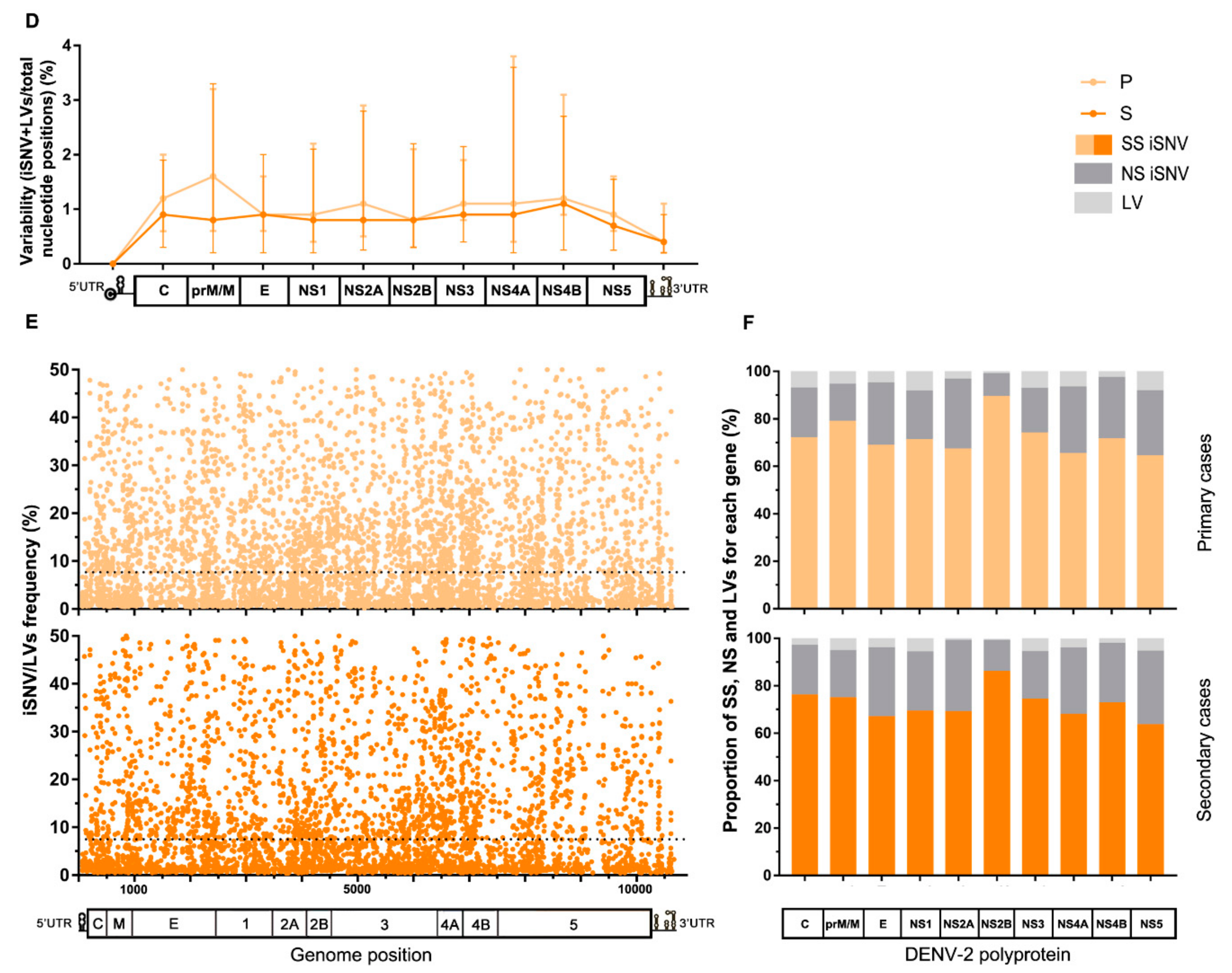
3.3. Intrahost Viral Population Structure
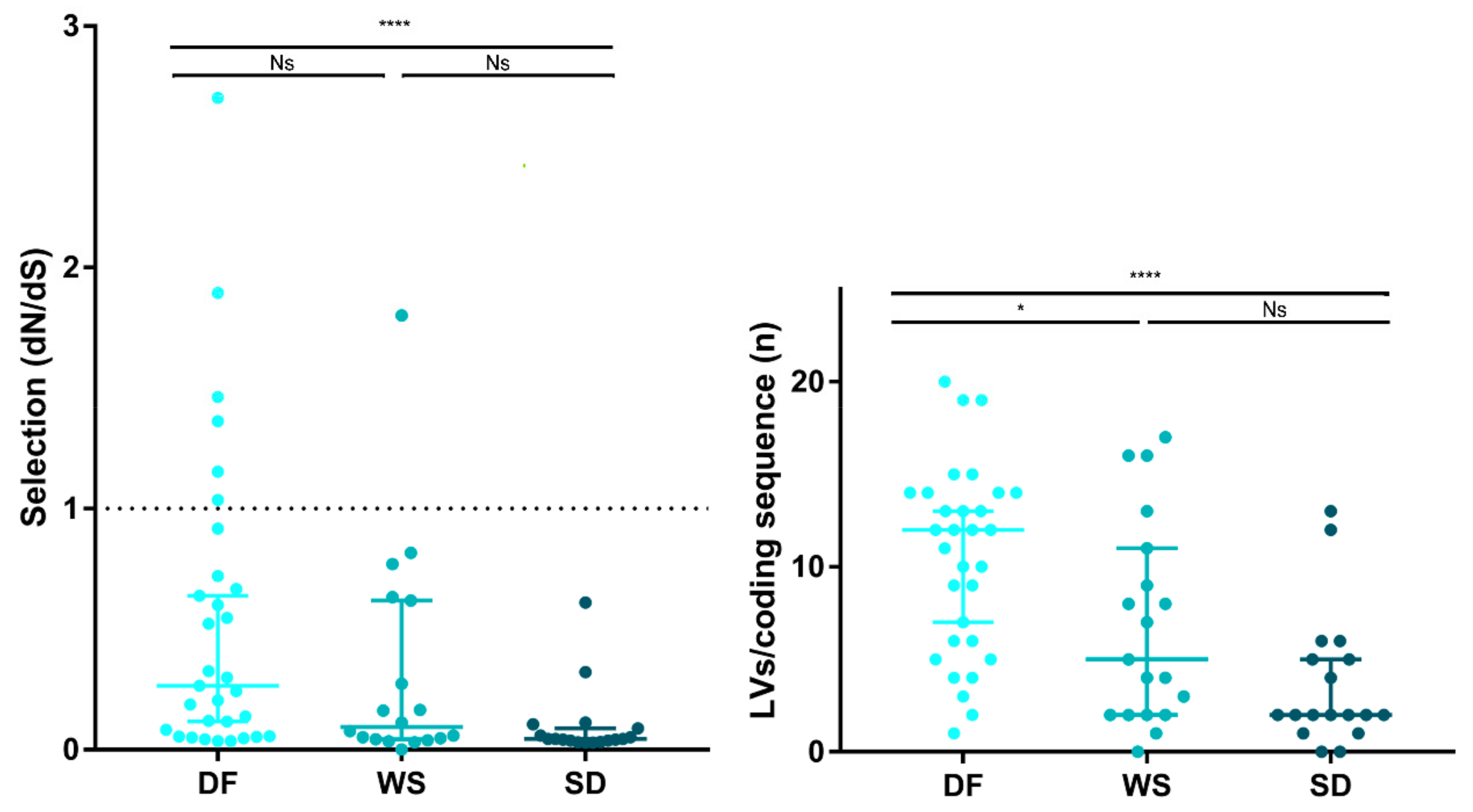
3.4. Intrahost Selective Pressure
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Dengue Guidelines for Diagnosis, Treatment, Prevention and Control. 2019. Available online: https://apps.who.int/iris/handle/10665/44188 (accessed on 17 June 2019).
