
Lentiviral Vector Bioprocessing


1
The Advanced Centre for Biochemical Engineering, Department of Biochemical Engineering, University College London, Gower St, London WC1E 6BT, UK
2
Division of Advanced Therapies, National Institute for Biological Standards and Control, South Mimms EN6 3QG, UK
*
Author to whom correspondence should be addressed.
†
Current address: Autolus Therapeutics, London W12 7FP, UK.
Viruses 2021, 13(2), 268; https://doi.org/10.3390/v13020268
Submission received: 7 January 2021
/
Revised: 2 February 2021
/
Accepted: 3 February 2021
/
Published: 9 February 2021
(This article belongs to the Special Issue Lentiviral Vectors)
Abstract
:Lentiviral vectors (LVs) are potent tools for the delivery of genes of interest into mammalian cells and are now commonly utilised within the growing field of cell and gene therapy for the treatment of monogenic diseases and adoptive therapies such as chimeric antigen T-cell (CAR-T) therapy. This is a comprehensive review of the individual bioprocess operations employed in LV production. We highlight the role of envelope proteins in vector design as well as their impact on the bioprocessing of lentiviral vectors. An overview of the current state of these operations provides opportunities for bioprocess discovery and improvement with emphasis on the considerations for optimal and scalable processing of LV during development and clinical production. Upstream culture for LV generation is described with comparisons on the different transfection methods and various bioreactors for suspension and adherent producer cell cultivation. The purification of LV is examined, evaluating different sequences of downstream process operations for both small- and large-scale production requirements. For scalable operations, a key focus is the development in chromatographic purification in addition to an in-depth examination of the application of tangential flow filtration. A summary of vector quantification and characterisation assays is also presented. Finally, the assessment of the whole bioprocess for LV production is discussed to benefit from the broader understanding of potential interactions of the different process options. This review is aimed to assist in the achievement of high quality, high concentration lentiviral vectors from robust and scalable processes.
| Contents | ||
| 1 | Introduction · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 2 |
| 2 | Bioprocessing of Lentiviral Vectors · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 2 |
| 2.1 Pseudotyped Envelope Proteins · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 4 | |
| 3 | Upstream Bioprocessing of Lentiviral Vectors · · · · · · · · · · · · · · · · · · · · · · · | 8 |
| 3.1 Cell Lines for LV Production · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 8 | |
| 3.2 Transient, Stable and Induced Production · · · · · · · · · · · · · · · · · · · · · · | 8 | |
| 3.3 Upstream Culture to Produce Lentiviral Vectors · · · · · · · · · · · · · · · · | 11 | |
| 3.3.1 Adherent Culture · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 12 | |
| 3.3.2 Suspension Culture · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 13 | |
| 3.3.3 Perfusion Culture · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 14 | |
| 3.3.4 Cell Media and Supplements · · · · · · · · · · · · · · · · · · · · · · · · · · · | 14 | |
| 4 | Downstream Processing of Lentiviral Vectors · · · · · · · · · · · · · · · · · · · · · · | 15 |
| 4.1 Vector Filtration: Initial Clarification · · · · · · · · · · · · · · · · · · · · · · · · · | 15 | |
| 4.2 Vector Filtration: Sterile Filtration · · · · · · · · · · · · · · · · · · · · · · · · · · · | 17 | |
| 4.3 Non-Chromatographic Purification · · · · · · · · · · · · · · · · · · · · · · · · · · | 17 | |
| 4.4 Nucleic Acid Reduction · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 18 | |
| 4.5 Chromatographic Purification · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 18 | |
| 4.5.1 Anion Exchange Chromatography · · · · · · · · · · · · · · · · · · · · · · | 22 | |
| 4.5.2 Affinity Chromatography · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 23 | |
| 4.5.3 Size Exclusion Chromatography · · · · · · · · · · · · · · · · · · · · · · · · | 23 | |
| 4.5.4 Steric Exclusion Chromatography · · · · · · · · · · · · · · · · · · · · · · · | 24 | |
| 4.6 Concentration and Buffer Exchange by Tangential Flow Filtration · | 24 | |
| 4.7 Formulation · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 28 | |
| 5 | Vector Characterisation and Quality Control · · · · · · · · · · · · · · · · · · · · · · · | 29 |
| 6 | Whole-Bioprocess Assessment of LV Production · · · · · · · · · · · · · · · · · · · · | 31 |
| 7 | Conclusions · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 34 |
| References | · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · · | 35 |
1. Introduction
Lentiviral vectors (LV) are commonly used in cell and gene therapies for the transfer and integration of transgenes of interest into recipient cells for therapeutic benefit [1]. As vectors, they are capable of transducing dividing and non-dividing cells such as neurons, haematopoietic stem cells and those of the immune system, notably T-cells, delivering transgenes of up to 11 kilobases (kb) in size. LVs represent a major vector of interest for the treatment of monogenic diseases and adoptive cell therapy trials where gene delivery is required, being present in 57% of ex vivo UK Advanced Therapy Medicinal Products (ATMP) [2]. Over 100 ongoing clinical trials in the US, China, EU and Canada are employing lentiviral vectors both for ex vivo modification of cells or in vivo therapy [3]. Overall, the market for LV production is predicted to grow up to $800 M by 2026 [4] as a result of its popularity in clinical trials and the market approval of recent CAR-T therapies, Kymriah and Yescarta.
With continued interest in lentiviral vectored-therapies, demand for efficient LV bioprocessing is growing. Problems during scale-up and production could delay the adoption of lentiviral vectors for clinical and commercial use. Some bioprocessing challenges encountered today are the inability to produce sufficient titres in the upstream coupled with generally low recoveries during downstream processing, resulting in many companies unable to provide enough capacity to satisfy demand at scale [5]. Despite the current issues with developing suitable quantities of vectors, the applications of viral vectors and their bioprocessing is a valuable enterprise. Considering that only the transgene needs to be changed to pivot to another product, the rise of a universal production process is likely. This can be in the form of a packaging cell line whereby the cell constitutively expresses vector components and an envelope protein of choice, awaiting a suitable transfer cassette for stable or transient expression [6]. For established platforms, a producer cell line may be valuable, whereby the cell constitutively expresses all components relevant to vector generation [7]. Such cell lines lend favourable commercial properties in theory, due to the lack of plasmid DNA and transfection step required. Once the optimised upstream and downstream are designed, the viability of developing a platform for rapid transgene exchange and validation is high. Intensified and continuous processing, such as that seen in recombinant protein production, may be beneficial for cost effective vector production.
2. Bioprocessing of Lentiviral Vectors
Lentiviral vectors are unique as a result of their physiological and physico-chemical characteristics. LVs are typically based on HIV-1 and share many of its features, such as its spherical shape at 80–120 nm in diameter [8], capsid core and functional enzymes with an envelope derived from the host cell membrane. The innate complexity and sensitivity of the LV particles impose challenges during processing such as particle thermostability (e.g., half life in the range of 7–8 h at 37 °C [9]), sensitivity to freeze–thaw cycles [10,11] requiring rapid processing, salt [8], pH [9,12], shear [13] and buffer osmolarity [14]. Here, the loss of transduction capability due to non-packaged vector or loss of enzyme function or damaged/absent envelope proteins, is problematic as defective particles can co-purify alongside functional vector [9], reducing process capacity. Furthermore, the producer cell type and their viability during production have been implicated in vector stability [15] and thus cellular health must be maintained for high yields.
The development of lentiviral vectors from the wild type virus has required the evaluation of the viral genome and the selection of viral components to ensure safe and efficient gene transfer. The HIV genome is organised into trans elements that code for functional, structural and accessory proteins, such as Gag-Polypeptide, envelope protein and functional enzymes, whilst non-coding cis elements such as long-terminal repeats (LTR) assist in the transcription and packaging of the viral RNA genome into viral particles and indicates the regions for reverse-transcription and integration into the recipient cell [16,17]. As a result, the initial development of lentiviral vectors has seen the separation of cis- and trans-acting elements of HIV-1 onto separate plasmids with the exception of HIV-1 envelope, which was pseudotyped with an alternative envelope protein such as murine leukaemia virus (MLV) amphotropic envelope protein or VSV-G on a third plasmid [1]. Subsequent generations have incorporated successive improvements to enhance titre and safety with removal of unnecessary viral sequences, such as the accessory proteins vif, vpr, vpu and nef [18]. In the modern third generation packaging system, tat was removed and a constitutive promoter replaced U3 in the 5’ LTR of the transfer plasmid to allow transcription without Tat for improved biosafety [19]. To further maintain safety and maintain vector titres, rev was provided on an additional plasmid [20]. Therefore, current practice features 3–4 plasmids, with a self-inactivating transfer plasmid containing a transgene of interest flanked by HIV-1 LTRs with a rev response element. This is co-transfected with a packaging plasmid containing the essential trans element Gag-Pol, a third envelope plasmid containing an envelope protein, and a fourth for HIV-1 rev if not included elsewhere.
The requirements of LV processing vary with application, but high titres and purity is of main importance. Most impurities in vector production will be that of residual DNA, transfection reagents, host cell proteins and media components (see Table 1). With cell therapies, where the final product is transduced cells, the purity profiles of the vector are less stringent than that for in vivo therapies as Quality Assurance/Quality Control (QA/QC) requirements for product release lie at the cell stage. For in vivo therapies, the requirements will be pertaining to the vector itself. Thus, for an in vivo therapy process, the critical quality attributes would be weighted towards quality and concentration, whereas ex vivo would be quality and yield [21].
The scale of production depends on the number of expectant patients, the quantity of cells needed to be transduced and which multiplicity of infection (MOI) is suitable, inclusive of repeated doses, potency, disease and whether delivery is in vivo or ex vivo. Ideally, MOI should be kept low to ensure single transgene integrations to limit the risk of cytotoxicity and tumorigenic potential [22,23,24]. Ultimately, there is no apparent fixed titre of vector required, but annual transducing units (TU) from manufacturing processes can range from to TU depending on patient numbers and dosing [25]. The sensitivity of autologous cell therapies to scalable viral production is indicated in some cost of goods analysis, with up to 26% contribution to cost when viral titre is poor [26]. Likewise, cost per dose is further shown to be dependent on titre at harvest and the production method employed [27].
2.1. Pseudotyped Envelope Proteins
LVs are commonly pseudotyped whereby the envelope protein of the vector is exchanged with that of another virus (see Table 2), in effect, enrobing the viral particle with proteins from other viruses [28,29] which provide varying characteristics to the vector, affecting its tropism and intended cell target as well as possibly impacting success in bioprocessing. When transducing a cell, the envelope protein must contact and bind to a surface receptor on the recipient cell. The choice of envelope protein, its frequency on the vector surface and the availability of the receptor are crucial for efficient transduction whilst restricting off-target interactions. In LV bioprocessing, the envelope proteins must be suitably expressed in producer cells and incorporated into the vector. As the proteins belong to the vector’s surface, they interact directly with the bulk media and are therefore affected by physico-chemical conditions applied during processing such as shear forces, salt concentrations and pH. As a result, the selection of envelope protein is important for vector design in the function of the vector and its intended target but also on how the vector is processed in terms of its initial expression during upstream processing, its effect on producer cells and its impact during unit operations in downstream processing.
Many viral envelope proteins can be pseudotyped onto LV particles. However, the most commonly pseudotyped envelope protein, and widely considered the gold standard, is the VSV-G glycoprotein from the vesicular stomatitis virus Indiana. This protein is routinely used in part due to its broad tropism, interacting with low-density lipoprotein receptors (LDL-R) which are ubiquitous in most cell types [68,69]. However, from a vector design perspective VSV-G has the disadvantage of displaying cytotoxicity, resulting in cell instability in LV producer cell lines when highly expressed [13] and limiting its application to transient transfection modalities. Although stable expression of VSV-G has been illustrated [37], it is likely the effect of vector superinfection or reduced expression resulting in poor results. While inducible promoters for VSV-G may avoid cytotoxicity from constitutive expression as described in [70], this may be problematic during processing due to the addition or removal of inducing agents (see Section 3.2). VSV-G LVs are typically applied in ex vivo therapies due to their inactivation by complement in human serum [71]. Additional immune responses against VSV-G are apparent in vivo [72] which can inhibit subsequent LV administration by adaptive immune responses [73]. Moreover, despite the ubiquity of LDL-R, clinically valuable resting lymphocytes for CAR-T therapies have particularly low expression of the receptor, and thus transduction is inefficient unless activated prior [74]. However, due to the high functional titres VSV-G provides, its application is widespread and is a common envelope protein used.
Envelope proteins are impacted by physico-chemical conditions that need to be considered in downstream processing. LVs with VSV-G envelopes have been observed to inactivate when pH diverges from pH 7 [9], and when treated with 1 M NaCl, where 50% of functional titre is lost within 1 h [8]. The implications for vector processing are particularly pronounced in ultracentrifugation, whereby LVs with the envelope protein VSV-G outperform those with influenza envelope proteins [75] or HIV-1 envelopes over VSV-G [13,76] and appear to be resistant to shear [13,76,77]. Robustness of envelope proteins during processing will need to be considered and to account for discrepancies across vector envelope stability.
Other envelope proteins have been investigated and applied for LV pseudotyping. The glycoprotein Cocal-G deriving from the same vesiculovirus family as VSV shares 71.5% of amino acids sequences to VSV-G, and therefore has similar characteristics [36]. Transduction by Cocal-G enveloped LVs has been inhibited by soluble LDL-R, similar to VSV-G, and is therefore likely to infect via the same LDL-R or similar receptors [36,68]. Unlike VSV-G, Cocal-G is not inactivated by human serum and can be expressed constitutively allowing the potential for stable expression in vector producing cell lines [36,78], although this does lead to superinfection and cell instability [37]. Additional vesiculovirus family glycoproteins have been examined for stable producer cell line generation, such as those from PIRY, Chandipura and VSV-New Jersey, which displayed titres of to TU/mL and robustness during concentration and freeze–thaw [37,79]. In addition, the viral envelope protein RD114 has been applied in vector production. Deriving from a feline endogenous retrovirus, the RD114 envelope shows less cytotoxicity, is not inactivated by complement and allows for usage in stable producer cell lines [30,32,80,81]. These envelope proteins are capable of transducing CD34+ cells and show robustness during ultracentrifugation with 50–70% yield with 100–200-fold concentration [30,82,83]. RD114 has further been modified, with the insertion at the R peptide cleavage site of the HIV matrix/capsid cleavage sequence, thus giving rise to the RDPro envelope protein, which has shown a log order higher titre in transient production compared to RD114 [34,84]. These RD114 derived envelope proteins further prevent the action of superinfection, whereby a produced vector is blocked from transducing the cell line it originated from [37] and has found application in the transduction of lymphocytes for CAR-T therapy [85,86] and CD34+ progenitor cells [35,87]. Other envelopes, such as Sendai, require trypsin treatment to activate the envelope by protein cleavage [88,89], although the supplementation of an additional unit operation will need to be considered. Moreover, Sendai activated envelopes perform worse during ultracentrifugation, dropping to 20% recovery compared to 50% of non-cleaved envelope [11].
With pseudotyping of viral vector, theoretically, any viral component that is expressed on a cell surface can be used. However, such methods rely on viral proteins, which can raise concerns on safety and development costs particularly on screening. A method that has been applied is “plug and play” envelope proteins, whereby LVs are targeted to specific cell types by covalently bonding cell targeting proteins onto their surface via a disulphide bond [90] or by biotinylated targeting ligands [91]. Such work may improve the possible targeting mechanism for vectors, although this may require additional unit operations during processing to bond targeting components fully.
3. Upstream Bioprocessing of Lentiviral Vectors
The focus of upstream processing is to mass produce LV in bulk while allowing efficient downstream purification. There are several important factors to manage which are dependent on the production mode, either by transient transfection of cells by plasmids or using a stable or inducible producer cell line. A fundamental factor is the scalable expansion of cells, their viability and achieving suitable cell densities for optimal LV generation. Therefore, the type of cell line, either adherent or suspension, the bioreactor for expansion, the type of cell media with suitable supplementation and the method of transient transfection if applicable are important factors to consider.
3.1. Cell Lines for LV Production
A suitable cell line is required to produce LV particles at a high titre. Commonly, the cell line of choice is the human embryonic kidney cell 293 (HEK-293) [92], specifically the derived 293T line [93]. HEK-293Ts have the SV40 T-antigen, which is implicated in inhibiting p53 [94] and preventing the activation of the intracellular innate immune response, and it has been shown to boost titres of LVs [95]. This latter cell line has shown several advantages over its precursors, notably a shorter doubling time, higher transfection efficiencies and higher vector titre [96,97] as well as adaptation to suspension culture [98]. The ability to grow dense cultures of cells is highly beneficial for high vector titres, as each cell is theoretically a production unit for LV [99]. This however requires a degree of optimisation in terms of the overall process, as high cell density is tied with lower cell viabilities, which will contribute to greater masses of contaminants to be removed during processing. This caveat may negate any benefits to titre that greater cell numbers provide [100].
3.2. Transient, Stable and Induced Production
The dominant LV production mode is the transient transfection of cells with a variety of plasmids for LV expression and packaging. Initially developed to minimise the risk of replication competent vectors, this method has seen generational development to improve yield and safety. Plasmids are co-transfected with chemical transfection agents, and typically give titres in the range of to TU/mL non-purified [97], although bigger transgenes give lower titres [101].
Despite this, contamination of the feed with residual plasmid DNA and transfection reagent is of concern and removal of these are necessary [102,103,104]. Media exchange post-transfection is normally mandatory due to cytotoxicity issues by transfection agents, requiring additional equipment for pumps and tanks to mix plasmid DNA, transfection reagents and media, in addition to increasing overall media consumption [105]. Moreover, many factors can affect the efficiency of transfection, including plasmid concentration, reagent:DNA ratio, cell density, incubation time, mixing regime, temperature and pH [106,107], some of which are challenging on scale up. As a result, significant batch-to-batch variability with vector titre is apparent [99]. The cost of clinical grade plasmids and transfection agents adds significant expense to a production run, with at least 1µg plasmid DNA per million cells being used typically [99].
There are a multitude of transfection reagents available for LV production, the choice of which will drastically affect the processing step and cost of goods for a production run. Calcium phosphate (CaPi) is an economical transfection reagent commonly used at lab or small scale that entails the formation of fine precipitates of calcium-phosphate and DNA upon mixing calcium chloride and DNA with HEPES buffered saline. The precipitates settle and are endocytosed by cells [1,108]. However, this method is difficult to scale due to the mixing of two buffers to produce consistent precipitates; with variables such as component concentrations, temperature and aggregation time requiring optimising [109]. The process is further complicated due to slight variations in pH which negatively affect transfection efficiency [110]. In addition, CaPi transfection may cause cytotoxicity in cells without the protective action of serum or albumin in the cell culture [111,112].
Cationic lipids such as lipofectamine have also been used [113]. They form liposomes that entrap plasmid DNA by complexing with its negative charge. The liposomes fuse with the cell membrane releasing its contents for expression. Although capable of transfecting many cell types, lipofectamine is cytotoxic and can lower viability [114] and thus media exchanges are required after application [115], and are therefore typically used only at development scale.
Polyethylenimine (PEI) is more suited to scalable transfection and offers an efficient and cost-effective choice by forming polyplexes with DNA that are endocytosed by cells [106,116]. The formation and degradation of polyplexes is a function of time, and therefore there is an optimal window for use. Despite this, PEI is considered simpler than CaPi and does not require as strict controls for optimal transfection whilst also using less plasmid DNA [117]. Recombinant baculoviruses have also been used as transfection reagents to transfer viral components [118,119], although this may incur greater costs to separate baculovirus from lentiviral vector in downstream processing (DSP). Regardless of method, vector production typically peaks within 48hr post-transfection and declines from 72 h onwards providing a limited time window for harvesting. Ideally, the vector population would be of similar quality, although, due to variable transfection efficiencies on a per cell scale and their impact on viability, the vector population is typically heterologous [15].
An alternative to transient transfection can be found in stable producer or packaging cell lines (see Table 3). In these, the viral components are constitutively expressed within the cell and do not require additional plasmids and transfection reagents, except for packaging which only requires the transfer plasmid [6]. This simplifies the upstream culture and avoids the additional cost and contaminant load from residual plasmid. Moreover, stable cell line producers may generate a greater quality of particles, due to the clonal source of production allowing near homologous particles to be produced. Furthermore, such harvests tend to be cleaner [120], likely due to less envelope-based vesicles present, in addition to absent plasmid and transfection reagent load. Such stable cell lines have the potential to offer a cost effective, scalable and reproducible vector run with less batch to batch variability as each cell would maintain consistent productivity.
High performing stable producers have been created via genome tagging, screening for active loci and replacing it with lentiviral components [127,128]. Cell lines such as WinPac [7] and LentiPro26 [121], which provide titres in the TU/mL/day range, have been developed with the former utilising a retroviral tagging and recombinase mediated cassette exchange for high Gag-Pol expression and the latter applying a less active mutated viral protease to maintain cell viability and improve titres. These stable producer cell lines can be arduous to develop, requiring the isolation and evaluation of individual clones for component expression that require culturing in selection antibiotics to maintain titres. Efforts have been developed to streamline this process such as the use of bacterial artificial chromosomes which incorporate all vector components on a singular construct [129,130,131] or developing high-throughput screening methods such as the co-culturing of singular isolated producer cells encoding vectors with partial GFP fragments and cells expressing the complementary GFP fragment and monitoring for GFP reconstitution to identify high producers [132]. However, critically, stable producer cells offer titres lower than those of transient led methods, and therefore their adoption is problematic. This is particularly due to the cytotoxic effects that viral protein expression can have on cells, such as the protease, which has been linked to cleaving pro-apoptotic proteins [133], and the limits on choice of envelope protein. As an example, VSV-G, a highly effective envelope protein, is difficult to express constitutively. Potentially cytotoxic transgenes are also problematic for long term expression in stable producers. This has been remedied via a bacterial tryptophan RNA-binding attenuation protein which blocks transgene expression by binding upstream of the ribosome initiation site [134], limiting its expression within the producer cell line.
