
Examination of Staphylococcus aureus Prophages Circulating in Egypt

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Prophage Prediction and Identification
2.2. Cluster Analysis
2.3. Pangenome Analysis
2.4. Gene Annotation
2.5. Phylogenetic Analyses
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lowy, F.D. Staphylococcus aureus Infections. N. Engl. J. Med. 1998, 339, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus Infections: Epidemiology, Pathophysiology, Clinical Manifestations, and Management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Hassan, A.M.; Ibrahim, O.; El Guinaidy, M. Surveillance of antibiotic use and resistance in Orthopaedic Department in an Egyptian University Hospital. Int. J. Infect. Control 2011, 7. [Google Scholar] [CrossRef]
- Boucher, H.W.; Corey, G.R. Epidemiology of Methicillin-Resistant Staphylococcus aureus. Clin. Infect. Dis. 2008, 46, S344–S349. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.A.; Sharma-Kuinkel, B.K.; Maskarinec, S.A.; Eichenberger, E.M.; Shah, P.P.; Carugati, M.; Holland, T.L.; Fowler, V.G. Methicillin-resistant Staphylococcus aureus: An overview of basic and clinical research. Nat. Rev. Microbiol. 2019, 17, 203–218. [Google Scholar] [CrossRef]
- Malachowa, N.; DeLeo, F.R. Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 2010, 67, 3057–3071. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Goerke, C.; Papenberg, S.M.Y.; Dasbach, S.; Dietz, K.; Ziebach, R.; Kahl, B.C.; Wolz, C. Increased Frequency of Genomic Alterations in Staphylococcus aureus during Chronic Infection Is in Part Due to Phage Mobilization. J. Infect. Dis. 2004, 189, 724–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahánková, J.; Pantůček, R.; Goerke, C.; Růžičková, V.; Holochová, P.; Doškař, J. Multilocus PCR typing strategy for differentiation of Staphylococcus aureus siphoviruses reflecting their modular genome structure. Environ. Microbiol. 2010, 12, 2527–2538. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Henry, T.; van Rooijen, W.J.M.; Perret, M.; Badiou, C.; Aerts, P.C.; Kemmink, J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. The Staphylococcal Toxin Panton-Valentine Leukocidin Targets Human C5a Receptors. Cell Host Microbe 2013, 13, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Vandenesch, F.; Naimi, T.; Enright, M.C.; Lina, G.; Nimmo, G.R.; Heffernan, H.; Liassine, N.; Bes, M.; Greenland, T.; Reverdy, M.-E.; et al. Community-Acquired Methicillin-Resistant Staphylococcus aureus Carrying Panton-Valentine Leukocidin Genes: Worldwide Emergence. Emerg. Infect. Dis. 2003, 9, 978–984. [Google Scholar] [CrossRef]
- Zanger, P.; Nurjadi, D.; Schleucher, R.; Scherbaum, H.; Wolz, C.; Kremsner, P.G.; Schulte, B. Import and spread of Panton-Valentine Leukocidin-positive Staphylococcus aureus through nasal carriage and skin infections in travelers returning from the tropics and subtropics. Clin. Infect. Dis. 2012, 54, 483–492. [Google Scholar] [CrossRef]
- Shallcross, L.J.; Fragaszy, E.; Johnson, A.M.; Hayward, A.C. The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: A systematic review and meta-analysis. Lancet Infect. Dis. 2013, 13, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Saeed, K.; Gould, I.; Esposito, S.; Ahmad-Saeed, N.; Ahmed, S.S.; Alp, E.; Bal, A.M.; Bassetti, M.; Bonnet, E.; Chan, M.; et al. Corrigendum to ‘Panton-Valentine Leucocidin (PVL) Staphylococcus aureus a position statement from the International Society of Chemotherapy’ (International Journal of Antimicrobial Agents 51/1 (2018) 16–25). Int. J. Antimicrob. Agents 2018, 52, 125. [Google Scholar] [CrossRef] [PubMed]