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef] [Green Version]
- Rothman, A.L. Immunity to dengue virus: A tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 2011, 11, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Lindenbarch, B.D.; Thiel, H.J.; Rice, C.M. Flaviviridae: Their Viruses and Their Replication. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Publishers: Philadelphia, PA, USA, 2007; pp. 1101–1152. [Google Scholar]
- Holmes, E. The RNA Virus Quasispecies. In The Evolution and Emergence of RNA Viruses; Harvey, P.H., May, R.M., Eds.; Oxford Series in Ecology and Evolution; Oxford University Press: Oxford, UK, 2009; pp. 87–103. [Google Scholar]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef]
- Pfeiffer, J.K.; Kirkegaard, K. A single mutation in poliovirus RNA-dependent RNA polymerase confers resistance to mutagenic nucleotide analogs via increased fidelity. Proc. Natl. Acad. Sci. USA 2003, 100, 7289–7294. [Google Scholar] [CrossRef] [Green Version]
- Fitzsimmons, W.J.; Woods, R.J.; McCrone, J.T.; Woodman, A.; Arnold, J.J.; Yennawar, M.; Evans, R.; Cameron, C.E.; Lauring, A.S. A speed-fidelity trade-off determines the mutation rate and virulence of an RNA virus. PLoS Biol. 2018, 16, e2006459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.Y.; Perelson, A.S.; Park, S.C.; Leitner, T. Dynamic correlation between intrahost HIV-1 quasispecies evolution and disease progression. PLoS Comput. Biol. 2008, 4, e1000240. [Google Scholar] [CrossRef] [Green Version]
- Sullivan, D.G.; Bruden, D.; Deubner, H.; McArdle, S.; Chung, M.; Christensen, C.; Hennessy, T.; Homan, C.; Williams, J.; McMahon, B.J.; et al. Hepatitis C virus dynamics during natural infection are associated with long-term histological outcome of chronic hepatitis C disease. J. Infect. Dis. 2007, 196, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignuzzi, M.; López, C.B. Defective viral genomes are key drivers of the virus-host interaction. Nat. Microbiol. 2019, 4, 1075–1087. [Google Scholar] [CrossRef]
- Cuypers, L.; Libin, P.J.K.; Simmonds, P.; Nowé, A.; Muñoz-Jordán, J.; Alcantara, L.C.J.; Vandamme, A.M.; Santiago, G.A.; Theys, K. Time to Harmonise Dengue Nomenclature and Classification. Viruses 2018, 10, 569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, R.M.; Miagostovich, M.P.; Lampe, E.; Schatzmayr, H.G. Isolation of dengue virus type 2 in Rio de Janeiro. Mem. Inst. Oswaldo Cruz 1990, 85, 253. [Google Scholar] [CrossRef] [Green Version]
- Sistema de Vigilância em Saúde (SVS). Dengue Epidemiological Bulletin: Weeks 1 to 52 of 2009. Ministério da Saúde, Secretaria de Vigilância em Saúde. Brasília/DF. 2009. Available online: http://portal.saude.gov.br/portal/arquivos/pdf/informe_epidemiologico_semana_1a52_09_revisado.pdf (accessed on 14 June 2019).