Typically, the greatest challenge for producer or packaging cells is managing the cytotoxic or cytostatic effects while maintaining high titre. Whilst this can be mitigated with inducible systems and have been accomplished with LV [126,135,136], these methods are currently not wholly practical. Tet-on induction will require the addition of tetracycline which must be removed at a later point, whereas tet-off induction may require extended culture time for expression to peak as well as requiring a complete media exchange, which may be uneconomical. In all inducible cases, the risk of “leaky” expression is also apparent with non-controlled production of LV being possible.
Nonetheless, with further development, the application of stable producers would be greatly beneficial in the production of LVs. Such improvements can be in the form of high throughput automated clonal isolation and evaluation in addition to improved cassette design, envelope choices and cell line development. Possible strategies to optimise LV production is to up-regulate anti-apoptotic genes, downregulate intracellular sensing and optimise the protein and lipid generation pathway for efficient vector production.
3.3. Upstream Culture to Produce Lentiviral Vectors
To produce high titres of LVs, high quantities of cells are required. There are various solutions to expand the number of producing cells (see Table 4) for LVs, pursuant of adherent or suspension cells at varying scales. During development, small batch sizes are desired for flexibility of production and are typically dominated by adherent cultures primarily due to HEK-293T being innately adherent, the high cell densities offered and ease of access. This is usually fulfilled by culture flasks, dishes and bottles. With scale up, where larger and more consistent batch sizes are desired, production transitions to bioreactors in the form of stirred tanks, rocking waves and fixed bed bioreactors with multi-layer flasks straddling the intermediate scales. In terms of efficient cost of goods production, a recent study indicates single-use stirred tanks as most cost-efficient where suspension culture is available, otherwise fixed beds offer greater savings than adherent flask culture [27]. Many upstream culture vessels have also transitioned onto single use disposables. These not only assist in reducing change-over times, but also aid in validation by reducing the cleaning and sterilisation stages for equipment.
3.3.1. Adherent Culture
At development stages, adherent cultures are ideal, however they become limiting with scale. Here, either scale out with multiple flasks or scale up vertically into multilayer flasks is employed. These flasks offer a range of surface areas available for cell attachment and growth with the 1–40 layer CellSTACKS (Corning, 636–25,440cm) and the 1–40 layer cell factories (Nunc, 632–25,280 cm) commonly used [97]. In addition, the HYPERflasks (1720 cm) and HYPERStacks (6000–18,000 cm) (both Corning) offer gas-permeable plastic for the mass transfer of O and CO, and therefore do not require as large of a headspace for vector production [138]. The scale out or scale up of these systems become increasingly bulky and cumbersome to handle at high surface areas, requiring greater incubator space and transport considerations, often needing additional equipment to assist. With scale-out, there is increased manual handling and risk of contamination from open manipulations, particularly problematic as batches are often pooled for downstream processing [96]. In addition, transfection stages are multiplied, increasing the chance of variable vector production, and adding complexity to technician workload. Adherent culture in flasks and roller bottles do not allow for culture management and are batch-mode in nature, with no in-line ability to monitor and control for dissolved oxygen, pH, waste products and nutrient replenishment.
An alternative culture method for adherent cells are microcarriers, which have been applied to HEK-293 and HEK-293T for retroviral vectors [150] and LV [151]. Such microcarriers can be porous or solid, allowing cell growth within or on the surface. However, microcarriers have been linked to clumping and cell detachment [151]. Furthermore, microenvironments can occur within the centre of the microcarriers, whereby cells on the outer surface limit the mass transfer in of nutrients and oxygen, while limiting the release of toxic metabolites, CO and viral particles [152], resulting in low LV titres [150].
For scaling purposes, LV production from adherent cells is often transitioned into fixed bed bioreactors. As an alternative to flask-based or bottle culture, cells are expanded on a 3D matrix composed of highly porous microfibre carriers that offer substantial surface areas at economical volumes. Such reactors have been applied to retroviral and adenoviral vectors [153,154]. Examples of commercialised fixed bed bioreactors are Pall’s iCELLis or Univercells’ Scale-X bioreactors offering surface areas of 0.53–500 and 2.4–600 m, respectively. Such bioreactors allow for in-line monitoring and control of culture parameters for vector production. In-line monitoring offers greater insight from the bioprocessing environment and raises applications in in silico modelling off-line [155] for future optimisation. Furthermore, the Scale-X process can be intensified as part of Univercells’ Nevoline that offers in-line concentration by Tangential Flow Filtration (TFF) and modular downstream processing options such as clarification and chromatography. The iCELLis has been used to produce transient LV in a perfusion system at fixed perfusion rates or targeting specific glucose targets, ultimately achieving titres above TU in a 4 m packed bed in perfusion mode (total volume 5.5 L) [144]. Although this work demonstrated that optimisation is still required, as fixed bed reactors are affected by poor cell distribution and may not optimally expand nor allow efficient transfection particularly at greater compaction, ultimately being less productive per square cm than adherent flasks [144]. Similar titres were produced in Scale-X bioreactors where cell distribution was more homogenous [149]. A side-by-side comparison of LVs from iCellis Nano with LVs manufactured under cGMP using 10-layer cell factories has also demonstrated similar transduction efficiencies [156]. Nonetheless, the singular run of an iCellis Nano in this study was equivalent to 30 triple flasks, demonstrating the impact of scale up options within the upstream.
3.3.2. Suspension Culture
Suspension cultures are commonly applied in the production of various recombinant proteins due to their ease of scaling, control over culture parameters and broad industrial familiarity. For LV production, HEK-293T cultures can be adapted to suspension cultures and used in conventional stirred tank bioreactors and rocking bags [99,140]. Suspension cultures provide a solution for scale up, minimising manual handling, allowing perfusion culture, automation, in-line monitoring and control in addition to simplified application of transfection reagents. However, cells must be adapted to suspension culture for use in these bioreactors. This may be difficult to accomplish without loss of productivity, with titres being in the TU/mL range and below [157]. Only recently have titres been reported to achieve TU/mL via gradual adaptation in suspension media [98]. In general, the cell density is much lower than in adherent, thus vector will be naturally diluted in such methods. Furthermore, additional consideration for initial clarification is required to remove suspended cells.
Stirred tank bioreactors provide the simplest scaling methodology applied to LV production. These units can incorporate development scale productions, such as in Ambr bioreactors [146] for HEK-293T growth optimisation, to larger scale with 10 L Biostat [147], with the latter bioreactor reported to scale up to 10,000 L. Additionally, the Pall Allegro reports titres of TU/L [158]. Scale up of stirred tank bioreactors follow typical scaling parameters, such as following kLa volumetric mass transfer coefficients, power volume (P/V) ratios and tip speeds. As mammalian cells typically require less kLa than bacterial cells [159,160], the scaling parameter is favoured towards P/V and tip speed to prevent shear damage to producer cells. Although in terms of scale up, limitations are evident in transient transfection production strategies whereby media replacements may be required which is more difficult with suspension cells [105].
Rocking bioreactors offer a method for the expansion of producer cells in single use inflated disposable bags wherein the cells and media are rocked gently on a platform to promote mixing at scales of 10–200 L. The method offers high mass transfer of gasses whilst giving a low shear environment for cells due to the absence of impellers. Such bioreactors have been used to produce vectors from stable producer cell lines at clinical scale [140], although this run utilised Fibra-Cel disks as growth matrix in this instance, although suspension cells are noted in patent documentation [161].
3.3.3. Perfusion Culture
The option of perfusion culture can remove cell waste products as they develop and maintain nutrient levels for producer cells. The vector residence time within the culture vessel is also reduced, meaning less non-functional vectors are present due to time-sensitive losses, improving yield. Such perfusion methods have allowed up to 15× improvement over batch runs [162] with up to cumulative TU per litre [163] and better successes in fixed bed reactors [144]. Furthermore, due to the temperature sensitivity of vectors, perfusion can allow vectors to be absconded to a cooler environment away from the destabilising warmer culture temperatures for cells. Although perfusion cultures require high media consumption depending on set perfusion rate which may increase cost, some monoclonal antibodies (mAb) processes indicate potential cost savings compared to fed-batch methods [164]. A perfusion culture can hypothetically be integrated to lead directly into any downstream process, such as chromatography, for continuous purification and intensified processes.
3.3.4. Cell Media and Supplements
The choice of media directly correlates to the expansion and viability of the producer cells and success during downstream processing as the media is responsible for the vector’s stability and forms the initial vector containing feed to process.
There is a drive to remove serum from the production of LV [98]. Many LV production strategies use serum to assist in optimal cell growth and for the stability of the vector itself, which seems to benefit from high protein solutions [165] likely due to albumin and lipid related stabilisation [166]. Supplementation with albumin shows stabilisation of the vector in DSP processing [167]. However, most serum used are bovine derived and thus bovine related disease transmission and the threat of global shortage are of concern. Whilst recombinant human albumin may possibly act as a substitute, this would add to the cost of production. Moreover, the high protein load adds stress to the downstream process which must remove most of the serum contaminants. Such pressures have led to the use of serum-free media for cell culture, and it is now well established in LV production with resulting titres comparable to serum containing media [98,126,168,169]; HyCell TransFX, FreeStyle 293 and SFM4TransFx-293 are just a few examples of commercially available media in use [170]. Such serum-free expression of vector typically involves the sequential reduction of serum in culture until cells are adapted to low or zero serum concentrations. Otherwise, a common upstream tactic is to expand the cells in serum containing media and exchanging to a serum-free alternative prior to vector harvest.
Supplementation of the cell culture has been connected to improvements of LV titre. Sodium butyrate addition is associated with improved recombinant protein production, via the inhibition of histone deacetylase, leading to improved transcription factor function resulting in increased RNA copies from viral LTRs and improved LV titres [171,172]. Additional supplementation with cholesterol and lipids has been linked to improved titre, likely due to stabilisation of lipid rafts on cell surfaces where virions bud [173,174,175]. However, a study that utilised Sendai F/HN envelope proteins and different media types has found no significant improvement to yield with additional supplementation, but it is still suggested that smaller cumulative effects may be of benefit [176]. There has been some improvements to yield due to the presence of caffeine in cell media post-transfection [177], although such results have not been replicated. Furthermore, it has been reported that decreasing the pH to 6 prior to harvesting leads to a tripling in titre [178]. In this experiment, VSV-G envelope protein was used, and at pH 6 its infectivity is likely reduced, thus potentially reducing the impact of superinfection. It has also been shown that high cell media osmolality is associated with improved retroviral stability via lower cholesterol to phospholipid ratios in the viral membrane, leading to improved stability [14,179].
4. Downstream Processing of Lentiviral Vectors
The downstream bioprocessing of LV material concerns itself with maximising vector recovery and minimising components which may negatively impact the efficacy or safety of the product whilst remaining economically viable. Whilst the ultimate product profile is dependent on whether vectors are applied in vivo or ex vivo [21], and the final amount of transducing units is dependent on verified treatment doses, in general, bioprocess operations must reduce the amount of impurities while maintaining viral efficacy to ensure end-user safety. These impurities can be residual DNA (from host cells and plasmid), host cell proteins, serum, proteoglycans and process related impurities such as nucleases and leachables.
The complete downstream processing of LVs is composed of numerous individual unit operations which aim to achieve purification, concentration and stability of the vector material (also reviewed in [180]). Briefly, a typical process, involves sequential purification steps consisting of removing cells and their debris followed by enrichment of vector and the removal of host cell or serum proteins, nucleic acids and lipids. The vector product may be further concentrated prior to exchanging into a suitable formulation buffer for stability and then finally undergoing sterile filtration prior to storage or application (please see Section 6 for whole bioprocess assessment).
4.1. Vector Filtration: Initial Clarification
Clarification and sterile filtration processes aim to remove cells and cell debris amongst other large particulate impurities. Clarification typically occurs early in the process, and therefore deals with crude bioprocess material with high load of cells and cell debris and dilute concentration of LVs while sterile filtration occurs in the later stages of the bioprocess and deals with lower concentration of cells or cell debris and higher concentration of LV particles. The initial downstream stages used in clarification are typically microfiltration and/or centrifugation. Cells and bulk particles can be sedimented and removed by low-speed centrifugation [181]. Alternatively, acoustic filters can be used to entrap suspended cells in soundwaves to return to the bioreactor during harvest [162]. Both are followed by microfiltration through a membrane or depth filter. The initial centrifugation acts as a pre-filtration step to prevent premature fouling of the filter. In larger production runs, the capacity of the centrifuge will need to be considered due to capacity considerations. The application of continuous centrifuges, such as disc stack centrifuges, which are prevalent in monoclonal antibody and recombinant protein processing is yet to be seen in LV production given the low-volume processing (litres to hundreds of litres) and the typical batch mode of these processes which need to be balanced with the centrifuge’s high capital cost compared to filtration options.
Filtration techniques, which are effective and scalable, dominate initial clarification, with disposable systems offering simplified cleaning and validation. The use of cascading filters, whereby the feed is filtered with progressively finer filters can prevent premature fouling of the end filter that may result in titre loss due to vector exclusion and extended process times due to reduced flux [182,183]. The choice of filter impacts the efficiency of the clarification step. With membrane filters, the feed passes through a flat or pleated sheet of inert polymer punctuated with pores of a specific diameter according to the membrane’s absolute retention ratings. Depth filters are a class of filters of a sponge-like texture, wherein particles are retained throughout the entire filter bed that is typically composed of polymer, binder and a filtration aid such as diatomaceous earth. For some depth filters, the media bears a charge for electrostatic retention of host cell proteins and DNA [184,185], with diatomaceous earth showing improved feed capacity although at high concentration it can lower LV titre [186]. In some small-scale studies, the use of multiple depth filter media, including cellulose-based and synthetic media (such as polyacrylic fibre with silica filter aids), has shown 90% turbidity reductions whilst maintaining high recovery of non-enveloped vectors, such as simian adenovirus based vectors [187]. In LV applications, depth filters without filter aids such as diatomaceous earth have recovered >95% of titre while reducing >85% HCP at 50 L scale [188]. Typically, most retention ratings are within the microfiltration range from 10 to 0.2 m in a cascading fashion either applying membranes only or a sequence of a depth filter and a membrane for finer filtration [145,183,189]. At 80–120 nm in diameter, LVs can pass through filters at small retention ratings, although the typical end point of 0.22 m does see loss in titre [141]. Some examples of available filters for clarification are listed in Table 5.
The performance of the vector is dependent on the retention ratings of the filter, quality of the feed and the load challenge expected at that scale. With highly fouled filters, high transmembrane pressures may result in impurities potentially transferring into the filtrate, likely due to rupture of cells or fragments on the filter’s surface. Clarification is affected by upstream production, with suspension culture necessitating removal of whole cells, whilst adherent culture is likely to be composed of cellular fragments. Moreover, transient transfection for LV production generates different host cell protein profiles compared to stable producer cell lines and can differ depending on envelope protein used [120]. In all cases, some degree of LV loss is to be expected due to adsorption onto the filter or onto a component excluded by the filter, in addition to losses in the filtration system’s hold-up volume. For many clarification processes, flushing the filters with buffer post-process is ideal to dislodge any adsorbed vectors to improve recovery and to clear dead volumes [145].
While commonly applied in concentration and diafiltration (see Section 4.6), tangential flow membrane microfiltration can also be applied in clarification. Any cake layer that builds up on the membrane surface is removed due to the tangential feed flow maintaining flux, as opposed to traditional dead-end filtration modes previously discussed. Recently, a new filter media, a 2–5 m depth filter from Repligen was used in tangential flow filtration to separate suspension cells and LVs at harvest [191] with reported LV yields of 90%. Such methods can separate suspension cells from LV harvests, allowing the opportunity to recover producer cells for continued culture and sequential harvests.
4.2. Vector Filtration: Sterile Filtration
Sterile filtration is typically the final step of the downstream process, wherein the viral vectors, having been purified, concentrated and formulated are passed through a fine filter to remove any adventitious agents such as bacteria or fungi while maintaining LV titres. Titre loss of 30–50% during sterile filtration with a 0.22 m syringe membrane filter has been reported [145,190,192]. This is partially due to insufficient formulation or poor purification leading to excessive aggregation and vector loss. Due to low vector recoveries and the risk of adventitious agents being present, sterile filtration can be run at an early stage with closed systems or can be revoked wholly, although this would be under close scrutiny of regulatory agencies [97,193].
4.3. Non-Chromatographic Purification
The purification of LVs seeks to exploit the differences of the vector and other media components, namely size, density, charge or specificity to a stationary phase. This can be accomplished via numerous methods, with common unit operations being ultracentrifugation and chromatography (see Section 4.5).
Ultracentrifugation is a unit operation that purifies and concentrates vector and is a common application during research and development stages of vector production. This technique has also been broadly used in retroviral, adenoviral and adeno-associated vector purification by pelleting viral particles under large g forces [8,194,195]. Due to the large size of the vector compared to free proteins in solution, the vector is pelleted initially, and proteins remain in the supernatant. Alternatively, a gradient is used [196] (typically, sucrose, ficoll or iodixanol is layered in progressively denser layers), and the vector may appear in a specific band, or pellet, with media contaminants in preceding layers, although vesicles or impurities of similar density can be co-sedimented with vector in addition to inhibiting proteoglycans [197,198,199,200]. However, vectors produced by sucrose-gradient display less immunogenic effect in mice, presumably from less serum contamination [201]. The vector pellets can be resuspended, and re-spun for greater purification, with up to four rounds providing relatively clean material [77]. In addition, some envelope proteins may be negatively affected by shear during operation, with VSV-G being particularly resistant [13,76,77]. Further, the particle itself may be damaged by osmolarity of a gradient [202]. Moreover, any gradient composition will likely require removal in sequential processing. High speed centrifuges (10,000× g) can also be used in purification, although this may require extended spin times with up to 4 h spin time reported with sucrose [203]. However, centrifugation for the purpose of vector purification can be time-consuming, scale-restricted (linked to capacity of ultracentrifuge unless scale-out is pursued) and labour intensive. Therefore, its application is commonly limited to research, development or early clinical trial phases.
LVs can be precipitated by the addition of excipients into the vector-containing media which can be utilised for concentration purposes. Precipitating agents such as polyethylene glycol (PEG) [65,204], poly-L-lysine [205], a mixture of chondroitin sulphate C and protamine sulphate [206] and calcium phosphate [207] have been employed. In most cases, the vector is incubated with the material before pelleting by centrifugation prior to resuspension and dissociating the precipitates followed by further processing such as TFF or dialysis. Mode of action is a decrease in solubility due to reduced solvent availability leading to vector precipitation or aggregation due to charges. However, the use of precipitating agents may require additional processing for their removal, as well as extended time to fully precipitate the vector particles and to pellet the precipitate with up to 16 h incubation common [65,204,206]. Furthermore, it may be problematic to resuspend and dissociate precipitates, with EDTA treatment required when using calcium phosphate [207]. Despite this, functional recoveries have been reported between 50% and 100% [65,207] across this unit operation.
4.4. Nucleic Acid Reduction
Nucleic acid impurities originate from either the plasmid DNA from transfection or host cell DNA from the producer cell lines. Nucleic acids need to be reduced during processing to improve purity and prevent the risk of any deleterious effect in recipient cells or patient, with limits of residual DNA being set out at as <200 bp in length and <10 ng per dose [208,209]. This can be done by the addition of nucleases, such as benzonase, at some point during the process [102,190,210]. Typically, the nuclease is added directly to LV culture in the bioreactor prior to or shortly after harvesting or after clarification. However, due to the greater volumes at these stages, more units of enzyme are required to be effective which incurs costs. An alternative is for the producer or helper cells to secrete the nuclease such as the SecNuc development by Oxford Biomedica [211], wherein a plasmid coding for the nuclease is co-transfected with vector components or the producer cells are co-cultured with nuclease-expressing cells. Otherwise, the nuclease can be added after a concentration step to lower enzyme requirements. Regardless of when the enzymes are added, they must be removed further downstream, and the effect of a high nucleic acid burden should be accounted for, such as co-elution and/or reduced binding capacity in chromatography and the formation of nucleic acid complexes [212,213]. In addition, the composition of the buffer the vector is in must be considered for optimal digestion as some nucleases require magnesium ions, elevated temperature and specific pH ranges to be effective [214] in addition to the required incubation time, which may contribute to vector inactivation. Nucleic acids can also be reduced with careful selection of ion exchange or mixed mode flow-through chromatography (see Section 4.5), or TFF, using stable producer cells (Section 3.2) and whole media replacement in the upstream (Section 3.3).
4.5. Chromatographic Purification
Chromatography is a unit operation that separates the components of a mixture via their interactions with stationary and mobile phases. It is a scalable solution for the purification and concentration of particles, in addition to being relatively quick compared to centrifugation and is easily automated for consistent and reliable results. It has seen broad application in the purification of many biologically derived materials such as mAbs, recombinant proteins, industrial enzymes and nucleic acids. Typically, the interactions exploited to resolve feeds are component size in size exclusion chromatography (SEC), charge in ion exchange (IEX), hydrophobicity and affinity to stationary or mobile phases. This can be run in capture and elute mode, or flow-through mode for polishing and refinement. For the purification of LV, multiple types of chromatography can be applied with varying recoveries observed (see Table 6).
The development of stationary phases for chromatography has been focused on the purification of small molecules. This has led to the broad application of porous bead based stationary phases to maximise surface area for high dynamic binding capacity. Due to the small sizes of the components, mass transfer is diffusion dominant. This is problematic for viral particles; whereby, due to their size, the pores of beads are too small and therefore their internal surface areas are inaccessible, thus lowering the binding capacity and performance for vector capture [21,218,219,220]. In addition, due to diffusion-based mass transfer, flow rates through the stationary phases tend to be slow, decreasing throughput, which may lead to greater loss of titre due to increased process time. Given these limitations, new stationary phases have been developed that rely on convective based mass transfer for larger particles (see Figure 1). Such phases allow greater flow rates and throughput while maintaining available surface area for binding capacity. The simplest are macroporous stationary phases, such as in monoliths, where numerous channels form a sponge-like structure for feed to flow through. Monoliths are characterised by their large surface area to volume ratios and have been successful in purifying multiple types of virus [221] including LV with different envelopes [119,222]. Alternatively, membrane stationary phases also offer convective mass transfer. These follow a typical flat or woven sheet, possibly stacked on one another to increase capacity, with pores for feed to flow-through and allowing the capture of vector [223,224]. Recently developed are nanofibre spun stationary phases, which offer anion exchange chromatography (AEX) and yield approximately 90% LV recovery [216] as developed by Puridify, now owned by Cytiva (formerly GE Healthcare Life Sciences). Such phases are defined by thin electrospun threads by which the mobile phase can flow past and retention is on the fibres themselves. Such nanofibers have seen utility in the bind-and-elute of LV [216] and adenovirus [225], although commercial capacity is currently focused on the mAb market.