- Mccarthy, A.J.; Witney, A.A.; Lindsay, J.A. Staphylococcus aureus Temperate Bacteriophage: Carriage and Horizontal Gene Transfer is Lineage Associated. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rountree, P.M. The Serological Differentiation of Staphylococcal Bacteriophages. J. Gen. Microbiol. 1949, 3, 164–173. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.E.O.; Rippon, J.E. Bacteriophage typing of Staphylococcus Aureus. J. Hyg. 1952, 50, 320–353. [Google Scholar] [CrossRef]
- Rippon, J.E. The classification of bacteriophages lysing staphylococci. J. Hyg. 1956, 54, 213–226. [Google Scholar] [CrossRef] [Green Version]
- Goerke, C.; Pantucek, R.; Holtfreter, S.; Schulte, B.; Zink, M.; Grumann, D.; Bröker, B.M.; Doskar, J.; Wolz, C. Diversity of prophages in dominant Staphylococcus aureus clonal lineages. J. Bacteriol. 2009, 191, 3462–3468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirtz, C.; Witte, W.; Wolz, C.; Goerke, C. Transcription of the phage-encoded Panton–Valentine leukocidin of Staphylococcus aureus is dependent on the phage life-cycle and on the host background. Microbiology 2009, 155, 3491–3499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.X.; Ito, T.; Kondo, Y.; Cho, M.; Yoshizawa, Y.; Kaneko, J.; Katai, A.; Higashiide, M.; Li, S.; Hiramatsu, K. Two Different Panton-Valentine Leukocidin Phage Lineages Predominate in Japan. J. Clin. Microbiol. 2008, 46, 3246–3258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wamel, W.J.B.; Rooijakkers, S.H.M.; Ruyken, M.; van Kessel, K.P.M.; van Strijp, J.A.G. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 2006, 188, 1310–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraushaar, B.; Hammerl, J.A.; Kienöl, M.; Heinig, M.L.; Sperling, N.; Dinh Thanh, M.; Reetz, J.; Jäckel, C.; Fetsch, A.; Hertwig, S. Acquisition of virulence factors in livestock-associated MRSA: Lysogenic conversion of CC398 strains by virulence gene-containing phages. Sci. Rep. 2017, 7, 2004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Nielsen, L.N.; Hvitved, A.; Haaber, J.K.; Wirtz, C.; Andersen, P.S.; Larsen, J.; Wolz, C.; Ingmer, H. Commercial Biocides Induce Transfer of Prophage Φ13 from Human Strains of Staphylococcus aureus to Livestock CC398. Front. Microbiol. 2017, 8, 2418. [Google Scholar] [CrossRef] [Green Version]
- Borg, M.A.; de Kraker, M.; Scicluna, E.; van de Sande-Bruinsma, N.; Tiemersma, E.; Monen, J.; Grundmann, H. Prevalence of methicillin-resistant Staphylococcus aureus (MRSA) in invasive isolates from southern and eastern Mediterranean countries. J. Antimicrob. Chemother. 2007, 60, 1310–1315. [Google Scholar] [CrossRef] [Green Version]
- Falagas, M.E.; Karageorgopoulos, D.E.; Leptidis, J.; Korbila, I.P. MRSA in Africa: Filling the Global Map of Antimicrobial Resistance. PLoS ONE 2013, 8, e68024. [Google Scholar] [CrossRef] [Green Version]
- Abouelfetouh, A. The Status of Methicillin Resistance Among Egyptian Staphylococcus aureus Isolates: An Overview. Infect. Disord. Drug Targets 2017, 17, 67–69. [Google Scholar] [CrossRef]
- Mashaly, G.E.-S.; El-Mahdy, R.H. Vancomycin heteroresistance in coagulase negative Staphylococcus blood stream infections from patients of intensive care units in Mansoura University Hospitals, Egypt. Ann. Clin. Microbiol. Antimicrob. 2017, 16, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montelongo Hernandez, C.; Mores, C.R.; Putonti, C.; Wolfe, A.J.; Abouelfetouh, A. Phylogenomic study of Staphylococcus aureus and Staphylococcus haemolyticus isolates from Egypt. Microb. Genom. under review.