- Nunes, P.C.; Sampaio, S.A.; da Costa, N.R.; de Mendonça, M.C.; Lima, M.R.; Araujo, S.E.; dos Santos, F.B.; Simões, J.B.; Gonçalves, B.S.; Nogueira, R.M.; et al. Dengue severity associated with age and a new lineage of dengue virus-type 2 during an outbreak in Rio De Janeiro, Brazil. J. Med. Virol. 2016, 88, 1130–1136. [Google Scholar] [CrossRef] [PubMed]
- Descloux, E.; Cao-Lormeau, V.M.; Roche, C.; De, L.X. Dengue 1 diversity and microevolution, French Polynesia 2001–2006: Connection with epidemiology and clinics. PLoS Negl. Trop. Dis. 2009, 3, e493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, K.T.; Henn, M.R.; Zody, M.C.; Tricou, V.; Nguyet, N.M.; Charlebois, P.; Lennon, N.J.; Green, L.; de Vries, P.J.; Hien, T.T.; et al. High-resolution analysis of intrahost genetic diversity in dengue virus serotype 1 infection identifies mixed infections. J. Virol. 2012, 86, 835–843. [Google Scholar] [CrossRef] [Green Version]
- Parameswaran, P.; Charlebois, P.; Tellez, Y.A.; Ryan, E.M.; Malboeuf, C.M.; Levin, J.Z.; Lennon, N.J.; Balmaseda, A.E.; Henn, M.R. Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity. J. Virol. 2012, 86, 8546–8558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Roche, R.; Blanc, H.; Bordería, A.V.; Díaz, G.; Henningsson, R.; Gonzalez, D.; Santana, E.; Alvarez, M.; Castro, O.; Fontes, M.; et al. Increasing Clinical Severity during a Dengue Virus Type 3 Cuban Epidemic: Deep Sequencing of Evolving Viral Populations. J. Virol. 2016, 90, 4320–4333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miagostovich, M.P.; Nogueira, R.M.R.; Araujo, E.S.M.; Santos, B.F.; Schatzmayr, V. Evaluation of an IgG enzyme linked immunosorbent assay for dengue diagnosis. J. Clin. Virol. 1999, 14, 183–189. [Google Scholar] [CrossRef]
- Santiago, G.A.; Vergne, E.; Quiles, Y.; Cosme, J.; Vazquez, J.; Medina, J.F.; Medina, F.; Colón, C.; Margolis, H.; Muñoz-Jordán, J.L. Analytical and clinical performance of the CDC real time RT-PCR assay for detection and typing of dengue virus. PLoS Negl. Trop. Dis. 2013, 7, e2311. [Google Scholar] [CrossRef]
- Freire, M.; Marchevsky, R.; Almeida, L.; Yamamura, A.; Caride, E.; Brindeiro, P.; Motta, M.; Nogueira, R.; Kubelka, C.; Bonaldo, M.; et al. Wild dengue virus types 1, 2 and 3 viremia in rhesus monkeys. Mem. Inst. Oswaldo Cruz 2007, 102, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Matranga, C.B.; Gladden-Young, A.; Qu, J.; Winnicki, S.; Nosamiefan, D.; Levin, J.Z.; Sabeti, P.C. Unbiased Deep Sequencing of RNA Viruses from Clinical Samples. J. Vis. Exp. 2016, 113, e54117. [Google Scholar] [CrossRef] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kiełbasa, S.M.; Wan, R.; Sato, K.; Horton, P.; Frith, M.C. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011, 21, 487–493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabherr, MG.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, D.J.; Dudas, G.; Wohl, S.; Goba, A.; Whitmer, S.L.; Andersen, K.G.; Sealfon, R.S.; Ladner, J.T.; Kugelman, J.R.; Matranga, C.B.; et al. Ebola Virus Epidemiology, Transmission, and Evolution during Seven Months in Sierra Leone. Cell 2015, 161, 1516–1526. [Google Scholar] [CrossRef] [Green Version]
- Wilm, A.; Aw, P.P.; Bertrand, D.; Yeo, G.H.T.; Ong, S.H.; Wong, C.H.; Khor, C.C.; Petric, R.; Hibberd, M.L.; Nagarajan, N. LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 2012, 40, 11189–11201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubaugh, N.D.; Fauver, J.R.; Rückert, C.; Weger-Lucarelli, J.; Garcia-Luna, S.; Murrieta, R.A.; Gendernalik, A.; Smith, D.R.; Brackney, D.E.; Ebel, G.D. Mosquitoes Transmit Unique West Nile Virus Populations during Each Feeding Episode. Cell Rep. 2017, 19, 709–718. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-Del Barrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 1, 3299–3302. [Google Scholar] [CrossRef]