Some hybrid mixed mode chromatography applications have also been developed. While AEX has been shown to be a useful tool for the purification of vector, some impurities are still present in the final elution, particularly DNA and proteoglycans. As a result, some mixed mode stationary phases such as CaptoCore have been developed. These combine size exclusion and AEX chromatography to purify vectors in a negative mode capacity. In this method, the vector is excluded from the internal core of the material which bears AEX ligands. Thus, vector flows through the column, whilst negatively charged DNA or other smaller proteins are captured and removed. This can be applied as a polishing step or contaminant reduction step for vectors and viruses [181,226,227].
4.5.1. Anion Exchange Chromatography
LVs have a net negative charge at neutral pH on their surface [228] due to the composition of the external envelope and their overall isoelectric point, therefore an AEX chromatography with quaternary amines (QA) or diethylaminoethyl (DEAE) ligands are viable options for their capture and elution. With retro or lentiviral vectors, elution tends to be stepped, with the sequential increases in salt concentrations firstly eluting loosely bound components to improve purity before a final high salt buffer elutes the vector. McNally utilised an initial elution of 0.3 M NaCl, before rising to 1.3 M for vector elution, maintaining a pH of 8 throughout Mustang Q AEX [223]. For a weak ligand such as DEAE, this profile can resemble pH 8 and NaCl concentrations of 0.1 and 0.65 M [190]. Stepped elution is particularly effective with vectors, whereby their large size and variable surface composition allow for interaction at multiple sites on the stationary phase and are thus retained to a greater extent compared to individual feed components [228]. This does, however, require greater salt concentrations or pH shift to elute, which necessitates immediate dilution to maintain stability due to osmotic effects [223] or envelope degradation [8,9]. Therefore, the concentration aspect of chromatography is attenuated, and increased buffer consumption for dilution must be considered. In addition, the transient production of LV may give rise to heterologous populations of vectors resulting in differing elution populations that may be attributed to varying compositions of the external envelope [145,229]. Envelope protein free vectors display a lower isoelectric point than envelope positive vectors with varying zeta-potentials which can affect the efficiency of chromatographic purification, likely due to multiple interactions with ligands of the chromatographic stationary phase [228]. Moreover, pre-treating the vector loading material with NaCl has been shown to satisfactorily inhibit bulk protein from binding to AEX material, improving purity [223].
4.5.2. Affinity Chromatography
Affinity chromatography resolves components of a mixture via their specificity to a ligand on the stationary phase. This can be a viable technique that can allow for high purity and concentration of a vector, without the risk of high salt concentrations for elution such as in IEX, thus preserving vector titre during downstream processing. As capture is based on affinity, ideally only vector is retained and eluted, with residual DNA and proteins remaining unbound within the column.
Previously, histidine tags have been expressed on LV envelopes to aid in its purification [215], however yield for this has been low at 46.7% with most presumably remaining attached to the column. Similarly, biotin can be expressed onto the surface of the vector, allowing its capture by immobilised streptavidin and elution by biotin addition [230,231] allowing vector concentration above 4500-fold. This has been further developed by addition of a biotin mimic on a CD8 stalk [167], although, despite high yields at small scale (60%), this declined to 20% on scale up. In addition, a similar method has been applied with Low Affinity Nerve Growth Factor Receptor (LNGFR) being passively incorporated onto the vector’s surface, and thus was able to be captured with an anti-LNGFR antibody on magnetic beads, resulting in excess of 85% recovery, although there was no release from the antibody beads and transduction recovery was only possible by mixing cells with vector-bound beads [232]. Applying affinity tags on the external surface of the vector envelope may also incur some regulatory apprehension if derived from non-mammalian sources, although this may be minimised for ex vivo therapies due to the less stringent requirements at the vector stage. Furthermore, it was observed that retroviral vectors were inhibited when mixed with soluble heparin [233,234,235], leading to the application of heparin affinity chromatography for the purification of LV, whereby heparin and the related heparin sulphate captures LV, leading to recoveries up to 53–61% of vector [157,236].
4.5.3. Size Exclusion Chromatography
Size exclusion chromatography (SEC) is a method to resolve the components of a feed based on size by the flow of material through packed porous beads. Inherently due to their large size in comparison to smaller particulates, LVs can be purified to a high standard with infectious recovery in excess of 70% and purity above 90% [202,237], although high molecular weight contaminants and DNA may remain. In addition, there are applications of SEC to buffer exchange vector into formulation buffer or to desalt after IEX [138]. However, due to the reduced linear flowrate, the overall throughput of the step is low. In addition, SEC is most effective when the feed volume is low, approximately 15% of the bed column, to maintain efficient separation and thus concentration is required prior. As the product of interest is diluted during elution, an additional round of concentration may be required. Ultimately, SEC can be utilised as a polishing step after the bulk of contaminating items are removed in prior stages.
4.5.4. Steric Exclusion Chromatography
Combinations with polyethylene glycol have led to the use of steric exclusion chromatography. The method is selective based on particle size and can be used on viruses as a quicker alternative to SEC. This method follows a similar mechanism as PEG precipitation, with the exception that a hydrophilic stationary phase is present for large particles to coalesce onto. Upon washing the column, the decrease in PEG concentration in the elution buffer releases bound vectors in a size-based selection. Whilst the method has been applied to influenza [238], recoveries at 60% are reported for LV [239].
4.6. Concentration and Buffer Exchange by Tangential Flow Filtration
Tangential flow filtration (TFF) is a unit operation often run in ultrafiltration/diafiltration (UF/DF) modalities that allows for volume reduction and buffer exchange in the processing of LV in addition to the removal of low molecular weight impurities while retaining vector particles [240,241]. It is used in various formats (Table 7) to differing parameters (Table 8). Here, tangential ultrafiltration allows volume reduction to concentrate the vector to appropriate titres and simplifies downstream processing with reduced volume feeds. Buffer exchange (diafiltration) often follows a concentration step for optimal replacement buffer consumption efficiency. The choice of buffer that the vector can be exchanged to is important for maintaining vector stability over time and during freeze–thaw (see Section 4.7) in addition to exchanging into optimal chromatography binding buffers or for efficient transduction of recipient cells. Outside of TFF, this can be accomplished by multiple rounds of centrifugation and resuspension in a buffer of choice [77] or SEC methods [237]. However, TFF offers greater autonomy and reduced manual handling while maintaining high vector concentration.
Some TFF systems utilise hollow fibre units, whereas others use cassettes and are composed of varying membranes such as cellulose-based (e.g., mixed cellulose esters) and polyethylene sulphone (PES). Both formats are scalable and applicable solutions for vector concentration and buffer exchange. Both offer high surface area to volume ratios, although flat sheet cassettes can offer higher fluxes than hollow fibres, although at the expense of higher shear rates on vector particles due to their circuitous flow paths and turbulence promoting screens.
Membranes utilising TFF allow the transfer of salts, buffers, and small molecule species across membranes in accordance to their retention rating (typically in kDa). The efficiency of clarification in TFF is related to the retention coefficients of the feed and each individual component. Some groups have utilised TFF as clarification and purification step as an alternative to chromatography, reaching up to 97% recovery [242] or when combined with ultracentrifugation allowing concentration up to 1800-fold [245]. As a result, TFF can be applied in multiple instances such as after chromatography to remove excessive salt from elution buffers, to buffer exchange into formulation buffers, into an optimised binding buffer for capture in chromatography or concentration prior to SEC. Furthermore, if the production of vector were to be intensified, tandem filtration [242] could be employed using hollow fibre membranes to concentrate product in successive hollow fibres of decreasing surface area or applying single-pass tangential flow filtrations, whereby the recirculation loop is absent, and the feed is flowed along a long circuitous membrane path [246]. Although, as of time of writing, no applications for LV have been published, single-pass TFF has seen adoption in mAbs and recombinant protein production [247,248]. As processes mature, the implementation of such unit operations is likely, particularly with perfusion-based bioreactors and stable producer cell lines allowing for optimal continuous production.
Despite this, the parameters of TFF need to be optimised depending on feed, as the flow rate and membrane type can lead to shear damage and loss of vector. In addition, the impact of transmembrane pressure, feed flow rates and the composition of feed affects flux due to fouling which may lead to extended processing times and require larger membranes which entail greater hold up volumes. Vectors can also adsorb or become trapped onto the surface of the membrane particularly if the pore size is of similar magnitude as the vector although larger pores offer better impurity removal and flux performance [145,216,249]. Therefore, membranes within the 100–750 kDa range tend to be employed, whereby larger pore sizes favour flux at the expense of vector adsorption, while lower cut-offs slow flux to maintain vector. Post-run washes can assist in the release of adsorbed vector and optimisation is dependent on the feed at that stage. Moreover, additives to the feed such as sucrose or similar viscosity enhancers may protect the vector from shear [243].
4.7. Formulation
The buffer formulation for LV is necessary to maintain the transduction capability of the vector throughout its processing and eventual application. The final formulation will ultimately vary depending on its application. An in vivo therapy for example would require more stringent formulations such as the use of water of injection for safety, compared with vectors for ex vivo applications or for temporary freezing during processing. For many ex vivo therapies, a vector is typically formulated into the cell media for culture in recipient cells [96]; this simplifies the process and no consideration of LV formulation on cell growth is required. However, this does not imply optimal formulation for LV, as the media components may not stabilise the vector satisfactorily or may allow for vector aggregation or adsorption onto containers. This is particularly problematic during freezing, which is required if vector needs to be transported over distances or stored for any amount of time, although the presence of protein and sugars in cell media can be of benefit here.
Typically, buffer excipients act as preservatives for vector formulations. These can feature recombinant proteins, such as human serum albumin, as some protein has been shown to stabilise vectors and sugars such as sucrose or trehalose which can act as cryopreservatives and osmolarity regulators [190]. A buffer species should be utilised to maintain a stable pH (ideally physiological) across a variety of temperatures, and thus species such as HEPES and histidine [250] have been used based on their effective buffering ranges and resistance to pH drift with temperature unlike common buffers such as TRIS which does see pH drift. Despite this, there has been success in the long-term storage of LV with lyophilisation in TRIS or phosphate buffered saline (PBS) with sucrose or trehalose being of benefit [251,252]. The latter demonstrates stability of vector while lyophilised for at least four weeks at 37 °C. Some NaCl may assist in stability, as does the addition of magnesium chloride to prevent aggregation [253]. Additional considerations is the impact of buffers on processing unit operations, for example, negative phosphate ions in common buffer systems interacts with positively charged ligands in AEX chromatography resins.
Kumru et al. [10] examined the effects of multiple excipients on the physical stability of LV, by observation of particle size, morphology and zeta potential where they indicated that the amino acids proline and lysine and the sugars mannose and lactose can minimise vector loss when incubated overnight in glass at 37 °C. Excipients which led to adsorptive losses onto glass were salts (e.g., calcium chloride), reducing agents, chelating agents and cationic peptides. These were also found to negatively affect LV during freeze thaw cycles [10], whereas sugars, polyols and cyclodextrins and certain amino acids such as leucine assisted. However, in this experiment, only physical stability was examined and not transduction capability.
5. Vector Characterisation and Quality Control
For applications in human trials, the LV batch should be extensively characterised, and QC tested before release. To maintain safety, the product must remain within a pre-determined specification backed up by suitably validated, precise and repeatable assay protocols. A typical LV batch will require specifications for purity, identity, safety and potency, and these must remain reasonably consistent from batch to batch as required by regulatory agencies, with stringency developing as a potential therapy extends through animal investigations to commercial release. Assays required can be typical and expected of most recombinant processes but can also extend to vector and transgene specific assays. For example, residual DNA can be assayed by Picogreen or quantitative polymerase chain reaction (qPCR), whereas host cell proteins can be analysed by enzyme linked immunosorbent assay (ELISA), SDS-PAGE or any form of total protein quantification such as Bradford, Lowry or bicinchoninic acid colorimetric assay, in addition to standard mycoplasma and endotoxin testing. Additives to the process such as nucleases can be titered by ELISA as can SV40 T-antigen from HEK-293T cells (qPCR for the antigen can signal host cell DNA impurity). Vector specific assays are more complicated due to the nature of the vector particle itself, containing nucleic acids, lipids and proteins, as well as differentiating functional titres and total particle numbers.
Validating quantification methods is essential for the development process and the QA/QC of LV for commercial supply. It is problematic to reach consensus with methodologies due to the variety of transgenes, envelope proteins and recipient cells available across various industrial or academic groups and even inter-group titres vary broadly depending on operator. Despite this, vector quantification is essential when characterising the effectiveness of a production run and is a requirement as a critical quality attribute for regulatory approval. The quantification of LV can broadly be separated into the quantification of various parts of the vector with some degree of crossover, these groupings can be listed as functionality, vector RNA quantification, vector protein, vector enzyme activity and physically counting said particles.
For functionality, the quantification method of choice is the transduction of a known quantity of cells and examining for transgene expression. This method typically requires a titration of the vector of interest across a variety of dilutions and mixing the vector solution and a known quantity of cells together [97]. Polybrene can be added to enhance transduction by minimising electrostatic repulsion between envelope protein and receptor [165]. After a period of time to allow cell expansion and to dilute out any episomal transgene expression, cells are examined for expression, typically by staining with antibodies or affinity-based dyes unless a marker gene is used (often GFP) and analysed with flow cytometry. The transducing units can be determined by knowing the per cent of transduced cells, the volume of vector solution added and the number of cells. However, this does not account for multiple integrations which may arise with high multiplicity of infections and thus titrations must be carried out. Moreover, the risk of overestimating titre due to transgene expression in episomes is apparent, and therefore suitable lengths of time between transduction and cell reading is recommended to dilute out non-integrated transgenes to better reflect long term cell culture for therapies. Furthermore, the total volume, density of cells, availability of cellular receptors and agitation may affect outcomes [254], and thus consistent titres between groups are difficult to compare directly. The transduced cells ideally should be of the same type as the target recipient cell, although typically HEK-293T are used. Moreover, the gating strategy during FACS analysis, the number of transgenes to stain and quality of the stain will need to be considered. In addition, the presence of transduction inhibitors, such as non-functional vector, free floating envelope proteins and proteoglycans, may cause the titre to be under reported in addition to the chance of vector never reaching the cell or available receptor.
A non-staining protocol for functional titre can be carried out via an integration assay where transduced cell genomic DNA (gDNA) is extracted and the provirus is quantified by qPCR and compared to a housekeeping gene. This assay can be unique to the transgene of interest, although a World Health Organization (WHO) standard has been produced for cross group comparison if sequences between the vector transgene and standard are shared [255]. qPCR can quantify multiple integrations although this is not an indicator of transgene functionality. In addition, quality is dependent on gDNA isolation and the lack of DNA contamination from plasmids, host cells and episomal forms [256], and thus expansion time and/or nucleases are required to minimise false positives. Considering the assay is still based on transduction efficiency, the practical applications are in cells where the transgene is difficult to stain for or for legacy sampling of transduced cell gDNA.
Nucleic acid quantification involves the quantification of vector RNA. This method requires the disruption of the vector, isolation of vector RNA, its reverse transcription to complementary DNA and then quantification. Of note, this method does not quantify vector function, and therefore its application is limited to an extent. There is a risk of plasmid DNA inflating the results which necessitates correction with non-reverse transcribed controls or DNase treatment. Furthermore, non-functional but packaged vector may cause over reporting. There is dependence on the efficiency of RNA extraction and its stability, although this can be controlled by a spiked RNA standard. However, its validity may be problematic for process development purposes, whereby varying inhibitors of qPCR or reverse transcriptase in samples may affect results, for example high salt from chromatography elution. qPCR can be further extended with digital droplet qPCR (ddqPCR), wherein individual qPCR reactions are separated by water–oil emulsion droplets at high dilution. By counting the number of positive droplets, the concentration of template can be calculated without a standard curve as responses follow a Poisson distribution. Such technology has been utilised with LV [257], can provide results even if templates are very low in abundance [258] and can be utilised to calculate the number of vector copies in recipient cells [259].
The p24 ELISA assay is a method to quantify the mass of the p24 HIV capsid protein from samples and can be purchased as regular commercial kits. The antibody-based assay can provide quantification of the protein over the course of a day compared to 2–3 days for functional infectivity. In the assay, the vector is disrupted by a detergent before incubation on a plate where either the protein binds by charge onto the plastic or the p24 is captured by a pre-immobilised antibody. An additional primary antibody is incubated and washed away before an enzyme linked secondary antibody is added. After washing, the bound enzyme allows for the colorimetric measurement of a change in a substrate which can be measured by absorbance or fluorescence, which directly corresponds to p24 quantity. This method has seen widespread adoption, with results typically reported as a ratio of the mass of p24 and transducing units (P:I ratio). This links particle mass to functionality and therefore acts as a measure of quality for vector, and even allowing groups to assume LV number by the estimate of particles of LV per pg of p24 [260]. However, p24 kits are reliant on the specificity of their antibodies, and in some cases over report due to the inclusion of non-processed p24 in the form of GAG, vector fragments and inactivated or immature virions.
The measurement of vector enzyme activity can offer an alternative quantification assay for viral proteins. In qPCR-based product enhanced reverse transcription assay (PERT), the vector is titrated and lysed with detergents before mixing with a standard RNA template [261,262]. A thermocycler is set-up with an initial incubation time for reverse transcription to occur, before a temperature rise leads to the enzyme’s inactivation. qPCR is then run to quantify the amount of RNA template converted to DNA, comparing to a known HIV-1 recombinant reverse transcriptase control. As it is dependent on the activity of reverse transcriptase, the assay will be sensitive to inhibitors of reverse transcriptase and may require its stabilisation by inert proteins typically provided in qPCR master mixes. This may appear problematic with processing samples which may have varying ranges of stabilisers or inhibitors. However, the method is rapid, providing results within 2 h and as a result can offer high throughput quantification for multiple samples within a day. The assay can also be more cost-effective than p24 assays due to the lack of specific antibodies and usage of common qPCR mixes.
Another technique is the counting of physical particles. Dynamic light scattering (DLS) is a method whereby the amount of light scattered from a beam when interrupted by a particle is quantified, and, by using the known viscosity of the sample, the hydrodynamic diameter of particles is calculated via the Stokes–Einstein equation. Results can be obtained within 30 min and estimate the mean particle size, polydispersity and a calculation for the particle size distribution. Although this too only determines physical particle numbers and not-functionality, the quality of the data from DLS is progressively unreliable with greater polydispersity, with increased particle numbers causing errant scattering which detracts from the particle of interest. This is problematic with process mixtures which may be reasonably high in polydispersity, in addition to varying viscosities which must be characterised for DLS accuracy. Unless a clean sample is provided, the method is mainly used for average particle sizes in a mixture and determining if a sample is aggregating. Although the DLS technique has been improved with multi-angle dynamic light scattering, which increases the number of detection angles for light scattering and offers more robust results with high polydispersity, accuracy for particle concentration may vary within 50% of a nominal value [263,264].
Alternative methods on similar principles as DLS can be found with nanoparticle tracking analysis methods. Here video clips are recorded through a microscope and the Brownian motion of small particles quantified by a tracking algorithm. Although unable to differentiate LV from other similar sized particles, some newer models allow for the staining and tracking of particles of a specific fluorescence and hence allows for the quantification of specific species. Similarly, tunable resistive pulse sensing detects the size and number of particles passing through a small pore and have been used with LV [265]. Other physical particle-based quantification can be accomplished by counting particles in electron microscopy with a negative stain, modern systems, e.g. the benchtop MiniTem from Vironova, offer automated electron microscopy and analysis [266]. In addition, the fluorescence of tagged LV can be compared with fluorescent beads in confocal microscopy. Both methods can be very time consuming to complete and insufficient for large numbers of samples. A recent addition, which utilises high-performance liquid chromatography (HPLC) with AEX resins, can elute bands of vector, and, based on their fluorescence, estimate the number of vector particles within to total particles per mL range [267]. This method has been used to differentiate DNA from vector and is comparable to ELISA and ddPCR and allows for the application of various samples from differing aspects of the process within 6.5 min. Although this method only quantifies vector particles and does not display functional units, such rapid and high throughput analytical considerations are of strong value for process development.
6. Whole-Bioprocess Assessment of LV Production
Figure 2 illustrates the different process options available for the manufacture of lentiviral vectors. There are process options which are only useful at small scale or for applications where low number of doses are required (ultracentrifugation, SEC and basket centrifugation) while mostly are scalable (e.g., filtration options, chromatography, etc). The final sequence of operations depends on the scale of application of the LV product, the technology used for LV generation, LV titres and the desired product and impurity profile. In this regard, the actual point in which certain operations need to be applied (e.g., concentration, diafiltration, DNA digestion, etc) will depend on these factors. For example, the application of benzonase, or similar products, for DNA digestion may be performed anywhere from the LV generation step [211], as part of the clarification step [97] or before or after chromatography [190].
In their review paper, McCarron et al. [270] provided an overview of the challenges of scaling-up lentiviral vector production. These are briefly summarised in Table 9. Among the challenges in LV production, bioprocess understanding is most relevant in addressing the low recoveries and loss of vector functionality. This starts with the understanding of the application of the final product, which define the final scale of operation and, the product and process specifications. In addition, determining the relevant parameters which impact the performance of process options will be important in the optimal selection of these options and their operation. Screening studies can provide crucial bioprocess information such as the level of transmembrane pressures [65] or crossflow rate [145] in TFF operations, column flow rate in AEX chromatography [145] or the right molecular weight cut-off (MWCO) or membrane material in TFF operation [65,145,243,244]. It is also important to determine the impact of using frozen-thawed materials in process development of unformulated LV products (e.g., [190,216]), as opposed to using fresh material, as this step may have a huge influence on the process performance rather than as result of the bioprocess operation itself.