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. ProgressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ’omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Chang, Y.; Shin, H.; Lee, J.-H.; Park, C.J.; Paik, S.-Y.; Ryu, S. Isolation and genome characterization of the virulent Staphylococcus aureus bacteriophage SA97. Viruses 2015, 10, 5225–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, J.E.S.; Wireman, J.; Hostetler, J.; Forberger, H.; Borman, J.; Gill, J.; Sanchez, S.; Mankin, A.; Lamarre, J.; Lindsay, J.A.; et al. Major families of multiresistant plasmids from geographically and epidemiologically diverse Staphylococci. G3 Genes Genomes Genet. 2011, 1, 581–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Z.H.; Lee, C.Y. Cloning, sequencing, and genetic characterization of regulatory genes, rinA and rinB, required for the activation of staphylococcal phage phi 11 int expression. J. Bacteriol. 1993, 175, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, M.D.; Quiles-Puchalt, N.; Harwich, M.D.; Tormo-Más, M.Á.; Campoy, S.; Barbé, J.; Lasa, I.; Novick, R.P.; Christie, G.E.; Penadés, J.R. RinA controls phage-mediated packaging and transfer of virulence genes in Gram-positive bacteria. Nucleic Acids Res 2011, 39, 5866–5878. [Google Scholar] [CrossRef]