- Martínez Torres, E. Dengue. Estudos Avançados 2008, 22, 33–52. [Google Scholar] [CrossRef]
- Drumond, B.P.; Mondini, A.; Schmidt, D.J.; de Morais Bronzoni, R.V.; Bosch, I.; Nogueira, M.L. Circulation of different lineages of Dengue virus 2, genotype American/Asian in Brazil: Dynamics and molecular and phylogenetic characterisation. PLoS ONE 2013, 8, e59422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jesus, J.G.; Dutra, K.R.; Sales, F.C.D.S.; Claro, I.M.; Terzian, A.C.; Candido, D.D.S.; Hill, S.C.; Thézé, J.; Torres, C.; D’Agostini, T.L.; et al. Genomic detection of a virus lineage replacement event of dengue virus serotype 2 in Brazil, 2019. Mem. Inst. Oswaldo Cruz 2020, 115, e190423. [Google Scholar] [CrossRef]
- Akey, D.L.; Brown, W.C.; Dutta, S.; Konwerski, J.; Jose, J.; Jurkiw, T.J.; Del Proposto, J.; Ogata, C.M.; Skiniotis, G.; Kuhn, R.J.; et al. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 2014, 343, 881–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotcheff, S.; Routh, A. Understanding Flavivirus Capsid Protein Functions: The Tip of the Iceberg. Pathogens 2020, 9, 42. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S.; Pierson, T.C. Molecular Insight into Dengue Virus Pathogenesis and Its Implications for Disease Control. Cell 2015, 162, 488–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.C.; Soto-Acosta, R.; Bradrick, S.S.; Garcia-Blanco, M.A.; Ooi, E.E. The 5′ and 3′ Untranslated Regions of the Flaviviral Genome. Viruses 2017, 9, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andino, R.; Domingo, E. Viral quasispecies. Virology 2016, 479, 46–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, S.; Aw, P.P.; Wilm, A.; Teoh, G.; Hue, K.D.; Nguyen, N.M.; Nagarajan, N.; Simmons, C.P.; Hibberd, M.L. Tracking Dengue Virus Intra-host Genetic Diversity during Human-to-Mosquito Transmission. PLoS Negl. Trop. Dis. 2015, 9, e0004052. [Google Scholar] [CrossRef] [Green Version]
- Grubaugh, N.D.; Weger-Lucarelli, J.; Murrieta, R.A.; Fauver, J.R.; Garcia-Luna, S.M.; Prasad, A.N.; Black, W.C., IV; Ebel, G.D. Genetic Drift during Systemic Arbovirus Infection of Mosquito Vectors Leads to Decreased Relative Fitness during Host Switching. Cell Host Microbe 2016, 19, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parameswaran, P.; Wang, C.; Trivedi, S.B.; Eswarappa, M.; Montoya, M.; Balmaseda, A.; Harris, E. Intrahost Selection Pressures Drive Rapid Dengue Virus Microevolution in Acute Human Infections. Cell Host Microbe 2017, 22, 400–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, H.Y.; Salem, G.M.; Chang, G.J.; Chao, D.Y. Application of Next-Generation Sequencing to Reveal How Evolutionary Dynamics of Viral Population Shape Dengue Epidemiology. Front. Microbiol. 2020, 11, 1371. [Google Scholar] [CrossRef]
- Gregori, J.; Perales, C.; Rodriguez-Frias, F.; Esteban, J.I.; Quer, J.; Domingo, E. Viral quasispecies complexity measures. Virology 2016, 493, 227–237. [Google Scholar] [CrossRef] [PubMed]
- León-Juárez, M.; Martínez-Castillo, M.; Shrivastava, G.; García-Cordero, J.; Villegas-Sepulveda, N.; Mondragón-Castelán, M.; Mondragón-Flores, R.; Cedillo-Barrón, L. Recombinant Dengue virus protein NS2B alters membrane permeability in different membrane models. Virol. J. 2016, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Jarabo, C.M.; Arias, A.; Baranowski, E.; Escarmís, C.; Domingo, E. Memory in viral quasispecies. J. Virol. 2000, 74, 3543–3547. [Google Scholar] [CrossRef] [Green Version]