Defining the steps in the bioprocess sequence of LV production requires a whole-bioprocess analysis due to the interaction among the different operations. For example, concentration and buffer exchange (TFF) followed by AEX chromatography produced an LV product that when used in transduction was not toxic to cells, despite the lower overall yield compared to just using a TFF step [65]. In another example, TFF operation, whether on its own or combined with ultracentrifugation, resulted in an LV product with higher functional titre not seen when this step was removed [245]. Finally, the location of DNA digestion within the process sequence [145] or the location of the TFF step [243] may be used to improve the following AEX chromatography.
The increased interest in viral vectored cell and gene therapies pushes the boundaries of what is currently done in bioprocessing. For larger scale LV production, the role of process shear needs to be investigated as large-scale production means using larger pumps or running pumps at higher flowrate and therefore, potentially higher shear rates. Furthermore, higher productivity requirements also mean increased flux requirements to shorten the time during TFF operation. This impacts LV production in several ways: the shorter time may be beneficial for LV stability while the increased flux may mean the need to run at higher crossflow rates [145] or higher transmembrane pressure [65]; both may result in exposure to high process shear [271,272]. Viral vectors in general are known to be fragile and therefore sensitive to shear. For example, Valkama et al. [145] mentioned that an increased recirculating flowrate, by-passing a column, resulted in the 20% loss of infective LVs. However, an early analysis of work in our lab found that some pseudotyped LVs have high recoveries even after exposure to very high process shear using ultra scale-down (USD) (unpublished data). This demonstrates that process shear may have different effects on different lentiviral vectors and that the design of bioprocess operations (e.g., TFF) should account for these in order to increase productivity and meet requirements at larger-scale manufacturing. We previously demonstrated the use of ultra scale-down devices to predict a larger scale TFF operation to produce monoclonal antibodies [273]. This larger-scale equipment is of similar type to that used in LV TFF processing [140,145]. Ultra scale-down approaches have also been used to evaluate other unit operations [272]. USD enables whole-bioprocess assessment because of the small amount of material required to perform the analysis. Lastly, as part of the whole-bioprocess analysis of the production of LVs, incorporating a process economic analysis would be beneficial as it could demonstrate the economic viability of bioprocess options [27].
7. Conclusions
We reviewed the basic unit operations, whole bioprocess options and other current developments in the bioprocessing of lentiviral vectors. The demand for LVs will remain high in the foreseeable future as the therapeutic benefits of cell and gene therapy are realised and transferred into the clinic with new applications being explored (e.g., as viral vaccine vectors [274]). Although current manufacturing capacity for LVs is low globally, and LV bioprocessing requires optimisation, efforts are apparent which are improving yields and recoveries. Such developments will lead to greater implementation of gene transfer agents to improve therapeutic outcomes. The fundamental understanding of the bioprocess requirements of lentiviral vectors is key in ensuring the translation of LV products from clinical development to use by patients. TFF and AEX chromatography are front-runners as unit operations of choice for scalable LV bioprocessing as does microfiltration. From what we already know of these operations, the solution environment (i.e., buffers, additives, excipients, etc.) as well as the solid-phase materials (e.g., membranes, resins or fibres) will have important contributions during processing of different pseudotyped LVs. The determination of key operational parameters and process conditions will be an essential activity in process development, along with a whole bioprocess assessment. This should lead to obtaining high LV concentrations and yields with minimal impurities in the LV product.
Author Contributions
C.P., Writing—original draft preparation, conceptualisation, visualisation and investigation; and A.C.M.E.R., Writing—review and editing, conceptualisation, validation and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by UK EPSRC CDT Bioprocess Engineering Leadership (EP/L01520X/1) at University College London and the National Institute for Biological Standards and Control (NIBSC).
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank: Dale Stibbs for comments on early draft; Joanne Del Rosario for proofreading the manuscript; Yasu Takeuchi for discussions and project supervision; Tarit Mukhopadhyay and Mary Collins, as former UCL supervisors.
Conflicts of Interest
One of the authors (A.C.M.E.R.) is supervising a on-going project on LV processing that is partly funded by Pall Biotech under the UCL—Pall Biotech Centre of Excellence (UCL—Pall COE) for research and training. The funders had no role in the design of the review; in the collection, analyses or interpretation of data; in the writing of the manuscript; or in the decision to publish of the review.
Abbreviations
The following abbreviations are used in this manuscript:
| AEX | Anion Exchange Chromatography |
| ATMP | Advanced Therapy Medicinal Products |
| CaPi | Calcium Phosphate |
| CAR-T | Chimeric Antigen Receptor T-Cell |
| Cocal-G | Cocal Glycoprotein |
| ddPCR | Digital Droplet Polymerase Chain Reaction |
| DEAE | Diethylaminoethyl |
| DLS | Dynamic Light Scattering |
| DMEM | Dulbecco’s Modified Eagle Medium |
| Elisa | Enzyme Linked Immunosorbent assay |
| GFP | Green Fluorescent Protein |
| HEK | Human embryonic Kidney |
| HIV | Human Immunodeficiency Virus |
| HPLC | High Performance Liquid Chromatography |
| IEX | Ion Exchange Chromatography |
| kb | Kilobase |
| LDL-R | Low-Density Lipoprotein Receptor |
| LMH | Litres/metres/hour |
| LNGFR | Low Affinity Nerve Growth Factor Receptor |
| LTR | Long Terminal Repeats |
| LV | Lentiviral Vector |
| mAb | Monoclonal Antibody |
| MLV | Murine leukemia virus |
| MOI | Multiplicity of Infection |
| MWCO | Molecular Weight Cut Off |
| p/v | Power /Volume |
| PBS | Phosphate Buffered Saline |
| PEG | Polyethylene glycol |
| PEI | Polyethylenimine |
| PERT | Product Enhanced Reverse Transcription |
| PES | Polyethersulphone |
| PVDF | Polyvinylidene Fluoride |
| QA | Quaternary Amine |
| QA/QC | Quality assurance/Quality Control |
| qPCR | Quantitative Polymerase Chain Reaction |
| SEC | Size Exclusion Chromatography |
| SF | Sterile Filtration |
| TFF | Tangential Flow Filtration |
| TMP | Transmembrane Pressure |
| TU | Transducing Unit |
| UC | Ultracentrifuge |
| UF/DF | Ultrafiltration/Diafiltration |
| USD | Ultra Scale Down |
| VSV-G | Vesicular Stomatitis Virus Glycoprotein |
| WHO | World Health Organization |
References
- Naldini, L.; Blömer, U.; Gallay, P.; Ory, D.; Mulligan, R.; Gage, F.H.; Verma, I.M.; Trono, D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science 1996, 272, 263–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cell and Gene Therapy Catapult. Cell and Gene Therapy Catapult Clinical Trials Database 2019; Technical Report; Cell and Gene Therapy Catapult: London, UK, 2020. [Google Scholar]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef] [PubMed]
- Southey, F. Oxford BioMedica to capture 25–30% of lentiviral vector market by 2026. Predicts CTO, 10 December 2018. [Google Scholar]
- Kolata, G. Gene Therapy Hits a Peculiar Roadblock: A Virus Shortage. The New York Times, 28 April 2016. [Google Scholar]
- Cosset, F.L.; Takeuchi, Y.; Battini, J.L.; Weiss, R.A.; Collins, M.K. High-titer packaging cells producing recombinant retroviruses resistant to human serum. J. Virol. 1995, 69, 7430–7436. [Google Scholar] [CrossRef] [Green Version]
- Sanber, K.S.; Knight, S.B.; Stephen, S.L.; Bailey, R.; Escors, D.; Minshull, J.; Santilli, G.; Thrasher, A.J.; Collins, M.K.; Takeuchi, Y. Construction of stable packaging cell lines for clinical lentiviral vector production. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Segura, M.D.L.M.; Garnier, A.; Kamen, A. Purification and characterization of retrovirus vector particles by rate zonal ultracentrifugation. J. Virol. Methods 2006, 133, 82–91. [Google Scholar] [CrossRef]
- Higashikawa, F.; Chang, L.J. Kinetic analyses of stability of simple and complex retroviral vectors. Virology 2001, 280, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Kumru, O.S.; Wang, Y.; Gombotz, C.W.R.; Kelley-Clarke, B.; Cieplak, W.; Kim, T.; Joshi, S.B.; Volkin, D.B. Physical Characterization and Stabilization of a Lentiviral Vector Against Adsorption and Freeze-Thaw. J. Pharm. Sci. 2018, 107, 2764–2774. [Google Scholar] [CrossRef]
- Kowolik, C.M.; Yee, J.K. Preferential Transduction of Human Hepatocytes with Lentiviral Vectors Pseudotyped by Sendai Virus F Protein. Mol. Ther. 2002, 5, 762–769. [Google Scholar] [CrossRef]
- Ye, K.; Dhiman, H.K.; Suhan, J.; Schultz, J.S. Effect of pH on infectivity and morphology of ecotropic moloney murine leukemia virus. Biotechnol. Prog. 2003, 19, 538–543. [Google Scholar] [CrossRef]
- Burns, J.C.; Friedmann, T.; Driever, W.; Burrascano, M.; Yee, J.K. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: Concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 1993, 90, 8033–8037. [Google Scholar] [CrossRef] [Green Version]
- Coroadinha, A.; Silva, A.; Pires, E.; Coelho, A.; Alves, P.; Carrondo, M. Effect of osmotic pressure on the production of retroviral vectors: Enhancement in vector stability. Biotechnol. Bioeng. 2006, 94, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Beer, C.; Meyer, A.; Müller, K.; Wirth, M. The temperature stability of mouse retroviruses depends on the cholesterol levels of viral lipid shell and cellular plasma membrane. Virology 2003, 308, 137–146. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, J.D.; Jayaraman, B.; Frankel, A.D. The HIV-1 Rev response element: An RNA scaffold that directs the cooperative assembly of a homo-oligomeric ribonucleoprotein complex. RNA Biol. 2012, 9, 6–11. [Google Scholar] [CrossRef] [Green Version]
- De Guzman, R.N. Structure of the HIV-1 Nucleocapsid Protein Bound to the SL3 -RNA Recognition Element. Science 1998, 279, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Zufferey, R.; Nagy, D.; Mandel, R.J.; Naldini, L.; Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 1997, 15, 871–875. [Google Scholar] [CrossRef]
- Kim, V.N.; Mitrophanous, K.; Kingsman, S.M.; Kingsman, A.J. Minimal Requirement for a Lentivirus Vector Based on Human Immunodeficiency Virus Type 1. J. Virol. 1998, 72, 811–816. [Google Scholar] [CrossRef] [Green Version]
- Dull, T.; Zufferey, R.; Kelly, M.; Mandel, R.J.; Nguyen, M.; Trono, D.; Naldini, L. A Third-Generation Lentivirus Vector with a Conditional Packaging System. J. Virol. 1998, 72, 8463–8471. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, T.; Carrondo, M.J.; Alves, P.M.; Cruz, P.E. Purification of retroviral vectors for clinical application: Biological implications and technological challenges. J. Biotechnol. 2007. [Google Scholar] [CrossRef]
- Hacein-Bey-Abina, S. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science 2003, 302, 415–419. [Google Scholar] [CrossRef]
- Moiani, A.; Paleari, Y.; Sartori, D.; Mezzadra, R.; Miccio, A.; Cattoglio, C.; Cocchiarella, F.; Lidonnici, M.R.; Ferrari, G.; Mavilio, F. Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts. J. Clin. Investig. 2012, 122, 1653–1666. [Google Scholar] [CrossRef] [Green Version]
- Themis, M.; Waddington, S.N.; Schmidt, M.; von Kalle, C.; Wang, Y.; Al-Allaf, F.; Gregory, L.G.; Nivsarkar, M.; Themis, M.; Holder, M.V.; et al. Oncogenesis Following Delivery of a Nonprimate Lentiviral Gene Therapy Vector to Fetal and Neonatal Mice. Mol. Ther. 2005, 12, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Masri, F.; Cheeseman, E.; Ansorge, S. Viral vector manufacturing: How to address current and future demands? Cell Gene Ther. Insights 2019, 5, 949–970. [Google Scholar] [CrossRef]
- Spink, K.; Steinsapir, A. The long road to affordability: A cost of goods analysis for an autologous CAR-T process. Cell Gene Ther. Insights 2018, 4, 1105–1116. [Google Scholar] [CrossRef]
- Comisel, R.M.; Kara, B.; Fiesser, F.H.; Farid, S.S. Lentiviral vector bioprocess economics for cell and gene therapy commercialization. Biochem. Eng. J. 2020, 107868. [Google Scholar] [CrossRef]
- Duvergé, A.; Negroni, M. Pseudotyping Lentiviral Vectors: When the Clothes Make the Virus. Viruses 2020, 12, 1311. [Google Scholar] [CrossRef]
- Gutierrez-Guerrero, A.; Cosset, F.L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef] [PubMed]
- Hanawa, H.; Kelly, P.F.; Nathwani, A.C.; Persons, D.A.; Vandergriff, J.A.; Hargrove, P.; Vanin, E.F.; Nienhuis, A.W. Comparison of Various Envelope Proteins for Their Ability to Pseudotype Lentiviral Vectors and Transduce Primitive Hematopoietic Cells from Human Blood. Mol. Ther. 2002, 5, 242–251. [Google Scholar] [CrossRef]
- Marin, M.; Lavillette, D.; Kelly, S.M.; Kabat, D. N-Linked Glycosylation and Sequence Changes in a Critical Negative Control Region of the ASCT1 and ASCT2 Neutral Amino Acid Transporters Determine Their Retroviral Receptor Functions. J. Virol. 2003, 77, 2936–2945. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, Y.; Cosset, F.L.; Lachmann, P.J.; Okada, H.; Weiss, R.A.; Collins, M.K. Type C retrovirus inactivation by human complement is determined by both the viral genome and the producer cell. J. Virol. 1994, 68, 8001–8007. [Google Scholar] [CrossRef] [Green Version]
- Tomás, H.A.; Mestre, D.A.; Rodrigues, A.F.; Guerreiro, M.R.; Carrondo, M.J.; Coroadinha, A.S. Improved GaLV-TR Glycoproteins to Pseudotype Lentiviral Vectors: Impact of Viral Protease Activity in the Production of LV Pseudotypes. Mol. Ther. Methods Clin. Dev. 2019, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, Y.; Takeuchi, Y.; Martin, F.; Cosset, F.L.; Mitrophanous, K.; Collins, M. Continuous high-titer HIV-1 vector production. Nat. Biotechnol. 2003, 21, 569–572. [Google Scholar] [CrossRef]
- Sandrin, V.; Boson, B.; Salmon, P.; Gay, W.; Negre, D.; Le Grand, R.; Trono, D.; Cosset, F.L. Lentiviral vectors pseudotyped with a modified RD114 envelope glycoprotein show increased stability in sera and augmented transduction of primary lymphocytes and CD34+ cells derived from human and nonhuman primates. Blood 2002, 100, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Humbert, O.; Gisch, D.W.; Wohlfahrt, M.E.; Adams, A.B.; Greenberg, P.D.; Schmitt, T.M.; Trobridge, G.D.; Kiem, H.P.P. Development of Third-generation Cocal Envelope Producer Cell Lines for Robust Lentiviral Gene Transfer into Hematopoietic Stem Cells and T-cells. Mol. Ther. 2016, 24, 1237–1246. [Google Scholar] [CrossRef] [Green Version]
- Tijani, M.; Munis, A.M.; Perry, C.; Sanber, K.; Ferraresso, M.; Mukhopadhyay, T.; Themis, M.; Nisoli, I.; Mattiuzzo, G.; Collins, M.K.; et al. Lentivector Producer Cell Lines with Stably Expressed Vesiculovirus Envelopes. Mol. Ther. Methods Clin. Dev. 2018, 10, 303–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olah, Z.; Lehel, C.; Anderson, W.B.; Eiden, M.V.; Wilson, C.A. The cellular receptor for gibbon ape leukemia virus is a novel high affinity sodium-dependent phosphate transporter. J. Biol. Chem. 1994, 269, 25426–25431. [Google Scholar] [CrossRef]
- O’Hara, B.; Johann, S.V.; Klinger, H.P.; Blair, D.G.; Rubinson, H.; Dunn, K.J.; Sass, P.; Vitek, S.M.; Robins, T. Characterization of a human gene conferring sensitivity to infection by gibbon ape leukemia virus. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1990, 1, 119. [Google Scholar]
- Christodoulopoulos, I.; Cannon, P.M. Sequences in the Cytoplasmic Tail of the Gibbon Ape Leukemia Virus Envelope Protein That Prevent Its Incorporation into Lentivirus Vectors. J. Virol. 2001, 75, 4129–4138. [Google Scholar] [CrossRef] [Green Version]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, M.; Tailor, C.S.; Nouri, A.; Kabat, D. Sodium-Dependent Neutral Amino Acid Transporter Type 1 Is an Auxiliary Receptor for Baboon Endogenous Retrovirus. J. Virol. 2000, 74, 8085–8093. [Google Scholar] [CrossRef] [Green Version]
- Humbert, J.M.; Frecha, C.; Bouafia, F.A.; N’Guyen, T.H.; Boni, S.; Cosset, F.L.; Verhoeyen, E.; Halary, F. Measles Virus Glycoprotein-Pseudotyped Lentiviral Vectors Are Highly Superior to Vesicular Stomatitis Virus G Pseudotypes for Genetic Modification of Monocyte-Derived Dendritic Cells. J. Virol. 2012, 86, 5192–5203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Schneider, I.C.; Gallet, M.; Kneissl, S.; Buchholz, C.J. Resting lymphocyte transduction with measles virus glycoprotein pseudotyped lentiviral vectors relies on CD46 and SLAM. Virology 2011, 413, 149–152. [Google Scholar] [CrossRef] [Green Version]
- Frecha, C.; Levy, C.; Costa, C.; Negre, D.; Amirache, F.; Buckland, R.; Russell, S.J.; Cosset, F.L.; Verhoeyen, E. Measles Virus Glycoprotein-Pseudotyped Lentiviral Vector-Mediated Gene Transfer into Quiescent Lymphocytes Requires Binding to both SLAM and CD46 Entry Receptors. J. Virol. 2011, 85, 5975–5985. [Google Scholar] [CrossRef] [Green Version]