- Oliveira, H.; Sampaio, M.; Melo, L.D.R.; Dias, O.; Pope, W.H.; Hatfull, G.F.; Azeredo, J. Staphylococci phages display vast genomic diversity and evolutionary relationships. BMC Genom. 2019, 20, 357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goerke, C.; Wirtz, C.; Flückiger, U.; Wolz, C. Extensive phage dynamics in Staphylococcus aureus contributes to adaptation to the human host during infection. Mol. Microbiol. 2006, 61, 1673–1685. [Google Scholar] [CrossRef]
- Frígols, B.; Quiles-Puchalt, N.; Mir-Sanchis, I.; Donderis, J.; Elena, S.F.; Buckling, A.; Novick, R.P.; Marina, A.; Penadés, J.R. Virus Satellites Drive Viral Evolution and Ecology. PLoS Genet. 2015, 11, e1005609. [Google Scholar] [CrossRef] [PubMed]
- Haaber, J.; Penadés, J.R.; Ingmer, H. Transfer of Antibiotic Resistance in Staphylococcus aureus. Trends Microbiol. 2017, 25, 893–905. [Google Scholar] [CrossRef]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senok, A.; Ehricht, R.; Monecke, S.; Al-Saedan, R.; Somily, A. Molecular characterization of methicillin-resistant Staphylococcus aureus in nosocomial infections in a tertiary-care facility: Emergence of new clonal complexes in Saudi Arabia. New Microbes New Infect. 2016, 14, 13–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khemiri, M.; Akrout Alhusain, A.; Abbassi, M.S.; El Ghaieb, H.; Santos Costa, S.; Belas, A.; Pomba, C.; Hammami, S. Clonal spread of methicillin-resistant Staphylococcus aureus-t6065—CC5-SCC mec V-agr II in a Libyan hospital. J. Glob. Antimicrob. Resist. 