- Briones, C.; de Vicente, A.; Molina-Paris, C.; Domingo, E. Minority memory genomes can influence the evolution of HIV-1 quasispecies in vivo. Gene 2006, 384, 129–138. [Google Scholar] [CrossRef]
- Moon, S.L.; Dodd, B.J.; Brackney, D.E.; Wilusz, C.J.; Ebel, G.D.; Wilusz, J. Flavivirus sfRNA suppresses antiviral RNA interference in cultured cells and mosquitoes and directly interacts with the RNAi machinery. Virology 2015, 485, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Syenina, A.; Vijaykrishna, D.; Gan, E.S.; Tan, H.C.; Choy, M.M.; Siriphanitchakorn, T.; Cheng, C.; Vasudevan, S.G.; Ooi, E.E. Positive epistasis between viral polymerase and the 3′ untranslated region of its genome reveals the epidemiologic fitness of dengue virus. Proc. Natl. Acad. Sci. USA 2020, 117, 11038–11047. [Google Scholar] [CrossRef] [PubMed]
- Bellone, R.; Lequime, S.; Jupille, H.; Göertz, G.P.; Aubry, F.; Mousson, L.; Piorkowski, G.; Yen, P.-S.; Gabiane, G.; Vazeille, M.; et al. Experimental adaptation of dengue virus 1 to Aedes albopictus mosquitoes by in vivo selection. Sci. Rep. 2020, 10, 18404. [Google Scholar] [CrossRef]
- Novella, I.S.; Zarate, S.; Metzgar, D.; Ebendick-Corpus, B.E. Positive selection of synonymous mutations in vesicular stomatitis virus. J. Mol. Biol. 2004, 342, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Nougairede, A.; De Fabritus, L.; Aubry, F.; Gould, E.A.; Holmes, E.C.; de Lamballerie, X. Random codon re-encoding induces stable reduction of replicative fitness of Chikungunya virus in primate and mosquito cells. PLoS Pathog. 2013, 9, e1003172. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Classification | Number of Cases [n (%)] | Gender [M: F (%)] | Age [Mean (Min–Max )] | Location [State (%)] | VL (pfu/mL) [Median (Min–Max )] | Days of Symptoms [Mean (Min–Max)] | Immune Response [n (%)] |
|---|---|---|---|---|---|---|---|
| DF | 31 (45.6%) | 15:16 (48.4–51.6%) | 39.2 (10–65) | RJ (51.6); MG (48.4) | 4.31 × 104 (2.47 × 102–8.38 × 106) | 2.2 (0–5) | p = 17 (54.8%) S = 14 (45.2%) |
| WS | 18 (27.9%) | 12:7 (63.2–36.8%) | 36.7 (8–85) | RJ (84.2); MG (10.5); SP (5.3) | 3.78 × 103 (4.68–1.32 × 106) | 2.9 (0–12) *a | p = 8 (42.1%) S = 11 (57.9%) |
| SD | 17 (26.5%) | 6:12 (33.3–66.7%) | 31.7 (7m-88) *a | RJ (77.3); SP (22.2) | 1.48 × 102 (2.86–4.94 × 105) | 7.1 (4–24) *b | p = 14 (77.8%) S = 4 (22.2%) |
| Total | 68 | 33:35 (48.5–51.5%) | 36.6 (7m-88) | RJ (67.6); MG (25.0); SP (7.4) | 1.21 × 104 (2.86–8.38 × 106) | 3.6 (0–24) * | p = 39 (57.4%) S = 29 (42.6%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres, M.C.; Lima de Mendonça, M.C.; Damasceno dos Santos Rodrigues, C.; Fonseca, V.; Ribeiro, M.S.; Brandão, A.P.; Venâncio da Cunha, R.; Dias, A.I.; Santos Vilas Boas, L.; Felix, A.C.; et al. Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes. Viruses 2021, 13, 349. https://doi.org/10.3390/v13020349
Torres MC, Lima de Mendonça MC, Damasceno dos Santos Rodrigues C, Fonseca V, Ribeiro MS, Brandão AP, Venâncio da Cunha R, Dias AI, Santos Vilas Boas L, Felix AC, et al. Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes. Viruses. 2021; 13(2):349. https://doi.org/10.3390/v13020349
Chicago/Turabian StyleTorres, Maria Celeste, Marcos Cesar Lima de Mendonça, Cintia Damasceno dos Santos Rodrigues, Vagner Fonseca, Mario Sergio Ribeiro, Ana Paula Brandão, Rivaldo Venâncio da Cunha, Ana Isabel Dias, Lucy Santos Vilas Boas, Alvina Clara Felix, and et al. 2021. "Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes" Viruses 13, no. 2: 349. https://doi.org/10.3390/v13020349