- Witting, S.R.; Vallanda, P.; Gamble, A.L. Characterization of a third generation lentiviral vector pseudotyped with Nipah virus envelope proteins for endothelial cell transduction. Gene Ther. 2013, 20, 997–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomares, K.; Vigant, F.; Van Handel, B.; Pernet, O.; Chikere, K.; Hong, P.; Sherman, S.P.; Patterson, M.; An, D.S.; Lowry, W.E.; et al. Nipah Virus Envelope-Pseudotyped Lentiviruses Efficiently Target ephrinB2-Positive Stem Cell Populations In Vitro and Bypass the Liver Sink When Administered in vivo. J. Virol. 2013, 87, 2094–2108. [Google Scholar] [CrossRef] [Green Version]
- Hislop, J.N.; Islam, T.A.; Eleftheriadou, I.; Carpentier, D.C.J.; Trabalza, A.; Parkinson, M.; Schiavo, G.; Mazarakis, N.D. Rabies Virus Envelope Glycoprotein Targets Lentiviral Vectors to the Axonal Retrograde Pathway in Motor Neurons. J. Biol. Chem. 2014, 289, 16148–16163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazarakis, N.D. Rabies virus glycoprotein pseudotyping of lentiviral vectors enables retrograde axonal transport and access to the nervous system after peripheral delivery. Hum. Mol. Genet. 2001, 10, 2109–2121. [Google Scholar] [CrossRef]
- Lentz, T.; Burrage, T.; Smith, A.; Crick, J.; Tignor, G. Is the acetylcholine receptor a rabies virus receptor? Science 1982, 215, 182–184. [Google Scholar] [CrossRef]
- Thoulouze, M.I.; Lafage, M.; Schachner, M.; Hartmann, U.; Cremer, H.; Lafon, M. The Neural Cell Adhesion Molecule Is a Receptor for Rabies Virus. J. Virol. 1998, 72, 7181–7190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuffereau, C. Low-affinity nerve-growth factor receptor (P75NTR) can serve as a receptor for rabies virus. EMBO J. 1998, 17, 7250–7259. [Google Scholar] [CrossRef]
- Mochizuki, H.; Schwartz, J.P.; Tanaka, K.; Brady, R.O.; Reiser, J. High-Titer Human Immunodeficiency Virus Type 1-Based Vector Systems for Gene Delivery into Nondividing Cells. J. Virol. 1998, 72, 8873–8883. [Google Scholar] [CrossRef] [Green Version]
- Sena-Esteves, M.; Tebbets, J.C.; Steffens, S.; Crombleholme, T.; Flake, A.W. Optimized large-scale production of high titer lentivirus vector pseudotypes. J. Virol. Methods 2004, 122, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Belot, L.; Ouldali, M.; Roche, S.; Legrand, P.; Gaudin, Y.; Albertini, A.A. Crystal structure of Mokola virus glycoprotein in its post-fusion conformation. PLoS Pathog. 2020, 16, e1008383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watson, D.J.; Kobinger, G.P.; Passini, M.A.; Wilson, J.M.; Wolfe, J.H. Targeted Transduction Patterns in the Mouse Brain by Lentivirus Vectors Pseudotyped with VSV, Ebola, Mokola, LCMV, or MuLV Envelope Proteins. Mol. Ther. 2002, 5, 528–537. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Kutner, R.H.; Bialkowska, A.; Marino, M.P.; Klimstra, W.B.; Reiser, J. Cell-specific targeting of lentiviral vectors mediated by fusion proteins derived from Sindbis virus, vesicular stomatitis virus, or avian sarcoma/leukosis virus. Retrovirology 2010, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.S.; Kuhn, R.J.; Strauss, E.G.; Ou, S.; Strauss, J.H. High-affinity laminin receptor is a receptor for Sindbis virus in mammalian cells. J. Virol. 1992, 66, 4992–5001. [Google Scholar] [CrossRef] [Green Version]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural Resistance-Associated Macrophage Protein Is a Cellular Receptor for Sindbis Virus in Both Insect and Mammalian Hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morizono, K.; Ku, A.; Xie, Y.; Harui, A.; Kung, S.K.P.; Roth, M.D.; Lee, B.; Chen, I.S.Y. Redirecting Lentiviral Vectors Pseudotyped with Sindbis Virus-Derived Envelope Proteins to DC-SIGN by Modification of N-Linked Glycans of Envelope Proteins. J. Virol. 2010, 84, 6923–6934. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Mohan Kumar, D.; Sax, C.; Schuler, C.; Akkina, R. Pseudotyping of lentiviral vector with novel vesiculovirus envelope glycoproteins derived from Chandipura and Piry viruses. Virology 2016, 488, 162–168. [Google Scholar] [CrossRef] [Green Version]
- Tenenhouse, H.S.; Gauthier, C.; Martel, J.; Gesek, F.A.; Coutermarsh, B.A.; Friedman, P.A. Na+-Phosphate Cotransport in Mouse Distal Convoluted Tubule Cells: Evidence for Glvr-1 and Ram-1 Gene Expression. J. Bone Miner. Res. 1998, 13, 590–597. [Google Scholar] [CrossRef]
- Bitzer, M.; Lauer, U.; Baumann, C.; Spiegel, M.; Gregor, M.; Neubert, W.J. Sendai virus efficiently infects cells via the asialoglycoprotein receptor and requires the presence of cleaved F0 precursor proteins for this alternative route of cell entry. J. Virol. 1997, 71, 5481–5486. [Google Scholar] [CrossRef] [Green Version]
- Okano, S.; Yonemitsu, Y.; Nagata, S.; Sata, S.; Onimaru, M.; Nakagawa, K.; Tomita, Y.; Kishihara, K.; Hashimoto, S.; Nakashima, Y.; et al. Recombinant Sendai virus vectors for activated T lymphocytes. Gene Ther. 2003, 10, 1381–1391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosales Gerpe, M.C.; van Lieshout, L.P.; Domm, J.M.; van Vloten, J.P.; Datu, J.; Ingrao, J.C.; Yu, D.L.; de Jong, J.; Moraes, T.J.; Krell, P.J.; et al. Optimized Pre-Clinical Grade Production of Two Novel Lentiviral Vector Pseudotypes for Lung Gene Delivery. Hum. Gene Ther. 2020, 31, 459–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.L.; Halbert, C.L.; Miller, A.D. Jaagsiekte Sheep Retrovirus Envelope Efficiently Pseudotypes Human Immunodeficiency Virus Type 1-Based Lentiviral Vectors. J. Virol. 2004, 78, 2642–2647. [Google Scholar] [CrossRef] [Green Version]
- Rai, S.K.; Duh, F.M.; Vigdorovich, V.; Danilkovitch-Miagkova, A.; Lerman, M.I.; Miller, A.D. Candidate tumor suppressor HYAL2 is a glycosylphosphatidylinositol (GPI)-anchored cell-surface receptor for jaagsiekte sheep retrovirus, the envelope protein of which mediates oncogenic transformation. Proc. Natl. Acad. Sci. USA 2001, 98, 4443–4448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munis, A.M.; Bentley, E.M.; Takeuchi, Y. A tool with many applications: Vesicular stomatitis virus in research and medicine. Expert Opinion Biol. Therapy 2020, 1–15. [Google Scholar] [CrossRef]
- Ory, D.S.; Neugeboren, B.A.; Mulligan, R.C. A stable human-derived packaging cell line for production of high titer retrovirus/vesicular stomatitis virus G pseudotypes. Proc. Natl. Acad. Sci. USA 1996, 93, 11400–11406. [Google Scholar] [CrossRef] [Green Version]
- DePolo, N.J.; Reed, J.D.; Sheridan, P.L.; Townsend, K.; Sauter, S.L.; Jolly, D.J.; Dubensky, T.W. VSV-G Pseudotyped Lentiviral Vector Particles Produced in Human Cells Are Inactivated by Human Serum. Mol. Ther. 2000, 2, 218–222. [Google Scholar] [CrossRef]
- Brown, B.D.; Sitia, G.; Annoni, A.; Hauben, E.; Sergi, L.S.; Zingale, A.; Roncarolo, M.G.; Guidotti, L.G.; Naldini, L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2007, 109, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Munis, A.M.; Mattiuzzo, G.; Bentley, E.M.; Collins, M.K.; Eyles, J.E.; Takeuchi, Y. Use of Heterologous Vesiculovirus G Proteins Circumvents the Humoral Anti-envelope Immunity in Lentivector-Based In Vivo Gene Delivery. Mol. Ther. Nucleic Acids 2019, 17, 126–137. [Google Scholar] [CrossRef] [Green Version]
- Amirache, F.; Lévy, C.; Costa, C.; Mangeot, P.E.E.; Torbett, B.E.; Wang, C.X.; Nègre, D.; Cosset, F.L.L.; Verhoeyen, E. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood 2014, 123, 1422–1424. [Google Scholar] [CrossRef]
- Kim, S.H.; Lim, K.I. Stability of retroviral vectors against ultracentrifugation is determined by the viral internal core and envelope proteins used for pseudotyping. Mol. Cells 2017, 40, 339–345. [Google Scholar] [CrossRef] [Green Version]
- Friedmann, T.; Yee, J. Pseudotyped Retroviral Vectors for Studies of Human Gene Therapy. Nat. Med. 1995, 1, 275. [Google Scholar] [CrossRef] [PubMed]
- Ichim, C.V.; Wells, R.A. Generation of high-titer viral preparations by concentration using successive rounds of ultracentrifugation. J. Transl. Med. 2011, 9, 137. [Google Scholar] [CrossRef] [Green Version]
- Trobridge, G.D.; Wu, R.A.; Hansen, M.; Ironside, C.; Watts, K.L.; Olsen, P.; Beard, B.C.; Kiem, H.P.P. Cocal-pseudotyped Lentiviral Vectors Resist Inactivation by Human Serum and Efficiently Transduce Primate Hematopoietic Repopulating Cells. Mol. Ther. 2010, 18, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Munis, A.M.; Tijani, M.; Hassall, M.; Mattiuzzo, G.; Collins, M.K.; Takeuchi, Y. Characterization of antibody interactions with the G protein of vesicular stomatitis virus Indiana strain and other vesiculovirus G proteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, P.F.; Vandergriff, J.; Nathwani, A.; Nienhuis, A.W.; Vanin, E.F. Highly efficient gene transfer into cord blood nonobese diabetic/severe combined immunodeficiency repopulating cells by oncoretroviral vector particles pseudotyped with the feline endogenous retrovirus (RD114) envelope protein. Blood 2000, 96, 1206–1214. [Google Scholar] [CrossRef]
- Strang, B.L.; Ikeda, Y.; Cosset, F.L.; Collins, M.K.; Takeuchi, Y. Characterization of HIV-1 vectors with gammaretrovirus envelope glycoproteins produced from stable packaging cells. Gene Ther. 2004, 11, 591–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatlin, J.; Melkus, M.W.; Padgett, A.; Kelly, P.F.; Garcia, J.V. Engraftment of NOD/SCID Mice with Human CD34+Cells Transduced by Concentrated Oncoretroviral Vector Particles Pseudotyped with the Feline Endogenous Retrovirus (RD114) Envelope Protein. J. Virol. 2001, 75, 9995–9999. [Google Scholar] [CrossRef] [Green Version]
- Ward, M.; Sattler, R.; Grossman, I.; Bell, A.J.; Skerrett, D.; Baxi, L.; Bank, A. A stable murine-based RD114 retroviral packaging line efficiently transduces human hematopoietic cells. Mol. Ther. 2003, 8, 804–812. [Google Scholar] [CrossRef]
- Bell, A.J.; Fegen, D.; Ward, M.; Bank, A. RD114 envelope proteins provide an effective and versatile approach to pseudotype lentiviral vectors. Exp. Biol. Med. 2010, 235, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Ghani, K.; Wang, X.; de Campos-Lima, P.O.; Olszewska, M.; Kamen, A.; Rivière, I.; Caruso, M. Efficient Human Hematopoietic Cell Transduction Using RD114- and GALV-Pseudotyped Retroviral Vectors Produced in Suspension and Serum-Free Media. Hum. Gene Ther. 2009, 20, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U.; et al. High Cytotoxic Efficiency of Lentivirally and Alpharetrovirally Engineered CD19-Specific Chimeric Antigen Receptor Natural Killer Cells Against Acute Lymphoblastic Leukemia. Front. Immunol. 2020, 10. [Google Scholar] [CrossRef]
- Relander, T.; Johansson, M.; Olsson, K.; Ikeda, Y.; Takeuchi, Y.; Collins, M.; Richter, J. Gene Transfer to Repopulating Human CD34+ Cells Using Amphotropic-, GALV-, or RD114-Pseudotyped HIV-1-Based Vectors from Stable Producer Cells. Mol. Ther. 2005, 11, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Ohuchi, M.; Homma, M. Trypsin action on the growth of Sendai virus in tissue culture cells. IV. Evidence for activation of sendai virus by cleavage of a glycoprotein. J. Virol. 1976, 18, 1147–1150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homma, M.; Ohuchi, M. Trypsin Action on the Growth of Sendai Virus in Tissue Culture Cells III. Structural Difference of Sendai Viruses Grown in Eggs and Tissue Culture Cells. J. Virol. 1973, 12, 1457–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasaraneni, N.; Chamoun-Emanuelli, A.M.; Wright, G.A.; Chen, Z. A simple strategy for retargeting lentiviral vectors to desired cell types via a disulfide-bond-forming protein-peptide pair. Sci. Rep. 2018, 8, 10990. [Google Scholar] [CrossRef] [Green Version]
- Milani, M.; Annoni, A.; Bartolaccini, S.; Biffi, M.; Russo, F.; Di Tomaso, T.; Raimondi, A.; Lengler, J.; Holmes, M.C.; Scheiflinger, F.; et al. Genome editing for scalable production of alloantigen-free lentiviral vectors for in vivo gene therapy. EMBO Mol. Med. 2017, 9, 1558–1573. [Google Scholar] [CrossRef]
- Lu, X.; Humeau, L.; Slepushkin, V.; Binder, G.; Yu, Q.; Slepushkina, T.; Chen, Z.; Merling, R.; Davis, B.; Chang, Y.N.; et al. Safe two-plasmid production for the first clinical lentivirus vector that achieves >99% transduction in primary cells using a one-step protocol. J. Gene Med. 2004, 6, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Naldini, L.; Blomer, U.; Gage, F.H.; Trono, D.; Verma, I.M. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc. Natl. Acad. Sci. USA 1996, 93, 11382–11388. [Google Scholar] [CrossRef] [Green Version]
- Ahuja, D.; Sáenz-Robles, M.T.; Pipas, J.M. SV40 large T antigen targets multiple cellular pathways to elicit cellular transformation. Oncogene 2005. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, C.B.; Sumner, R.P.; Rodriguez-Plata, M.T.; Rasaiyaah, J.; Milne, R.S.; Thrasher, A.J.; Qasim, W.; Towers, G.J. Lentiviral Vector Production Titer Is Not Limited in HEK293T by Induced Intracellular Innate Immunity. Mol. Ther. Methods Clin. Dev. 2020, 17, 209–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merten, O.W.; Charrier, S.; Laroudie, N.; Fauchille, S.; Dugué, C.; Jenny, C.; Audit, M.; Zanta-Boussif, M.A.; Chautard, H.; Radrizzani, M.; et al. Large-scale manufacture and characterization of a lentiviral vector produced for clinical Ex vivo gene therapy application. Hum. Gene Ther. 2011, 22, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Ausubel, L.J.; Hall, C.; Sharma, A.; Shakeley, R.; Lopez, P.; Quezada, V.; Couture, S.; Laderman, K.; McMahon, R.; Huang, P.; et al. Production of CGMP-grade lentiviral vectors. BioProcess Int. 2012, 10, 32–43. [Google Scholar]
- Bauler, M.; Roberts, J.K.; Wu, C.C.; Fan, B.; Ferrara, F.; Yip, B.H.; Diao, S.; Kim, Y.I.; Moore, J.; Zhou, S.; et al. Production of Lentiviral Vectors Using Suspension Cells Grown in Serum-free Media. Mol. Ther. Methods Clin. Dev. 2020, 17, 58–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ansorge, S.; Lanthier, S.; Transfiguracion, J.; Durocher, Y.; Henry, O.; Kamen, A. Development of a scalable process for high-yield lentiviral vector production by transient transfection of HEK293 suspension cultures. J. Gene Med. 2009, 11, 868–876. [Google Scholar] [CrossRef]
- Iammarino, M.; Nti-Gyabaah, J.; Chandler, M.; Roush, D.; Göklen, K. Impact of Cell Density and Viability on Primary Clarification of Mammalian Cell Broth an Analysis Using Disc-Stack Centrifugation and Charged Depth Filtration; Technical Report; Bioprocess International: New York, NY, USA, 2007. [Google Scholar]
- al Yacoub, N.; Romanowska, M.; Haritonova, N.; Foerster, J. Optimized production and concentration of lentiviral vectors containing large inserts. J. Gene Med. 2007, 9, 579–584. [Google Scholar] [CrossRef]
- Sastry, L.; Xu, Y.; Cooper, R.; Pollok, K.; Cornetta, K. Evaluation of Plasmid DNA Removal from Lentiviral Vectors by Benzonase Treatment. Hum. Gene Ther. 2004, 15, 221–226. [Google Scholar] [CrossRef]
- Shaw, A.; Bischof, D.; Jasti, A.; Ernstberger, A.; Hawkins, T.; Cornetta, K. Using pulmozyme dnase treatment in lentiviral vector production. Hum. Gene Ther. Methods 2012, 23, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Grassman, E.; Higashimoto, T.; Rohrbach, J.; Hall, D.; Williams, D.A.; Reeves, L.; Malik, P. Pulmozyme® Treatment of Vectors Produced by Transient Transfection Reduces Residual Plasmid DNA on Human CD34+ Hematopoietic Progenitor Cells without Loss of Transduction or Engraftment Efficiency. Blood 2008, 112, 4626. [Google Scholar] [CrossRef]
- Tuvesson, O.; Uhe, C.; Rozkov, A.; Lüllau, E. Development of a generic transient transfection process at 100 L scale. Cytotechnology 2008, 56, 123–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raymond, C.; Tom, R.; Perret, S.; Moussouami, P.; L’Abbé, D.; St-Laurent, G.; Durocher, Y. A simplified polyethylenimine-mediated transfection process for large-scale and high-throughput applications. Methods 2011, 55, 44–51. [Google Scholar] [CrossRef] [Green Version]
- Lesch, H.; Valkama, A.; Malinen, J.; Lipponen, E.; Leinonen, H. Large Scale Pei-Mediated Plasmid Transfection. U.S. Patent US2020165557, 28 May 2020. [Google Scholar]
- Chen, C.; Okayama, H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol. 1987, 7, 2745–2752. [Google Scholar] [CrossRef] [PubMed]
- Rhizobium, G.P.E. Complete Genome Sequence of the Sesbania Symbiont and Rice. Nucleic Acids Res. 2013, 1, 13–14. [Google Scholar] [CrossRef]
- Kuroda, H.; Kutner, R.H.; Bazan, N.G.; Reiser, J. Simplified lentivirus vector production in protein-free media using polyethylenimine-mediated transfection. J. Virol. Methods 2009, 157, 113–121. [Google Scholar] [CrossRef]
- Guo, L.; Wang, L.; Yang, R.; Feng, R.; Li, Z.; Zhou, X.; Dong, Z.; Ghartey-Kwansah, G.; Xu, M.M.; Nishi, M.; et al. Optimizing conditions for calcium phosphate mediated transient transfection. Saudi J. Biol. Sci. 2017, 24, 622–629. [Google Scholar] [CrossRef]
- Girard, P.; Porte, L.; Berta, T.; Jordan, M.; Wurm, F.M. Calcium phosphate transfection optimization for serum-free suspension culture. Cytotechnology 2001, 35, 175–180. [Google Scholar] [CrossRef]
- Fassati, A.; Takahara, Y.; Walsh, F.S.; Dickson, G. Production of high titre helper-free recombinant retroviral vectors by lipofection. Nucleic Acids Res. 1994, 22, 1117–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Larcher, L.M.; Ma, L.; Veedu, R.N. Systematic screening of commonly used commercial transfection reagents towards efficient transfection of single-stranded oligonucleotides. Molecules 2018, 23, 2564. [Google Scholar] [CrossRef] [Green Version]
- Invitrogen. Improve Lentiviral Production Using Lipofectamine 3000 Reagent, Application Note, Invitrogen. 2015. Available online: https://www.thermofisher.com/content/dam/LifeTech/global/life-sciences/CellCultureandTransfection/pdfs/Lipofectamine3000-LentiVirus-AppNote-Global-FHR.pdf (accessed on 7 January 2021).