2017, 10, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, J.; Kamio, Y. Bacterial Two-component and Hetero-heptameric Pore-forming Cytolytic Toxins: Structures, Pore-forming Mechanism, and Organization of the Genes. Biosci. Biotechnol. Biochem. 2004, 68, 981–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boakes, E.; Kearns, A.M.; Ganner, M.; Perry, C.; Hill, R.L.; Ellington, M.J. Distinct Bacteriophages Encoding Panton-Valentine Leukocidin (PVL) among International Methicillin-Resistant Staphylococcus aureus Clones Harboring PVL. J. Clin. Microbiol. 2011, 49, 684–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Hu, F.; Jin, S.; Xu, X.; Zou, Y.; Ding, B.; He, C.; Gong, F.; Liu, Q. Typing of Panton-Valentine Leukocidin-Encoding Phages and lukSF-PV Gene Sequence Variation in Staphylococcus aureus from China. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chavda, K.D.; Solanki, M.; Mediavilla, J.R.; Mathema, B.; Schlievert, P.M.; Kreiswirth, B.N. Genetic Variation among Panton-Valentine Leukocidin-Encoding Bacteriophages in Staphylococcus aureus Clonal Complex 30 Strains. J. Clin. Microbiol. 2013, 51, 914–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coombs, G.W.; Baines, S.L.; Howden, B.P.; Swenson, K.M.; O’Brien, F.G. Diversity of bacteriophages encoding Panton-Valentine leukocidin in temporally and geographically related Staphylococcus aureus. PLoS ONE 2020, 15, e0228676. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | No. Strains with Intact Prophages | No. Intact Prophages | Average No. Prophages/Strain | Max No. Prophages/Strain |
|---|---|---|---|---|
| Medical Microbiology Laboratory at AMUH isolates (n = 56) | 45 | 71 | 1.5 | 3 |
| Other MRSA isolates from Egypt (n = 17) | 8 | 16 | 2 | 3 |
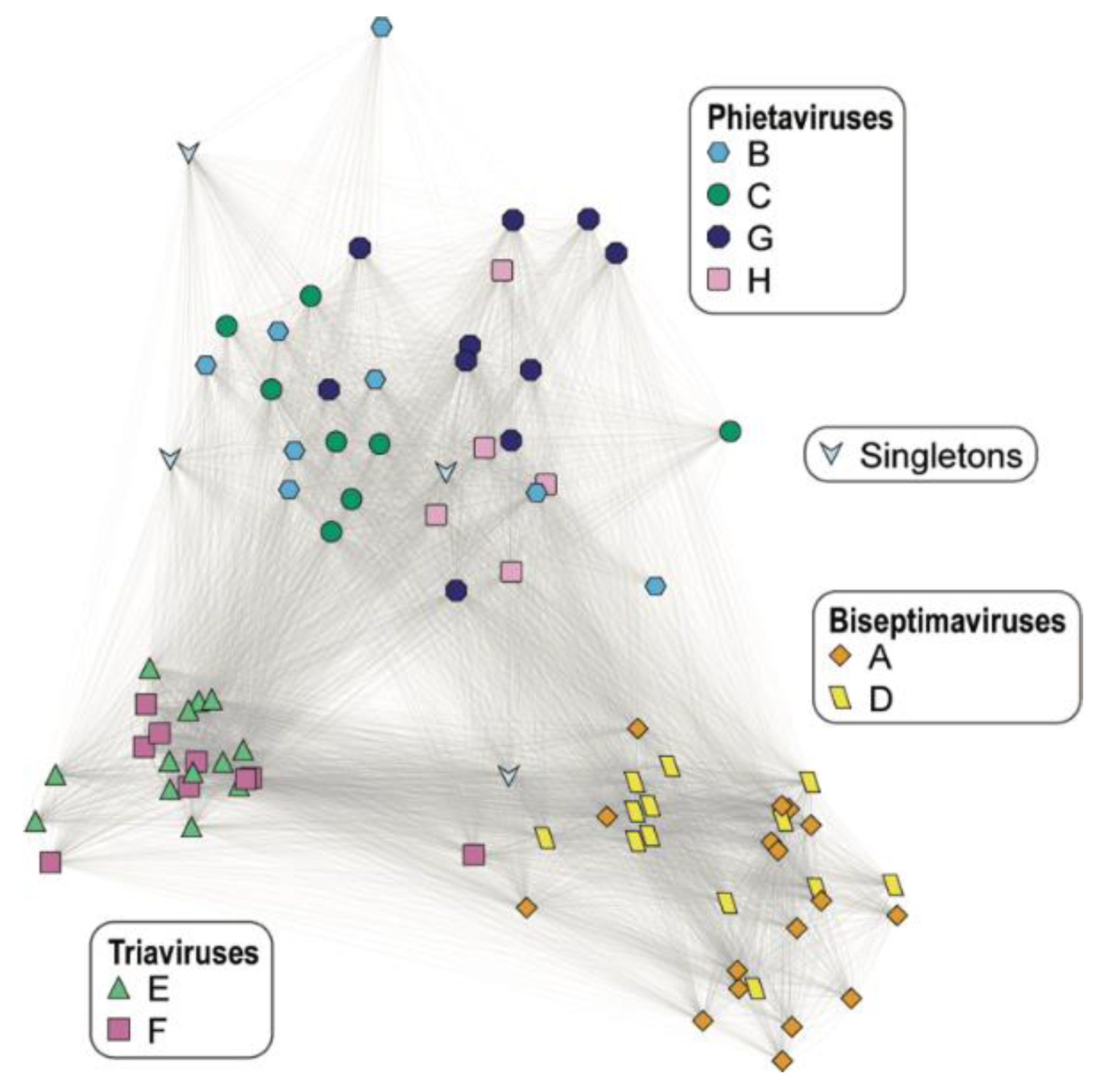
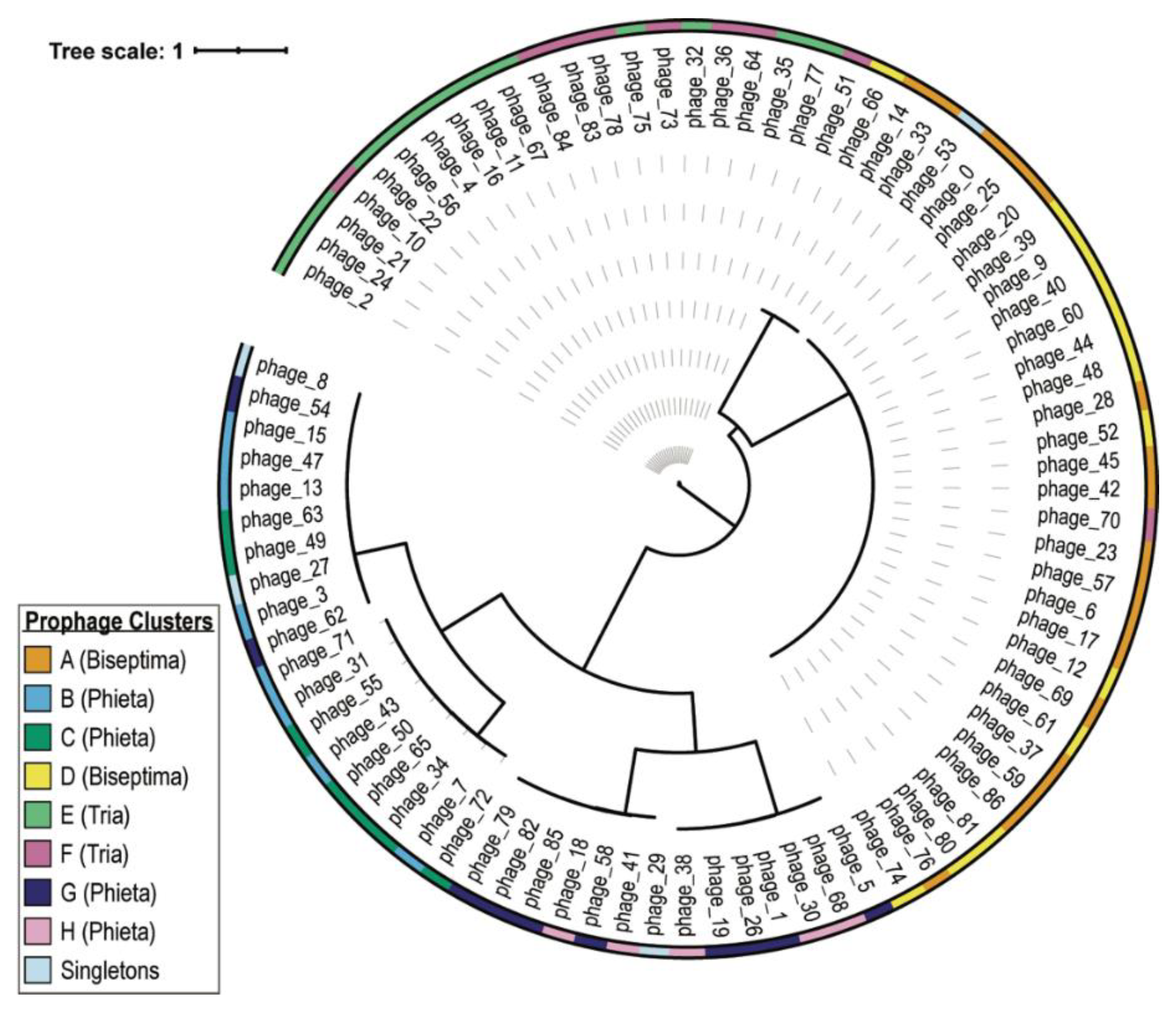
| Cluster ID | Cluster Size | ANI Score Range (%) | Predicted Genus |
|---|---|---|---|
| A | 17 | 87.93–100 | Biseptimavirus |
| B | 8 | 73.29–100 | Phietavirus |
| C | 8 | 82.92–99.86 | Phietavirus |
| D | 13 | 91.12–100 | Biseptimavirus |
| E | 13 | 96.09–100 | Triavirus |
| F | 9 | 88.39–100 a | Triavirus |
| G | 10 | 77.01–100 | Phietavirus |
| H | 5 | 92.72–97.19 | Phietavirus |
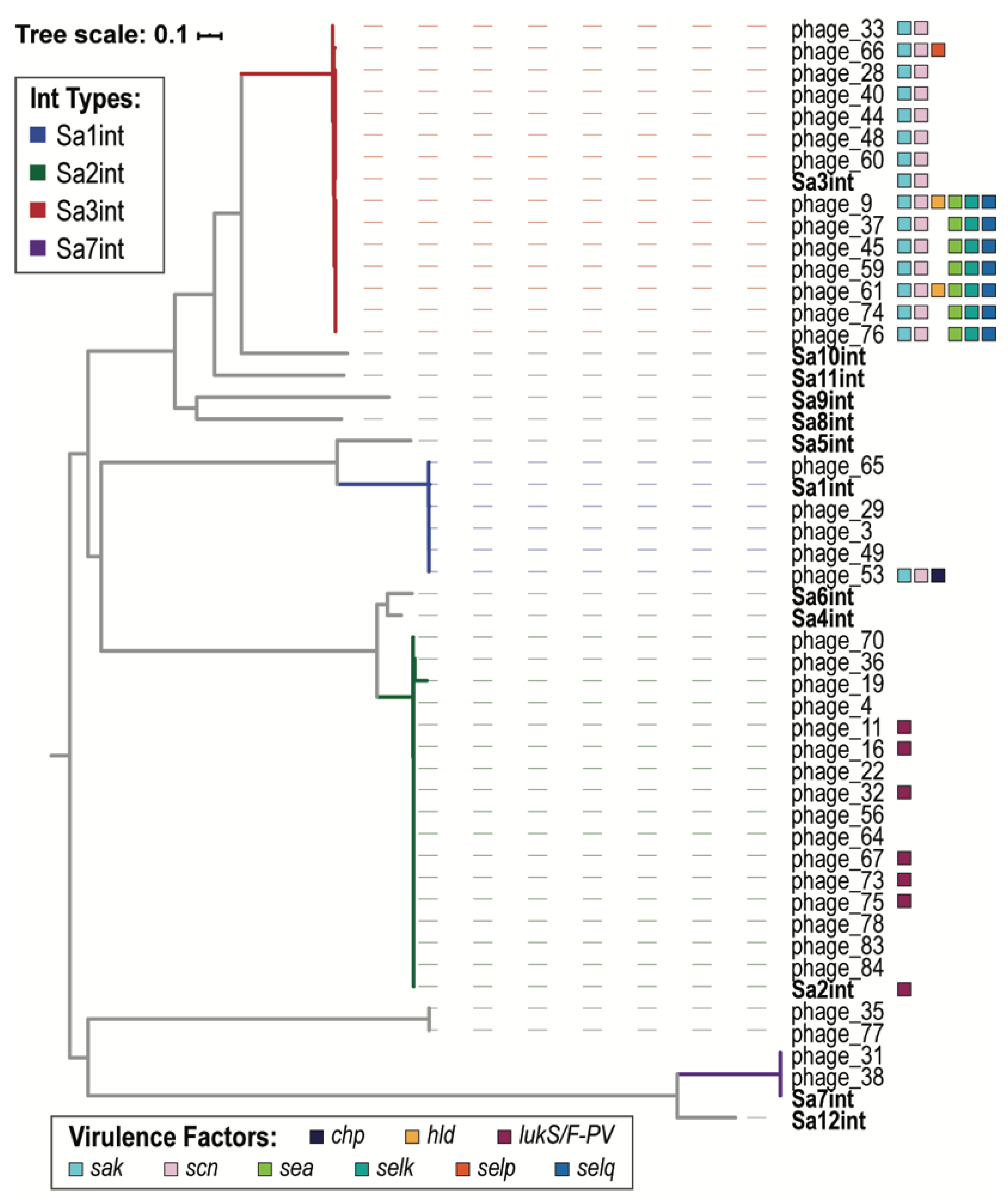
| VF Class | Virulence Factors | Related Genes | No. Prophages |
|---|---|---|---|
| Enzyme | Serine protease | splA | 2 |
| splB | 2 | ||
| splC | 2 | ||
| splD | 1 | ||
| splE | 1 | ||
| splF | 1 | ||
| Staphylokinase | sak | 30 | |
| Immune Evasion | CHIPS | chp | 1 |
| SCIN | scn | 30 | |
| Toxin | Delta hemolysin | hld | 2 |