- Boussif, O.; LezoualC’H, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: Polyethylenimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Garson, K.; Li, L.; Vanderhyden, B.C. Optimization of lentiviral vector production using polyethylenimine-mediated transfection. Oncol. Lett. 2015, 9, 55–62. [Google Scholar] [CrossRef]
- Lesch, H.P.; Turpeinen, S.; Niskanen, E.A.; Mähönen, A.J.; Airenne, K.J.; Ylä-Herttuala, S. Generation of lentivirus vectors using recombinant baculoviruses. Gene Ther. 2008, 15, 1280–1286. [Google Scholar] [CrossRef] [Green Version]
- Lesch, H.P.; Laitinen, A.; Peixoto, C.; Vicente, T.; Makkonen, K.E.; Laitinen, L.; Pikkarainen, J.T.; Samaranayake, H.; Alves, P.M.; Carrondo, M.J.; et al. Production and purification of lentiviral vectors generated in 293T suspension cells with baculoviral vectors. Gene Ther. 2011, 18, 531–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.; Wheeler, J.X.; Thorpe, R.; Collins, M.; Takeuchi, Y.; Zhao, Y. Mass spectrometry analysis reveals differences in the host cell protein species found in pseudotyped lentiviral vectors. Biologicals 2018, 52, 59–66. [Google Scholar] [CrossRef]
- Tomás, H.A.; Rodrigues, A.F.; Carrondo, M.J.; Coroadinha, A.S. LentiPro26: Novel stable cell lines for constitutive lentiviral vector production. Sci. Rep. 2018, 8, 5271. [Google Scholar] [CrossRef] [Green Version]
- Stornaiuolo, A.; Piovani, B.M.; Bossi, S.; Zucchelli, E.; Corna, S.; Salvatori, F.; Mavilio, F.; Bordignon, C.; Rizzardi, G.P.; Bovolenta, C. RD2-MolPack- Chim3, a Packaging Cell Line for Stable Production of Lentiviral Vectors for Anti-HIV Gene Therapy. Hum. Gene Ther. Methods 2013, 24, 228–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marin, V.; Stornaiuolo, A.; Piovan, C.; Corna, S.; Bossi, S.; Pema, M.; Giuliani, E.; Scavullo, C.; Zucchelli, E.; Bordignon, C.; et al. RD-MolPack technology for the constitutive production of self-inactivating lentiviral vectors pseudotyped with the nontoxic RD114-TR envelope. Mol. Ther. Methods Clin. Dev. 2016, 3, 16033. [Google Scholar] [CrossRef] [PubMed]
- Throm, R.E.; Ouma, A.A.; Zhou, S.; Chandrasekaran, A.; Lockey, T.; Greene, M.; De Ravin, S.S.; Moayeri, M.; Malech, H.L.; Sorrentino, B.P.; et al. Efficient construction of producer cell lines for a SIN lentiviral vector for SCID-X1 gene therapy by concatemeric array transfection. Blood 2009, 113, 5104–5110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wielgosz, M.M.; Kim, Y.S.; Carney, G.G.; Zhan, J.; Reddivari, M.; Coop, T.; Heath, R.J.; Brown, S.A.; Nienhuis, A.W. Generation of a lentiviral vector producer cell clone for human Wiskott-Aldrich syndrome gene therapy. Mol. Ther. Methods Clin. Dev. 2015, 2, 14063. [Google Scholar] [CrossRef]
- Broussau, S.; Jabbour, N.; Lachapelle, G.; Durocher, Y.; Tom, R.; Transfiguracion, J.; Gilbert, R.; Massie, B. Inducible packaging cells for large-scale production of lentiviral vectors in serum-free suspension culture. Mol. Ther. 2008, 16, 500–507. [Google Scholar] [CrossRef]
- Schucht, R.; Coroadinha, A.S.; Zanta-Boussif, M.A.; Verhoeyen, E.; Carrondo, M.J.; Hauser, H.; Wirth, D. A New Generation of Retroviral Producer Cells: Predictable and Stable Virus Production by Flp-Mediated Site-Specific Integration of Retroviral Vectors. Mol. Ther. 2006, 14, 285–292. [Google Scholar] [CrossRef]
- Coroadinha, A.S.; Schucht, R.; Gama-Norton, L.; Wirth, D.; Hauser, H.; Carrondo, M.J. The use of recombinase mediated cassette exchange in retroviral vector producer cell lines: Predictability and efficiency by transgene exchange. J. Biotechnol. 2006, 124, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Pallant, C.; Sampson, C.J.; Boiti, A.; Johnson, S.; Brazauskas, P.; Hardwicke, P.; Marongiu, M.; Marinova, V.M.; Carmo, M.; et al. Rapid Lentiviral Vector Producer Cell Line Generation Using a Single DNA Construct. Mol. Ther. Methods Clin. Dev. 2020, 19, 47–57. [Google Scholar] [CrossRef]
- Sabine, J.; Celeste, P.; Eirini, V.; Conrad, V. Stable Cell Lines For Retroviral Production. WIPO Patent WO2017/089308Al, 1 June 2017. [Google Scholar]
- Kara, B. Lentiviral Vector Manufacturing-Challenges and Solutions; Presentation: Cardiff, UK, 2017. [Google Scholar]
- Rodrigues, A.F.; Formas-Oliveira, A.S.; Guerreiro, M.R.; Tomás, H.A.; Alves, P.M.; Coroadinha, A.S. Single-step cloning-screening method: A new tool for developing and studying high-titer viral vector producer cells. Gene Ther. 2015, 22, 685–695. [Google Scholar] [CrossRef] [Green Version]
- Nie, Z.; Bren, G.D.; Vlahakis, S.R.; Schimnich, A.A.; Brenchley, J.M.; Trushin, S.A.; Warren, S.; Schnepple, D.J.; Kovacs, C.M.; Loutfy, M.R.; et al. Human Immunodeficiency Virus Type 1 Protease Cleaves Procaspase 8 in vivo. J. Virol. 2007, 81, 6947–6956. [Google Scholar] [CrossRef] [Green Version]
- Maunder, H.E.; Wright, J.; Kolli, B.R.; Vieira, C.R.; Mkandawire, T.T.; Tatoris, S.; Kennedy, V.; Iqball, S.; Devarajan, G.; Ellis, S.; et al. Enhancing titres of therapeutic viral vectors using the transgene repression in vector production (TRiP) system. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Stewart, H.J.; Leroux-Carlucci, M.A.; Sion, C.J.M.; Mitrophanous, K.A.; Radcliffe, P.A. Development of inducible EIAV-based lentiviral vector packaging and producer cell lines. Gene Ther. 2009, 16, 805–814. [Google Scholar] [CrossRef] [PubMed]
- Farson, D.; Witt, R.; McGuinness, R.; Dull, T.; Kelly, M.; Song, J.; Radeke, R.; Bukovsky, A.; Consiglio, A.; Naldini, L. A New-Generation Stable Inducible Packaging Cell Line for Lentiviral Vectors. Hum. Gene Ther. 2001, 12, 981–997. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.W.; Cruz, P.E.; Rochette, C.; Geny-Fiamma, C.; Bouquet, C.; Gonçalves, D.; Danos, O.; Carrondo, M.J.T. Comparison of Different Bioreactor Systems for the Production of High Titer Retroviral Vectors. Biotechnol. Prog. 2001, 17, 326–335. [Google Scholar] [CrossRef]
- Kutner, R.H.; Puthli, S.; Marino, M.P.; Reiser, J. Simplified production and concentration of HIV-1-based lentiviral vectors using HYPERFlask vessels and anion exchange membrane chromatography. BMC Biotechnol. 2009, 9, 10. [Google Scholar] [CrossRef] [Green Version]
- Tinch, S.; Szczur, K.; Swaney, W.; Reeves, L.; Witting, S.R. A scalable lentiviral vector production and purification method using mustang Q chromatography and tangential flow filtration. In Methods in Molecular Biology; Humana Press Inc.: Totoya, NJ, USA, 2019; Volume 1937, pp. 135–153. [Google Scholar] [CrossRef]
- Greene, M.R.; Lockey, T.; Mehta, P.K.; Kim, Y.S.S.; Eldridge, P.W.; Gray, J.T.; Sorrentino, B.P. Transduction of Human CD34 + Repopulating Cells with a Self-Inactivating Lentiviral Vector for SCID-X1 Produced at Clinical Scale by a Stable Cell Line. Hum. Gene Ther. Methods 2012, 23, 297–308. [Google Scholar] [CrossRef] [Green Version]
- Gándara, C.; Affleck, V.; Stoll, E.A. Manufacture of Third-Generation Lentivirus for Preclinical Use, with Process Development Considerations for Translation to Good Manufacturing Practice. Hum. Gene Ther. Methods 2018, 29, 1–15. [Google Scholar] [CrossRef] [Green Version]
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Transient Lentiviral Vector Production Using a Packed-Bed Bioreactor System. Hum. Gene Ther. Methods 2019, 30, 93–101. [Google Scholar] [CrossRef]
- Lesch, H.P. Back to the future: Where are we taking lentiviral vector manufacturing? Cell Gene Ther. Insights 2018, 4, 1137–1150. [Google Scholar] [CrossRef]
- Valkama, A.J.; Leinonen, H.M.; Lipponen, E.M.; Turkki, V.; Malinen, J.; Heikura, T.; Ylä-Herttuala, S.; Lesch, H.P. Optimization of lentiviral vector production for scale-up in fixed-bed bioreactor. Gene Ther. 2018, 25, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valkama, A.J.; Oruetxebarria, I.; Lipponen, E.M.; Leinonen, H.M.; Käyhty, P.; Hynynen, H.; Turkki, V.; Malinen, J.; Miinalainen, T.; Heikura, T.; et al. Development of Large-Scale Downstream Processing for Lentiviral Vectors. Mol. Ther. Methods Clin. Dev. 2020, 17, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Bollmann, F.; Riethmüller, D.; Johansson, E.; Tappe, A. Optimization of HEK293T Suspension Cultivation with a DoE-Approach in Ambr® 15 Microbioreactor. Advancing Manufacture of Cell and Gene Therapies VI, 2019. Available online: https://dc.engconfintl.org/cell_gene_therapies_vi/16 (accessed on 29 July 2020).
- Matet, A.; Kostic, C.; Bemelmans, A.P.; Moulin, A.; Rosolen, S.G.; Martin, S.; Mavilio, F.; Amirjanians, V.; Stieger, K.; Lorenz, B.; et al. Evaluation of tolerance to lentiviral LV-RPE65 gene therapy vector after subretinal delivery in non-human primates. Transl. Res. 2017, 188, 40–57.e4. [Google Scholar] [CrossRef] [Green Version]
- Rout-Pitt, N.; McCarron, A.; McIntyre, C.; Parsons, D.; Donnelley, M. Large-scale production of lentiviral vectors using multilayer cell factories. J. Biol. Methods 2018, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leinonen, H.M.; Lepola, S.; Lipponen, E.M.; Heikura, T.; Koponen, T.; Parker, N.; Ylä-Herttuala, S.; Lesch, H.P. Benchmarking of Scale-X Bioreactor System in Lentiviral and Adenoviral Vector Production. Hum. Gene Ther. 2020, 31, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.C.S.C.; Huang, G.L.G.Y.L.; Liu, J.H.J.H. Production of retrovirus and adenovirus vectors for gene therapy: A comparative study using microcarrier and stationary cell culture. Biotechnol. Prog. 2002, 18, 617–622. [Google Scholar] [CrossRef]
- Witting, S.R.; Jasti, A.; Dolan, S.; Cornetta, K. 46. Production of Lentivirus by Transient Transfection of HEK 293T Grown on Spherical, Polystyrene Microcarriers. Mol. Ther. 2009, 17, S19. [Google Scholar] [CrossRef]
- Yamaji, H.; Fukuda, H. Growth and death behaviour of anchorage-independent animal cells immobilized within porous support matrices. Appl. Microbiol. Biotechnol. 1992, 37, 244–251. [Google Scholar] [CrossRef]
- Lesch, H.P.; Heikkilä, K.M.; Lipponen, E.M.; Valonen, P.; Müller, A.; Räsänen, E.; Tuunanen, T.; Hassinen, M.M.; Parker, N.; Karhinen, M.; et al. Process Development of Adenoviral Vector Production in Fixed Bed Bioreactor: From Bench to Commercial Scale. Hum. Gene Ther. 2015, 26, 560–571. [Google Scholar] [CrossRef]
- Wang, X.; Olszewska, M.; Qu, J.; Wasielewska, T.; Bartido, S.; Hermetet, G.; Sadelain, M.; Rivière, I. Large-scale clinical-grade retroviral vector production in a fixed-bed bioreactor. J. Immunother. 2015, 38, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Baradez, M.O.; Churchwell, J.; Evie, I.; Dewhirst, O.; Thompson, S.; O’grady, C.; Williams, T.; Sanches, R.; Davies, L.; Nimmo, R.; et al. Process Analytical Technology Strategy For Lentiviral Manufacture Real-Time Monitoring of Lentiviral Manufacture; Technical Report; Cell and Gene Therapy Catapult: London, UK, 2020. [Google Scholar]
- Powers, A.D.; Drury, J.E.; Hoehamer, C.F.; Lockey, T.D.; Meagher, M.M. Lentiviral Vector Production from a Stable Packaging Cell Line Using a Packed Bed Bioreactor. Mol. Ther. Methods Clin. Dev. 2020, 19, 1–13. [Google Scholar] [CrossRef]
- Segura, M.M.; Garnier, A.; Durocher, Y.; Coelho, H.; Kamen, A. Production of lentiviral vectors by large-scale transient transfection of suspension cultures and affinity chromatography purification. Biotechnol. Bioeng. 2007, 98, 789–799. [Google Scholar] [CrossRef] [PubMed]
- PALL. White-Paper USD3244. Choice of Upstream Bioreactor Technologies for Industrial Scale Viral Manufacturing; Technical Report; PALL: Port Washington, NY, USA, 2018. [Google Scholar]
- Barrett, T.A.; Wu, A.; Zhang, H.; Levy, M.S.; Lye, G.J. Microwell engineering characterization for mammalian cell culture process development. Biotechnol. Bioeng. 2010, 105, 260–275. [Google Scholar] [CrossRef]
- Bareither, R.; Pollard, D. A review of advanced small-scale parallel bioreactor technology for accelerated process development: Current state and future need. Biotechnol. Prog. 2011, 27, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Marceau, N.; Gasmi, M. Scalable Lentiviral Vector Production System Compatible with Industrial Pharmaceutical Applications. EP Patent 2012/073645, 26 November 2011. [Google Scholar]
- Manceur, A.P.; Kim, H.; Misic, V.; Andreev, N.; Dorion-Thibaudeau, J.; Lanthier, S.; Bernier, A.; Tremblay, S.; Gélinas, A.M.; Broussau, S.; et al. Scalable Lentiviral Vector Production Using Stable HEK293SF Producer Cell Lines. Hum. Gene Ther. Methods 2017, 28, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, D.M.; Rodenbrock, A.; Lanthier, S.; Burney, E.; Spanjaard, R.; Manceur, A. Continuous Perfusion with a Stable Producer HEK293 Cell Line for Scaling Up Lentiviral Vector Production, 2020. In Proceedings of the ASGCT 23rd, Poster Presentation. 12–15 May 2020. Available online: https://www.asgct.org/global/documents/asgct20_abstracts_may8?_zs=S2i4b&_zl=U9052 (accessed on 29 July 2020).
- Pollock, J.; Ho, S.V.; Farid, S.S. Fed-batch and perfusion culture processes: Economic, environmental, and operational feasibility under uncertainty. Biotechnol. Bioeng. 2013, 110, 206–219. [Google Scholar] [CrossRef]
- Andreadis, S.; Palsson, B.O. Coupled effects of polybrene and calf serum on the efficiency of retroviral transduction and the stability of retroviral vectors. Hum. Gene Ther. 1997, 8, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo, M.; Alves, A.; Rodrigues, A.F.; Coroadinha, A.S.; Carrondo, M.J.T.; Alves, P.M.; Cruz, P.E. Stabilization of gammaretroviral and lentiviral vectors: From production to gene transfer. J. Gene Med. 2009, 11, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Mekkaoui, L.; Parekh, F.; Kotsopoulou, E.; Darling, D.; Dickson, G.; Cheung, G.W.; Chan, L.; MacLellan-Gibson, K.; Mattiuzzo, G.; Farzaneh, F.; et al. Lentiviral Vector Purification Using Genetically Encoded Biotin Mimic in Packaging Cell. Mol. Ther. Methods Clin. Dev. 2018, 11, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Li, M.; du Jeu, X.d.M. 462. Serum Free Clinical Grade Large Scale Lentiviral Production System for Gene Therapy Application. Mol. Ther. 2016, 24, S183. [Google Scholar] [CrossRef]
- Oberbek, A.; Matasci, M.; Hacker, D.L.; Wurm, F.M. Generation of stable, high-producing cho cell lines by lentiviral vector-mediated gene transfer in serum-free suspension culture. Biotechnol. Bioeng. 2011, 108, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Do Minh, A.; Tran, M.Y.; Kamen, A.A. Lentiviral Vector Production in Suspension Culture Using Serum-Free Medium for the Transduction of CAR-T Cells. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2020; Volume 2086, pp. 77–83. [Google Scholar] [CrossRef]
- Sakoda, T.; Kasahara, N.; Hamamori, Y.; Kedes, L. A High-Titer Lentiviral Production System Mediates Efficient Transduction of Differentiated Cells Including Beating Cardiac Myocytes. J. Mol. Cell. Cardiol. 1999, 31, 2037–2047. [Google Scholar] [CrossRef]
- Laughlin, M.A.; Zeichner, S.; Kolson, D.; Alwine, J.C.; Seshamma, T.; Pomerantz, R.J.; Gonzalez-Scarano, F. Sodium Butyrate Treatment of Cells Latently Infected with HIV-1 Results in the Expression of Unspliced Viral RNA. Virology 1993, 196, 496–505. [Google Scholar] [CrossRef]
- Chen, Y.; Ott, C.J.; Townsend, K.; Subbaiah, P.; Aiyar, A.; Miller, W.M. Cholesterol supplementation during production increases the infectivity of retroviral and lentiviral vectors pseudotyped with the vesicular stomatitis virus glycoprotein (VSV-G). Biochem. Eng. J. 2009, 44, 199–207. [Google Scholar] [CrossRef]
- Nguyen, D.H.; Hildreth, J.E.K. Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid-Enriched Membrane Lipid Rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [Green Version]
- van Til, N.P.; Heutinck, K.M.; van der Rijt, R.; Paulusma, C.C.; van Wijland, M.; Markusic, D.M.; Oude Elferink, R.P.; Seppen, J. Alteration of viral lipid composition by expression of the phospholipid floppase ABCB4 reduces HIV vector infectivity. Retrovirology 2008, 5, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gélinas, J.F.; Davies, L.A.; Gill, D.R.; Hyde, S.C. Assessment of selected media supplements to improve F/HN lentiviral vector production yields. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ellis, B.L.; Potts, P.R.; Porteus, M.H. Creating higher titer lentivirus with caffeine. Hum. Gene Ther. 2011, 22, 93–100. [Google Scholar] [CrossRef]
- Holic, N.; Seye, A.K.; Majdoul, S.; Martin, S.; Merten, O.W.; Galy, A.; Fenard, D. Influence of mildly acidic pH conditions on the production of lentiviral and retroviral vectors. Hum. Gene Ther. Clin. Dev. 2014, 25, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.I.; Coroadinha, A.S.; Merten, O.W.; Alves, P.M. Improving retroviral vectors production: Role of carbon sources in lipid biosynthesis. J. Biotechnol. 2008, 138, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Moreira, A.S.; Cavaco, D.G.; Faria, T.Q.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Advances in Lentivirus Purification. Biotechnol. J. 2020, 2000019. [Google Scholar] [CrossRef] [PubMed]
- Olgun, H.B.; Tasyurek, H.M.; Sanlioglu, A.D.; Sanlioglu, S. High-Grade Purification of Third-Generation HIV-Based Lentiviral Vectors by Anion Exchange Chromatography for Experimental Gene and Stem Cell Therapy Applications. In Methods in Molecular Biology; Humana Press Inc.: New York, NY, USA, 2018; Volume 1879, pp. 347–365. [Google Scholar] [CrossRef]
- Reeves, L.; Cornetta, K. Clinical retroviral vector production: Step filtration using clinically approved filters improves titers. Gene Ther. 2000, 7. [Google Scholar] [CrossRef] [Green Version]
- Raghavan, B.; Collins, M.; Walls, S.; Lambropoulos, A.; Bergheim-Pietza, S. Optimizing the clarification of industrial scale viral vector culture for gene therapy. Cell Gene Ther. Insights 2019, 5, 1311–1322. [Google Scholar] [CrossRef]
- Khanal, O.; Singh, N.; Traylor, S.J.; Xu, X.; Ghose, S.; Li, Z.J.; Lenhoff, A.M. Contributions of depth filter components to protein adsorption in bioprocessing. Biotechnol. Bioeng. 2018, 115, 1938–1948. [Google Scholar] [CrossRef] [PubMed]
- Yigzaw, Y.; Piper, R.; Tran, M.; Shukla, A.A. Exploitation of the Adsorptive Properties of Depth Filters for Host Cell Protein Removal during Monoclonal Antibody Purification. Biotechnol. Prog. 2006, 22, 288–296. [Google Scholar] [CrossRef]
- Labisch, J.J.; Bollmann, F.; Wolff, M.W.; Pflanz, K. A new simplified clarification approach for lentiviral vectors using diatomaceous earth improves throughput and safe handling. J. Biotechnol. 2021, 326, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fedosyuk, S.; Merritt, T.; Peralta-Alvarez, M.P.; Morris, S.J.; Lam, A.; Laroudie, N.; Kangokar, A.; Wright, D.; Warimwe, G.M.; Angell-Manning, P.; et al. Simian adenovirus vector production for early-phase clinical trials: A simple method applicable to multiple serotypes and using entirely disposable product-contact components. Vaccine 2019, 37, 6951–6961. [Google Scholar] [CrossRef]
- Marques, J.; Dillingham, M.; Beckett, P.; Cherradi, Y.; Paun, A.; Boumlic, A.; Carter, P. Optimizing Viral Vector Manufacturing for Gene Therapy. Pharmaceutical Technology Biologics and Sterile Drug Manufacturing eBook; Pharmaceutical Technology: Cranbury, NJ, USA, 2020. [Google Scholar]
- De Las Mercedes Segura, M.; Kamen, A.; Garnier, A. Purification of retrovirus particles using heparin affinity chromatography. Methods Mol. Biol. 2008, 434, 1–11. [Google Scholar] [CrossRef]
- Bandeira, V.; Peixoto, C.; Rodrigues, A.F.; Cruz, P.E.; Alves, P.M.; Coroadinha, A.S.; Carrondo, M.J.T. Downstream Processing of Lentiviral Vectors: Releasing Bottlenecks. Hum. Gene Ther. Methods 2012, 23, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Williams, T.; Goodyear, O.; Davies, L.; Knevelman, C.; Bransby, M.; Mitrophanous, K.; Miskin, J. Cell & Gene Therapy Insights Lentiviral vector manufacturing process enhancement utilizing TFDF™ technology. Cell Gene Ther. Insights 2020, 6, 455–467. [Google Scholar] [CrossRef]
- Cornetta, K.; Matheson, L.; Ballas, C. Retroviral vector production in the National Gene Vector Laboratory at Indiana University. Gene Ther. 2005, 12, S28–S35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nina, F.; Clive, G.; Joseph, V.H.; Pratik, J.; Debbie, K.; John, M.; Alan, M.; Josefina, N.; Steve, P.; Steven, P.; et al. Key Considerations in Gene Therapy Manufacturing for Commercialization; Cell Culture Dish: Port Washington, NY, USA, 2018. [Google Scholar]
- Hermens, W.T.; Brake, O.T.; Dijkhuizen, P.A.; Sonnemans, M.A.; Grimm, D.; Kleinschmidt, J.A.; Verhaagen, J. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 1999, 10, 1885–1891. [Google Scholar] [CrossRef]
- Ugai, H.; Yamasaki, T.; Hirose, M.; Inabe, K.; Kujime, Y.; Terashima, M.; Liu, B.; Tang, H.; Zhao, M.; Murata, T.; et al. Purification of infectious adenovirus in two hours by ultracentrifugation and tangential flow filtration. Biochem. Biophys. Res. Commun. 2005, 331, 1053–1060. [Google Scholar] [CrossRef]
- Transfiguracion, J.; Ansorge, S.; Tran, R.; Kamen, A. 361. Purification of Lentivirus Vector by Iodixanol Self-Forming Density Gradient. Mol. Ther. 2010, 18, S140. [Google Scholar] [CrossRef]
- Doux, J.M.L.; Morgan, J.R.; Yarmush, M.L. Removal of proteoglycans increases efficiency of retroviral gene transfer. Biotechnol. Bioeng. 1998, 58, 23–34. [Google Scholar] [CrossRef]
- Bess, J.W.; Gorelick, R.J.; Bosche, W.J.; Henderson, L.E.; Arthur, L.O. Microvesicles are a source of contaminating cellular proteins found in purified HIV-1 preparations. Virology 1997, 230, 134–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seppen, J.; Barry, S.; Lam, G.M.; Ramesh, N.; Osborne, W.R. Retroviral preparations derived from PA317 packaging cells contain inhibitors that copurify with viral particles and are devoid of viral vector RNA. Hum. Gene Ther. 2000, 11, 771–775. [Google Scholar] [CrossRef] [PubMed]
- Braas, G.; Searle, P.F.; Slater, N.K.; Lyddiatt, A. Strategies for the isolation and purification of retroviral vectors for gene therapy. Bioseparation 1996, 6, 211–228. [Google Scholar] [PubMed]
- Baekelandt, V.; Eggermont, K.; Michiels, M.; Nuttin, B.; Debyser, Z. Optimized lentiviral vector production and purification procedure prevents immune response after transduction of mouse brain. Gene Ther. 2003, 10, 1933–1940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, M.; Witte, O.; Pincus, T.; Weissman, I.L. Retrovirus purification: Method that conserves envelope glycoprotein and maximizes infectivity. J. Virol. 1978, 25. [Google Scholar] [CrossRef] [Green Version]
- Jiang, W.; Hua, R.; Wei, M.; Li, C.; Qiu, Z.; Yang, X.; Zhang, C. An optimized method for high-titer lentivirus preparations without ultracentrifugation. Sci. Rep. 2015, 5, 13875. [Google Scholar] [CrossRef] [Green Version]
- Kohno, T.; Mohan, S.; Goto, T.; Morita, C.; Nakano, T.; Hong, W.; Sangco, J.C.E.; Morimatsu, S.; Sano, K. A new improved method for the concentration of HIV-1 infective particles. J. Virol. Methods 2002, 106, 167–173. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, H.Q.; Cleghorn, G.; Gobe, G.; West, M.; Wei, M.Q. A highly efficient and consistent method for harvesting large volumes of high-titre lentiviral vectors. Gene Ther. 2001, 8, 1745–1751. [Google Scholar] [CrossRef]
- Lee, J.Y.; Lee, H.H. A new chemical complex can rapidly concentrate lentivirus and significantly enhance gene transduction. Cytotechnology 2018, 70, 193–201. [Google Scholar] [CrossRef] [PubMed]
- L, P.; H, Y.; FL, C.; SJ, R.; KW, P. Concentration of viral vectors by co-precipitation with calcium phosphate. J. Gene Med. 2001, 3. [Google Scholar] [CrossRef]
- WHO. WHO Requirements for the Use of Animal Cells asin vitroSubstrates for the Production of Biologicals (Requirements for Biological Susbstances No. 50). Biologicals 1998, 26, 175–193. [Google Scholar] [CrossRef]
- FDA. Guidance for Industry-Characterization and Qualification of Cell Substrates and Other Biological Materials Used in the Production of Viral Vaccines for Infectious Disease Indications; Technical Report; FDA: Rockville, MD, USA, 2010. [Google Scholar]
- Konz, J.O.; Lee, A.L.; Lewis, J.A.; Sagar, S.L. Development of a purification process for adenovirus: Controlling virus aggregation to improve the clearance of host cell DNA. Biotechnol. Prog. 2005, 21, 466–472. [Google Scholar] [CrossRef]
- Oxford Biomedica. SecNuc for AAV, Lentiviral Vectors and Adenovirus; Oxford Biomedica: Oxford, UK, 2019. [Google Scholar]
- Nian, R.; Zhang, W.; Tan, L.; Lee, J.; Bi, X.; Yang, Y.; Gan, H.T.; Gagnon, P. Advance chromatin extraction improves capture performance of protein A affinity chromatography. J. Chromatogr. A 2016, 1431, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nian, R.; Gagnon, P. Advance chromatin extraction enhances performance and productivity of cation exchange chromatography-based capture of Immunoglobulin G monoclonal antibodies. J. Chromatogr. A 2016, 1453, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Jia, P.; Sharif, R.; Li, Z.; Li, Y.; Chen, P. High-Level Production of DNA-Specific Endonuclease AsEndI with Synonymous Codon and its Potential Utilization for Removing DNA Contamination. Appl. Biochem. Biotechnol. 2018, 185, 641–654. [Google Scholar] [CrossRef] [PubMed]
- Cheeks, M.C.; Kamal, N.; Sorrell, A.; Darling, D.; Farzaneh, F.; Slater, N.K. Immobilized metal affinity chromatography of histidine-tagged lentiviral vectors using monolithic adsorbents. J. Chromatogr. A 2009, 1216, 2705–2711. [Google Scholar] [CrossRef]
- Ruscic, J.; Perry, C.; Mukhopadhyay, T.; Takeuchi, Y.; Bracewell, D.G. Lentiviral Vector Purification Using Nanofiber Ion-Exchange Chromatography. Mol. Ther. Methods Clin. Dev. 2019, 15, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Marino, M.P.; Panigaj, M.; Ou, W.; Manirarora, J.; Wei, C.H.; Reiser, J. A scalable method to concentrate lentiviral vectors pseudotyped with measles virus glycoproteins. Gene Ther. 2015, 22, 280–285. [Google Scholar] [CrossRef] [PubMed]
- Jungbauer, A. Chromatographic media for bioseparation. J. Chromatogr. A 2005, 1065, 3–12. [Google Scholar] [CrossRef]
- Barut, M.; Podgornik, A.; Brne, P.; Štrancar, A. Convective Interaction Media short monolithic columns: Enabling chromatographic supports for the separation and purification of large biomolecules. J. Sep. Sci. 2005, 28, 1876–1892. [Google Scholar] [CrossRef]
- Trilisky, E.I.; Lenhoff, A.M. Sorption processes in ion-exchange chromatography of viruses. J. Chromatogr. A 2007, 1142, 2–12. [Google Scholar] [CrossRef]
- Krajacic, M.; Ravnikar, M.; Štrancar, A.; Gutiérrez-Aguirre, I. Application of monolithic chromatographic supports in virus research. Electrophoresis 2017, 38, 2827–2836. [Google Scholar] [CrossRef]
- Zimmermann, K.; Scheibe, O.; Kocourek, A.; Muelich, J.; Jurkiewicz, E.; Pfeifer, A. Highly efficient concentration of lenti- and retroviral vector preparations by membrane adsorbers and ultrafiltration. BMC Biotechnol. 2011, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNally, D.J.; Darling, D.; Farzaneh, F.; Levison, P.R.; Slater, N.K. Optimised concentration and purification of retroviruses using membrane chromatography. J. Chromatogr. A 2014, 1340, 24–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orr, V.; Zhong, L.; Moo-Young, M.; Chou, C.P. Recent advances in bioprocessing application of membrane chromatography. Biotechnol. Adv. 2013. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, J.; Wright, B.; Green, N.K.; Tarrant, R.; Roberts, I.; Hardick, O.; Bracewell, D.G. Adenovirus 5 recovery using nanofiber ion-exchange adsorbents. Biotechnol. Bioeng. 2019, 116, 1698–1709. [Google Scholar] [CrossRef]
- James, K.T.; Cooney, B.; Agopsowicz, K.; Trevors, M.A.; Mohamed, A.; Stoltz, D.; Hitt, M.; Shmulevitz, M. Novel High-throughput Approach for Purification of Infectious Virions. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Weigel, T.; Solomaier, T.; Peuker, A.; Pathapati, T.; Wolff, M.W.; Reichl, U. A flow-through chromatography process for influenza A and B virus purification. J. Virol. Methods 2014, 207, 45–53. [Google Scholar] [CrossRef]
- Rodrigues, T.; Alves, A.; Lopes, A.; Carrondo, M.J.; Alves, P.M.; Cruz, P.E. Removal of envelope protein-free retroviral vectors by anion-exchange chromatography to improve product quality. J. Sep. Sci. 2008, 31, 3509–3518. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; McCarty, D.M.; Madden, V.J.; Walsh, C.E. Lentivirus vector purification using anion exchange HPLC leads to improved gene transfer. BioTechniques 2003, 34, 1074–1080. [Google Scholar] [CrossRef] [Green Version]
- Chan, L.; Nesbeth, D.; MacKey, T.; Galea-Lauri, J.; Gäken, J.; Martin, F.; Collins, M.; Mufti, G.; Farzaneh, F.; Darling, D. Conjugation of Lentivirus to Paramagnetic Particles via Nonviral Proteins Allows Efficient Concentration and Infection of Primary Acute Myeloid Leukemia Cells. J. Virol. 2005, 79, 13190–13194. [Google Scholar] [CrossRef] [Green Version]
- Nesbeth, D.; Williams, S.L.; Chan, L.; Brain, T.; Slater, N.K.; Farzaneh, F.; Darling, D. Metabolic biotinylation of lentiviral pseudotypes for scalable paramagnetic microparticle-dependent manipulation. Mol. Ther. 2006, 13, 814–822. [Google Scholar] [CrossRef]
- Stornaiuolo, A.; Stefano, C.; Sergio, B.; Claudio, B.; Paolo, R.G.; Chiara, B. 520. A New Strategy for Retroviral Vector Purification Based on LNGR-Ab Immunomagnetic Selection. Mol. Ther. 2013, 21, S201. [Google Scholar]
- Walker, S.J.; Pizzato, M.; Takeuchi, Y.; Devereux, S. Heparin Binds to Murine Leukemia Virus and Inhibits Env-Independent Attachment and Infection. J. Virol. 2002, 76, 6909–6918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guibinga, G.H.; Miyanohara, A.; Esko, J.D.; Friedmann, T. Cell surface heparan sulfate is a receptor for attachment of envelope protein-free retrovirus-like particles and VSV-G pseudotyped MLV-derived retrovirus vectors to target cells. Mol. Ther. 2002, 5, 538–546. [Google Scholar] [CrossRef]
- Arai, T.; Matsumoto, K.; Saitoh, K.; Ui, M.; Ito, T.; Murakami, M.; Kanegae, Y.; Saito, I.; Cosset, F.L.; Takeuchi, Y.; et al. A new system for stringent, high-titer vesicular stomatitis virus G protein-pseudotyped retrovirus vector induction by introduction of Cre recombinase into stable prepackaging cell lines. J. Virol. 1998, 72, 1115–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segura, M.D.L.M.; Kamen, A.; Trudel, P.; Garnier, A.; de las Mercedes Segura, M.; Kamen, A.; Trudel, P.; Garnier, A. A novel purification strategy for retrovirus gene therapy vectors using heparin affinity chromatography. Biotechnol. Bioeng. 2005, 90, 391–404. [Google Scholar] [CrossRef]
- Transfiguracion, J.; Jaalouk, D.E.; Ghani, K.; Galipeau, J.; Kamen, A. Size-exclusion chromatography purification of high-titer vesicular stomatitis virus G glycoprotein-pseudotyped retrovectors for cell and gene therapy applications. Hum. Gene Ther. 2003, 14, 1139–1153. [Google Scholar] [CrossRef] [PubMed]
- Marichal-Gallardo, P.; Pieler, M.M.; Wolff, M.W.; Reichl, U. Steric exclusion chromatography for purification of cell culture-derived influenza A virus using regenerated cellulose membranes and polyethylene glycol. J. Chromatogr. A 2017, 1483, 110–119. [Google Scholar] [CrossRef]
- Marichal-Gallardo, P.; Genzel, Y.; Börner, K.; Grimm, D.; Wolff, M.; Reichl, U. A Single-Use Chromatographic Purification Platform for Viral Gene Transfer Vectors & Viral Vaccines. Advancing Manufacture of Cell and Gene Therapies VI, 2019. In Proceedings of the Advancing Manufacture of Cell and Gene Therapies VI. Available online: https://dc.engconfintl.org/cell_gene_therapies_vi/92 (accessed on 29 July 2020).
- Paul, R.W.; Morris, D.; Hess, B.W.; Dunn, J.; Overell, R.W. Increased viral titer through concentration of viral harvests from retroviral packaging lines. Hum. Gene Ther. 1993, 4, 609–615. [Google Scholar] [CrossRef]
- Miller, D.L.; Meikle, P.J.; Anson, D.S. A rapid and efficient method for concentration of small volumes of retroviral supernatant. Nucleic Acids Res. 1996, 24. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.R.; Patel, S.; Senadheera, S.; Plath, K.; Kohn, D.B.; Hollis, R.P. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J. Virol. Methods 2011, 177, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Boudeffa, D.; Bertin, B.; Biek, A.; Mormin, M.; Leseigneur, F.; Galy, A.; Merten, O.W. Toward a Scalable Purification Protocol of GaLV-TR-Pseudotyped Lentiviral Vectors. Hum. Gene Ther. Methods 2019, 30, 153–171. [Google Scholar] [CrossRef]
- Soldi, M.; Sergi Sergi, L.; Unali, G.; Kerzel, T.; Cuccovillo, I.; Capasso, P.; Annoni, A.; Biffi, M.; Rancoita, P.M.V.; Cantore, A.; et al. Laboratory-Scale Lentiviral Vector Production and Purification for Enhanced Ex Vivo and In Vivo Genetic Engineering. Mol. Ther. Methods Clin. Dev. 2020, 19, 411–425. [Google Scholar] [CrossRef]
- Geraerts, M.; Michiels, M.; Baekelandt, V.; Debyser, Z.; Gijsbers, R. Upscaling of lentiviral vector production by tangential flow filtration. J. Gene Med. 2005, 7, 1299–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marino, K.; Levison, P. Achieving Process Intensification with Single-Pass TFF. Proceedings of the Advancing Manufacture of Cell and Gene Therapies VI. 2017. Available online: https://www.genengnews.com/magazine/299/achieving-process-intensification-with-single-pass-tff/ (accessed on 30 July 2020).
- Dizon-Maspat, J.; Bourret, J.; D’Agostini, A.; Li, F. Single pass tangential flow filtration to debottleneck downstream processing for therapeutic antibody production. Biotechnol. Bioeng. 2012, 109, 962–970. [Google Scholar] [CrossRef] [PubMed]
- Casey, C.; Gallos, T.; Alekseev, Y.; Ayturk, E.; Pearl, S. Protein concentration with single-pass tangential flow filtration (SPTFF). J. Membr. Sci. 2011, 384, 82–88. [Google Scholar] [CrossRef]
- Makino, M.; Ishikawa, G.; Yamaguchi, K.; Okada, Y.; Watanabe, K.; Sasaki-Iwaki, Y.; Manabe, S.; Honda, M.; Komuro, K. Concentration of live retrovirus with a regenerated cellulose hollow fiber, BMM. Arch. Virol. 1994, 139, 87–96. [Google Scholar] [CrossRef]
- Fan, J.; Jiang, L.; Zhou, Z. Recombinant Lentiviral Vector Formulation. Patent EP2829285A1, 2017. Available online: https://patents.google.com/patent/EP2829285A1/en (accessed on 10 August 2020).
- Delacroix, N.; Ouengue Mbele, G.; Deroyer, J.; Deplaine, G.; Bauche, C. 671. Development of a Successful Lyophilization Process for Lentiviral Vector Clinical Batches. Mol. Ther. 2015, 23, S267. [Google Scholar] [CrossRef] [Green Version]
- Mather, S.T.; Wright, E.; Scott, S.D.; Temperton, N.J. Lyophilisation of influenza, rabies and Marburg lentiviral pseudotype viruses for the development and distribution of a neutralisation -assay-based diagnostic kit. J. Virol. Methods 2014, 210, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Driss, B.; Otto-Wilhelm, M.; David, F. Method for Purifying Enveloped Viruses or Viral Vectors. U.S. Patent US20170002332A1, 5 January 2015. [Google Scholar]
- Blaha, B.A.F.; Toufexi, A.; Berger, G.; Hassan, E.; Gaddum, N.R. Increasing Lentiviral Transduction Efficiency: Towards Cost-Effective T Cell Therapy Manufacturing. Mol. Therapy 2019. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Stepto, H.; Schneider, C.K. Development of the First World Health Organization Lentiviral Vector Standard: Toward the Production Control and Standardization of Lentivirus-Based Gene Therapy Products. Hum. Gene Ther. Methods 2017, 28, 205–214. [Google Scholar] [CrossRef]
- Chen, J.; Reeves, L.; Sanburn, N.; Croop, J.; Williams, D.A.; Cornetta, K. Packaging cell line DNA contamination of vector supernatants: Implication for laboratory and clinical research. Virology 2001, 282, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bergelson, S.; Feschenko, M. Determination of lentiviral infectious titer by a novel droplet digital PCR method. Hum. Gene Ther. Methods 2018, 29, 96–103. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Cerbo, V.D.; Bagnati, M.; Santeramo, I.; Bagnati, M. Single Cell Analysis of Lentiviral Transduction to Support Ex-Vivo Gene-Modified Cell Therapies; Technical Report; Cell and Gene Therapy Catapult: London, UK, 2020. [Google Scholar]
- Briggs, J.A.G.; Simon, M.N.; Gross, I.; Kräusslich, H.G.; Fuller, S.D.; Vogt, V.M.; Johnson, M.C. The stoichiometry of Gag protein in HIV-1. Nat. Struct. Mol. Biol. 2004, 11, 672–675. [Google Scholar] [CrossRef] [PubMed]
- Pizzato, M.; Erlwein, O.; Bonsall, D.; Kaye, S.; Muir, D.; McClure, M.O. A one-step SYBR Green I-based product-enhanced reverse transcriptase assay for the quantitation of retroviruses in cell culture supernatants. J. Virol. Methods 2009, 156, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Vermeire, J.; Naessens, E.; Vanderstraeten, H.; Landi, A.; Iannucci, V.; van Nuffel, A.; Taghon, T.; Pizzato, M.; Verhasselt, B. Quantification of Reverse Transcriptase Activity by Real-Time PCR as a Fast and Accurate Method for Titration of HIV, Lenti- and Retroviral Vectors. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Naiim, M.; Boualem, A.; Ferre, C.; Jabloun, M.; Jalocha, A.; Ravier, P. Multiangle dynamic light scattering for the improvement of multimodal particle size distribution measurements. Soft Matter 2015, 11, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.; Minelli, C.; Hamilton, D.; Wywijas, M.; Jones, H.J. Nanoparticle number concentration measurements by multi-angle dynamic light scattering. J. Nanopart. Res. 2020, 22, 1–15. [Google Scholar] [CrossRef]
- Heider, S.; Muzard, J.; Zaruba, M.; Metzner, C. Integrated Method for Purification and Single-Particle Characterization of Lentiviral Vector Systems by Size Exclusion Chromatography and Tunable Resistive Pulse Sensing. Mol. Biotechnol. 2017, 59, 251–259. [Google Scholar] [CrossRef]
- Turkki, V.; Lesch, H.P.; Ryner, M.; Nilsson, J. Viral Vector Particle Integrity and Purity Analyses in Early Process Development. BioProcess Int. 2017, 15, 9. [Google Scholar]
- Transfiguracion, J.; Tran, M.Y.; Lanthier, S.; Tremblay, S.; Coulombe, N.; Acchione, M.; Kamen, A.A. Rapid in-process monitoring of lentiviral vector particles by high performance liquid chromatography. Mol. Therapy Methods Clin. Dev. 2020. [Google Scholar] [CrossRef] [PubMed]
- Merten, O.W.; Schweizer, M.; Chahal, P.; Kamen, A. Manufacturing of viral vectors: Part II. Downstream processing and safety aspects. Pharm. Bioprocess. 2014, 2, 237–251. [Google Scholar] [CrossRef]
- Truran, R.; Buckley, R.; Radcliffe, P.; Miskin, J.; Mitrophanous, K. Virus Purification. U.S. Patent US9169491B2, 2009. Available online: https://patents.google.com/patent/US9169491B2/en (accessed on 16 August 2020).
- McCarron, A.; Donnelley, M.; McIntyre, C.; Parsons, D. Challenges of up-scaling lentivirus production and processing. J. Biotechnol. 2016, 240, 23–30. [Google Scholar] [CrossRef]
- Fernandez-Cerezo, L.; Rayat, A.C.M.E.; Chatel, A.; Pollard, J.M.; Lye, G.J.; Hoare, M. An ultra scale-down method to investigate monoclonal antibody processing during tangential flow filtration using ultrafiltration membranes. Biotechnol. Bioeng. 2019, 116, 581–590. [Google Scholar] [CrossRef] [Green Version]
- Rayat, A.C.; Chatel, A.; Hoare, M.; Lye, G.J. Ultra scale-down approaches to enhance the creation of bioprocesses at scale: Impacts of process shear stress and early recovery stages. Curr. Opin. Chem. Eng. 2016, 14, 150–157. [Google Scholar] [CrossRef]
- Fernandez-Cerezo, L.; Rayat, A.C.; Chatel, A.; Pollard, J.M.; Lye, G.J.; Hoare, M. The prediction of the operating conditions on the permeate flux and on protein aggregation during membrane processing of monoclonal antibodies. J. Membr. Sci. 2020, 596, 117606. [Google Scholar] [CrossRef]
- Vrba, S.M.; Kirk, N.M.; Brisse, M.E.; Liang, Y.; Ly, H. Development and Applications of Viral Vectored Vaccines to Combat Zoonotic and Emerging Public Health Threats. Vaccines 2020, 8, 680. [Google Scholar] [CrossRef]
Figure 1.
Illustrative examples of chromatography resins in use for lentiviral vector purification with increasing convective mass transfer from left to right.
Figure 1.
Illustrative examples of chromatography resins in use for lentiviral vector purification with increasing convective mass transfer from left to right.

Figure 2.
Bioprocess options in the production of lentiviral vectors. Some studies have shown sequence of membrane filtration of different pore sizes or inclusion of low-speed centrifugation prior [145,183,189]. Sequence of filtration processes would be an option depending on scale and cell density and product and impurity profile. These are examples of sequences of operations used in pre-clinical and clinical investigations [145,268,269]. Benzonase may be added at a variety of steps within the downstream process.
Figure 2.
Bioprocess options in the production of lentiviral vectors. Some studies have shown sequence of membrane filtration of different pore sizes or inclusion of low-speed centrifugation prior [145,183,189]. Sequence of filtration processes would be an option depending on scale and cell density and product and impurity profile. These are examples of sequences of operations used in pre-clinical and clinical investigations [145,268,269]. Benzonase may be added at a variety of steps within the downstream process.