| Enterotoxin A | sea | 17 | |
| Enterotoxin G | seg | 1 | |
| Enterotoxin I | sei | 1 | |
| Enterotoxin Yent1 | yent1 | 1 | |
| Enterotoxin Yent2 | yent2 | 1 | |
| Enterotoxin-like K | selk | 7 | |
| Enterotoxin-like M | selm | 1 | |
| Enterotoxin-like N | seln | 1 | |
| Enterotoxin-like O | selo | 1 | |
| Enterotoxin-like P | selp | 1 | |
| Enterotoxin-like Q | selq | 7 | |
| Gamma hemolysin | hlgA | 3 | |
| Leukotoxin D | lukD | 3 | |
| Leukotoxin E | lukE | 3 | |
| Panton-Valentine leukocidin | lukF-PV | 11 | |
| lukS-PV | 11 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ene, A.; Miller-Ensminger, T.; Mores, C.R.; Giannattasio-Ferraz, S.; Wolfe, A.J.; Abouelfetouh, A.; Putonti, C. Examination of Staphylococcus aureus Prophages Circulating in Egypt. Viruses 2021, 13, 337. https://doi.org/10.3390/v13020337
Ene A, Miller-Ensminger T, Mores CR, Giannattasio-Ferraz S, Wolfe AJ, Abouelfetouh A, Putonti C. Examination of Staphylococcus aureus Prophages Circulating in Egypt. Viruses. 2021; 13(2):337. https://doi.org/10.3390/v13020337
Chicago/Turabian StyleEne, Adriana, Taylor Miller-Ensminger, Carine R. Mores, Silvia Giannattasio-Ferraz, Alan J. Wolfe, Alaa Abouelfetouh, and Catherine Putonti. 2021. "Examination of Staphylococcus aureus Prophages Circulating in Egypt" Viruses 13, no. 2: 337. https://doi.org/10.3390/v13020337