{kind=link}
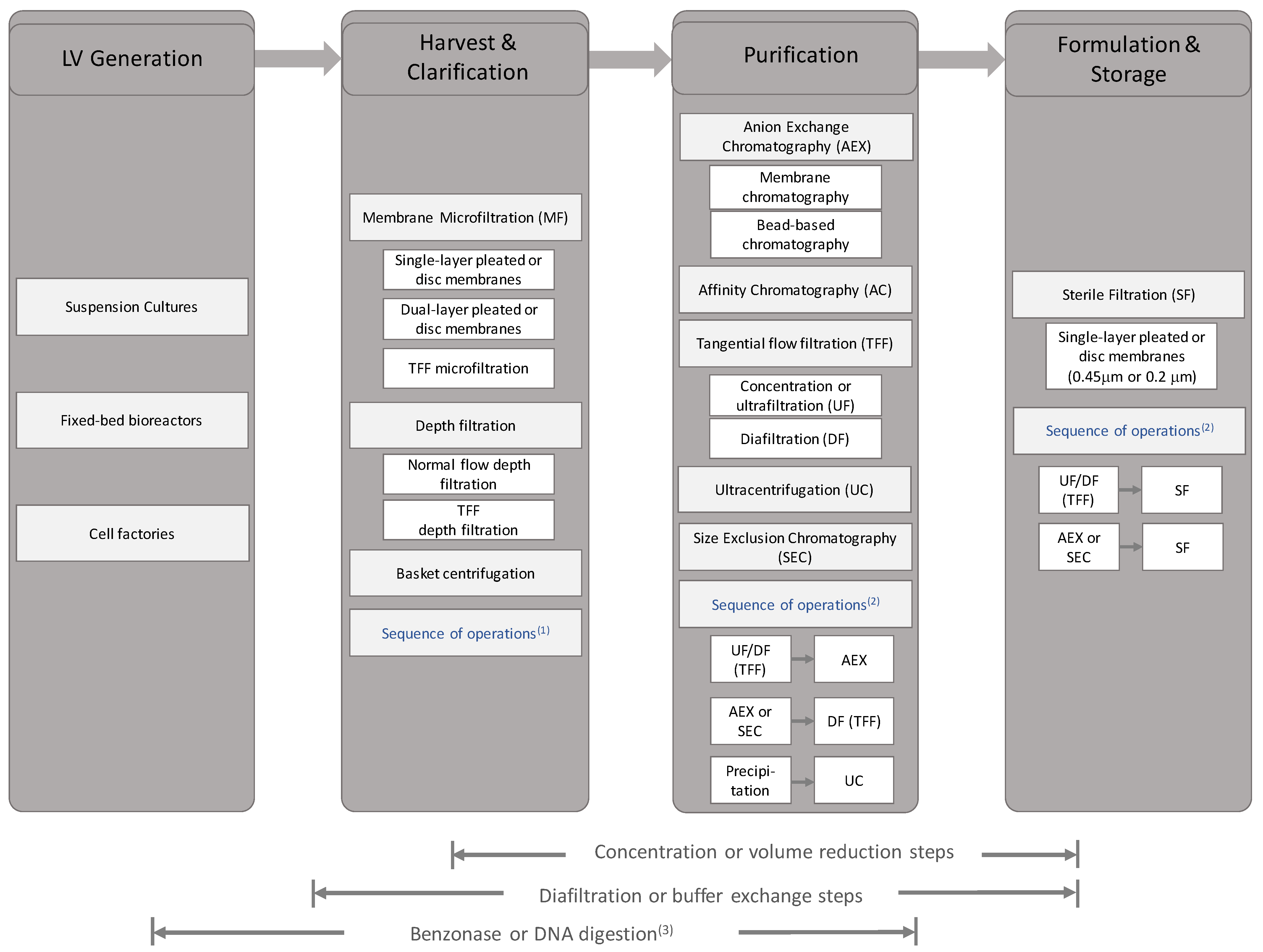{kind=link}
Table 1.
Common impurities within the processing of lentiviral vectors.
| Product Impurities | Process Impurities |
|---|---|
| Non-functional vector (broken, immature, insufficient envelope protein or non-packaged) | Host cell proteins, lipids, DNA and debris |
| Free Vector Envelopes | Plasmid DNA |
| Viral aggregates | Transfection Reagents |
| Free vector components | Expression Enhancers/Inducing Agents |
| Proteoglycans | Buffers |
| Salts | |
| Nucleases | |
| Culture Leachables | |
| Serum derived protein, amino acids, lipids and salts | |
| Media derived sugars, buffers and salts |
Table 2.
Examples of envelope proteins that have been pseudotyped with lentiviral vectors.
| Pseudotyped Envelope Protein | Virus Origin | Receptor | Cell Target | Reported Titre, Transgene, Assay Cell Target, Production Mode | Ref. |
|---|---|---|---|---|---|
| RD114 | Feline endogenous virus | ASCT-2 | T, B Cells | TU/mL, GFP, HeLa, transient [30] | [30,31,32] |
| RDPro | Feline endogenous virus | ASCT-2 | T, B Cells | TU/mL, GFP, 293T, transient [33], TU/mL, GFP, 293T, stable [7] | [7,31,33,34] |
| RD114-TR | Feline endogenous virus | ASCT-2 | T, B Cells | TU/mL, GFP, 293T, transient [33] | [31,33,35] |
| VSV-G | Vesicular Stomatitis Virus | LDL-R | Near ubiquity | TU/mL, GFP, HeLa, transient [30] | [13,30] |
| Cocal-G | Cocal | LDL-R | Near ubiquity | TU/mL, GFP, HT1080, transient [36], TU/mL, GFP, HT1080, stable [36]. TU/mL, GFP, 293T, stable [37] | [36,37] |
| GALV-TR | Gibbon ape leukaemia virus | SLC20A1 (GLVR1) | Hematopoietic Cells | TU/mL, GFP, 293T, transient [33] | [33,38,39,40] |
| BaEV | Baboon ape leukaemia virus | ASCT-2, ASCT-1 | Hematopoietic Cells | IU/mL, GFP, 293T, transient [41] | [31,41,42] |
| BaEV-TR | Baboon ape leukaemia virus | ASCT-2, ASCT-1 | Hematopoietic Cells | IU/mL, GFP, 293T, transient [41] | [31,41,42] |
| BaEVRLess | Baboon ape leukaemia virus | ASCT-2, ASCT-1 | Hematopoietic Cells | IU/mL, GFP, 293T, transient [41] | [31,41,42] |
| H/F | Measles | SLAM, CD46 | Lymphocyte | TU/mL (100× concentrated), GFP, 293T, transient [43] | [43,44,45] |
| G/F | Nipah | EphrinB2, EphrinB4 | Hematopoietic Stem Cells, endothelial cells | to TU/mL (truncation-dependent), GFP, 293, transient [46] | [46,47] |
| RabV | Rabies | p75NTR, CD56, nAchR | Neuronal | LacZ-forming units/mL (ultracentrifuge), LacZ, 293T, transient [48] | [48,49,50,51,52] |
| MOKV | Mokola | Likely similar to RabV | Neuronal | CFU/μg p24, -galactosidase, HOS, transient [53]. TU/mL GFP, 293T, transient [54] | [53,54,55,56] |
| SV | Sindbis | Integrin, heparin sulphate | 67LR, NRAMP2 | TU/mL, GFP, 293T, transient [57] | [57,58,59,60] |
| PRYV-G | Piry | Unknown | – TU/mL (ultracentrifuged 500–1000×), GFP 293T, transient, [61] | [37,61] | |
| CNV-G | Chandipura | Unknown | Neuroblastoma Cells | 6.9–9.1 TU/mL (ultracentrifuged 500–1000×), GFP 293T, transient, [61,61] | |
| Ampho | Murine Leukaemia Virus | SLC20A2 (GLVR2) | Hematopoietic Cells | TU/mL, GFP, HeLa, transient [30]. CFU/mL, -galactosidase, 293T, transient, [40] | [30,40,62] |
| F/HN | Sendai | ASGP-R | Hepatocytes, T lymphocytes | TU/mL, -galactosidase, HT1080, transient [11] | [11,63,64] |
| △GP Jenv | Jaagsiekte Sheep Retrovirus | hyaluronidase 2 | Lung Epithelial Cells | TU/mL, GFP, 293T, transient [65] | [65,66,67] |
Table 3.
Stable and inducible cell lines for Lentiviral vector production.
| Cell Line | Inducible | Vector Generation | Envelope | Transgene | Reported Titre | Adherent or Suspension | Notes | Ref. |
|---|---|---|---|---|---|---|---|---|
| LentiPro26 | N/A | 3rd Generation | MLV Amphotropic Envelope | GFP | TU/mL/Day | Adherent | Mutated HIV-1 protease for less cytotoxic activity. | [121] |
| WinPac | N/A | 3rd Generation | RDPro | GFP | TU/mL | Adherent | Retroviral Tagging and Recombinase Mediated Cassette Exchange for high Gag-Pol expression. | [7] |
| STAR | N/A | 2nd Generation | MLV Amphotropic Envelope | GFP | IU/mL | Adherent | HIV-1 Gag-Pol delivered by MLV vector. | [34,81] |
| STAR | N/A | 2nd Generation | RDPro | GFP | IU/mL | Adherent | HIV-1 Gag-Pol delivered by MLV vector. | [34,81] |
| RD2-MolPack | N/A | 2nd Generation | RD114-TF | GFP | TU/mL | Adherent | RD114 fused to cytoplasmic tail of MLV-ampho 4070. Baculo-AAV integration of Gag-Pro-Pol and Rev. | [122] |
| RD3-MolPack | N/A | 3rd Generation | RD114-TF | GFP | TU/mL | Adherent | RD114 fused to cytoplasmic tail of MLV-ampho 4070. Baculo-aav integration of Gag-Pro-Pol and rev. | [123] |
| GPRG-TL20-GFP | TET-OFF | 3rd Generation | VSV-G | GFP | TU/mL | Adherent | Concatemeric array transfection technique for SIN vector genome | [124] |
| GPRG-TL20-IL2RG | TET-OFF | 3rd Generation | VSV-G | IL2RG | TU/mL | Adherent | Concatemeric array transfection technique for SIN vector genome | [124] |
| EF1hWASp | TET-OFF | 3rd Generation | VSV-G | hWASp | IU/mL | Adherent | Based on GPRG | [125] |
| 293SF-PacLV | TET-ON, Cumate | 3rd Generation | VSV-G | GFP | TU/mL/day | Suspension | Serum Free | [126] |
Table 4.
Examples of upstream cell culturing for the production of lentviral vector.
| Supplier | Model | Volume/Growing Area | Cell Growth | Vessel Type | Ref. |
|---|---|---|---|---|---|
| Corning | CellCube | 8500 to 85,000 cm; 2.6–8 L | Adherent | Multilayer and Perfusion | [137] |
| Corning | CellSTACK | 636–25,440 cm (1–40 stacks) | Adherent | Multilayer | [97] |
| Corning | Hyperflask | 1720 cm; 500 mL | Adherent | Multilayer | [138] |
| Corning | HyperStack | 6000–18,000 cm (12–36 layers) | Adherent | Multilayer | [139] |
| Cytiva * | WAVE | 10–200 L | Suspension | Rocking Platform | [140] |
| Cytiva * | Xcellerex XDR | 4.5–2000 L | Suspension | Stirred Tank | [141] |
| Eppendorf | BioBLU 5p | Up to 180,000 cm | Adherent | Fixed Bed | [142] |
| Pall | Allegro STR | 20–2000 L | Suspension | Stirred Tank | [143] |
| Pall | iCELLIS | 0.53–4 m; 1 L (Nano). 66–500 m; 120 L (500 Model) | Adherent | Fixed Bed | [144,145] |
| Sartorius | AMBR | 10–250 mL | Suspension | Stirred Tank | [146] |
| Sartorius | BioStat | 1–2000 L | Suspension | Stirred Tank | [147] |
| ThermoFisher | NUNC Cell Factories | 632–25,280 cm; 1–40 Layers | Adherent | Multilayer | [148] |
| Univercells | Scale-X | 2.4 m (Hydro); 10–30 m (Carbo); 600 m (Nitro) | Adherent | Fixed Bed | [149] |
* Formerly GE Healthcare Life Sciences.
Table 5.
Examples of filtration units for initial clarification of lentiviral vectors.
| Chemistry | Cell Culture | Constant Flux or Constant Pressure | Load Challenge | Vector Recovery | Ref |
|---|---|---|---|---|---|
| PES | HEK-293T adherent | Constant Flux-(1200 LMH) | 112 L/m | 91 ± 6% | [190] |
| PES | HEK-293T adherent | Constant Flux-(220 LMH) (small scale); (146 LMH) (large scale) | 290 kg/m * (small scale); 215 kg/m * (first scale-up); 232 kg/m * (second scale-up) | [145] | |
| Cellulose Acetate | HEK-293T adherent | Constant Flux-(220 LMH) (small scale); (146 LMH) (large scale) | 290 kg/m * (small scale); 215 kg/m * (first scale-up); 232 kg/m * (second scale-up) | 75–90% functional particle, 100% total particles | [145] |
| PES | HEK-293T Adherent | Constant Pressure- (0.5 barg) | ≈175 L/m * | ≈71% * | [183] |
| PVDF | HEK-293T Adherent | Constant Pressure- (0.5 barg) | ≈360 L/m * | ≈78% * | [183] |
| Nylon 66 | HEK-293T Adherent | Constant Pressure- (0.5 barg) | ≈390 L/m * | ≈77% * | [183] |
| Resin-bonded GF | HEK-293T Adherent | Constant Pressure- (0.5 barg) | ≈1850 L/m * | ≈100% * | [183] |
| Polypropylene | HEK-293Tsa (suspension) | Constant Flux-(150 to 75 LMH) | 90 L/m | >70% physical titre (ELISA); >95% TU/mL | [188] |
* Calculated from data in text. PES, Polyethersulphone; PVDF, Polyvinylidene fluoride.
Table 6.
Example of chromatographic operations in lentiviral vector purification.
| Unit Name | Mode | Stationary Phase | Ligand Chemistry | Column Volume | Equilibration Buffer (Composition and CV) | Wash Buffer (Composition and CV) | Elution Buffer (Composition and CV) | Elution Type | Feed (Composition and CV) | Envelope Protein | Vector Recovery | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BIA CIM-IDA-NI | Affinity | Monolith | IMAC (with Histidine Tagged LV) | 0.34 mL | 5 CV PBS supplemented with 10 mM imidazole | 30CV, 3 mM imidazole, PBS | 150 mM imidazole | Isocratic | Up to 117 CV * DMEM, 10% FBS, 10 mM imidazole | VSV-G | 69% **** | [215] |
| CIM DEAE | AEX | Monolith | DEAE | 8 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | Up to 1.5 M NaCl, 10 mM Tris (pH 8) | Gradient | 18.75 CV * (150 mL) | VSV-G | 65% | [119] |
| CIM DEAE | AEX | Monolith | DEAE | 8 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | 0.1 M, 0.65 M NaCl (10 mM Tris (pH 8)) | Stepwise | DMEM, 10% FBS | VSV-G | 80% | [190] |
| CIM DEAE | AEX | Monolith | DEAE | 8 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | 0.1 M, 0.65 M NaCl, 50 mM HEPES (pH 8) | Stepwise | DMEM, 10% FBS | VSV-G | 71% | [190] |
| CIM DEAE | AEX | Monolith | DEAE | 8 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | 0.1 M, 0.65 M NaCl (10 mM PBS | Stepwise | DMEM, 10% FBS | VSV-G | 74% | [190] |
| CIMac Streptavidin | Affinity | Monolith | Streptavidin–Biotin Mimic | 0.1 mL | 50 CV PBS, 500 CV DMEM, 50CV PBS | 2 × 300 CV, PBS | 150 CV X-Vivo15 media supplemented 15 mM Biotin, 0.5% BSA | Isocratic | 500 CV, serum-free DMEM | RDPro | 20% | [167] |
| Fibro (Prototype) | AEX | Nanofiber | RC QA | 0.1 mL | PBS | PBS | 0–50% elution buffer over 100 CV, 60 CV step to 100%. PBS, and PBS +2 M NaCl | Gradient/Step | 4000 CV; DMEM, 10% FBS | RDPro | 90% | [216] |
| FractoGel | Affinity | Bead | Heparin | 1 mL | 150 mM NaCl. 20 mM Tris (pH 7.5) | 15 CV; 150 mM NaCl | 13 CV; 350 mM NaCl | Gradient | 35 CV; Freestyle | VSV-G | 53% | [157] |
| HiTrap Q | AEX | Bead | Quaternary Amine | 1 mL | 10 CV; 100 mM Tris (pH 7.5) | 100 mM Tris (pH 7.5) + 1 M NaCl | Gradient | 1 CV; 200× concentration LV in equilibration buffer | VSV-G ** | 53% *** after ultracentrifugation and desalting | [181] | |
| Mustang Q | AEX | Membrane | Quaternary Amine | 60 mL | 40 CV, 25 mM Tris-HCl (pH 8) | 40 CV, equilibration buffer | 20 mL 25 mM Tris-HCl (pH 8), 1.2 M NaCl | Isocratic | 100 CV, DMEM, 10% FBS, 1% sodium pyruvate | VSV-G | Final titre approximately to TU in 150 mL from 6 L non-concentrated | [139] |
| Mustang Q | AEX | Membrane | Quaternary Amine | 0.18 mL | 11 CV *, 25 mM Tris-HCl (pH 8), 0.6 M NaCl | 0.3 M to 1.5 M NaCl in 25 mM Tris- HCl (pH 8) | Stepwise | 2777 CV *. DMEM, 10% FBS, 1% GlutaMAX, 1% Pen/Strep adjusted to 25 mM Tris-HCl (pH 8), 0.6 M NaCl | VSV-G | 76% | [138] | |
| Mustang Q | AEX | Membrane | Quaternary Amine | 0.8 mL | 12.5 CV * 150 mM NaCl, 25 mM Tris-HCl (pH 7.4) | 12.5 CV * 150 mM NaCl, 25 mM Tris-HCl (pH 7.4) | 1M NaCl, 25 mM Tris-HCl (pH 7.4) | Stepwise | 675 CV; DMEM, 10% FBS ** | Measles M/V | 80% | [217] |
| Mustang Q | AEX | Membrane | Quaternary Amine | 0.8 mL | 12.5 CV * 150 mM NaCl, 25 mM Tris-HCl (pH 7.4) | 12.5 CV * 150 mM NaCl, 25 mM Tris-HCl (pH 7.4) | 0.2 to 0.4 NaCl, 25 mM Tris-HCl (pH 7.4) | Gradient | 675 CV; DMEM, 10% FBS ** | Measles M/V | 65% | [217] |
| Sartobind D | AEX | Membrane | DEAE | 2 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | Up to 1.5 M NaCl, 10 mM Tris (pH 8) | Gradient | 75 CV (150mL) | VSV-G | 29% | [119] |
| Sartobind D | AEX | Membrane | DEAE | 2.1 mL | 10 mM Tris (pH 8) | 10 mM Tris (pH 8) | 0.1 M, 0.65 M NaCl, 50 mM Tris (pH 8) | Stepwise | DMEM, 10% FBS | VSV-G | 28% | [190] |
| Sartobind Q | AEX | Membrane | Quaternary Amine | 400 mL | 50 mM HEPES, 300 mM NaCl (pH 7.5) | 50 mM HEPES, 300 mM NaCl (pH 7.5) | 12, 30, 45% elution. 1.5 M NaCl, 50 mM HEPES (pH 7.5) | Stepwise | 11 kg concentrated and Buffer exchanged | VSV-G | 33.1% | [145] |
* Calculated from text; ** assumption based on text; *** recovery based on integration copy numbers; **** elution efficiency: TU eluted/captured.
Table 7.
Examples of tangential flow filtration operations and membrane materials in lentiviral vector processing.
Table 7.
Examples of tangential flow filtration operations and membrane materials in lentiviral vector processing.
| Pseudotype | Step Before TFF | TFF Operation | Membrane Type. (MWCO, Total Area), Brand, Supplier | Membrane Format (TFF System) | Ref. |
|---|---|---|---|---|---|
| VSV-G | Clarification | UF (20× CF). DF (10 DV) | Cellulose. Ultracel type V (100 kDA or 300 kDa, 0.1 m), Pellicon 2, Merck Millipore | Flat Sheet Cassette (n.m.) | [145] |
| VSV-G | Clarification | UF/DF | PES. Biomax type V (100 kDA, 0.1 m), Pellicon 2, Merck Millipore | Flat Sheet Cassette (n.m.) | [145] |
| VSV-G | Clarification | UF/DF | Cellulose. Hydrosart (100 kDA, 0.11 m), Sartocon Slice, Sartorius | Flat Sheet Cassette (AKTA crossflow) | [145] |
| VSV-G | Clarification | UF/DF | Cellulose. Hydrosart (100 kDA, 0.11 m), Sartocon Slice, Sartorius | Flat Sheet Cassette (Cogent M) | [145] |
| VSV-G | Clarification | UF (13–18× CF) DF (9–10 DV) | Cellulose. Hydrosart (100 kDA, 5 × 0.6 m or 1 × 3 m + 2 × 0.6m) Sartocon or Sartocube, Sartorius | Flat Sheet Cassette (Mobius) | [145] |
| VSV-G | Clarification | UF (110× CF) DF (20 DV) (Tandem TFF—1 of 2) | Membrane type (n.m.). 320 fibres (0.5 mm internal diameter) with a total surface area of 615 cm and a 500 kDa cut-off | Hollow Fibre. (KrosFlo Research II TFF System) | [242] |
| VSV-G | TFF (Tandem TFF—1 of 2) | UF only (50× CF) (Tandem TFF—2 of 2) | Membrane type (n.m.). 12 fibres (0.5 mm internal diameter) with a total surface area of 40 cm and a 500 kDa cut-off | Hollow Fibre. (KrosFlo Research II TFF System) | [242] |
| VSV-G | Clarification | UF (10–22× CF) DF (10–11 DV) | 36 mPES fibres (0.5 mm diameter, 20cm) with a total surface area of 115 cm and 500 kDA cut-off | Hollow Fibre. (KrosFlo Research II TFF System, Spectrum Labs, Repligen) | [243] |
| VSV-G | Clarification | UF (10-22x CF) DF (10-11 DV) | Cytiva (former GE Healthcare) PES (1 mm diameter, 30 cm) with a total surface area of 110 cm and 750 kDA cut-off | Hollow Fibre. (KrosFlo Research II TFF System, Spectrum Labs, Repligen) | [243] |
| GalV-TR | Clarification | UF (10–22× CF) DF (10–11 DV) | Cytiva (former GE Healthcare) PES (1 mm dia., 30 cm) with a total surface area of 110 cm and 750 kDA cut-off | Hollow Fibre. (KrosFlo Research II TFF System, Spectrum Labs, Repligen) | [243] |
| VSV-G. | Clarification | UF only | Omega PES (200 cm). 50 kDA, 100 kDA, 300 kDA | Flat sheet cassette. (Centramate, Pall) | [65] |
| VSV-G | Clarification | UF only | Omega PES (200 cm). 50 kDA, 100 kDA, 300 kDA | Flat sheet cassette. (Centramate, Pall) | [65] |
| GP JenV | Clarification | UF only | Omega PES (50 kDa, 200 cm). | Flat sheet cassette. (Centramate, Pall) | [65] |
| GP JenV. | Clarification | UF only | Omega PES (50 kDa, 200 cm). | Flat sheet cassette. (Centramate, Pall) | [65] |
| RDPro | Clarification | DF only | 36 mPES fibres (0.5 mm diameter, 20 cm) with a total surface area of 115 cm and 500 kDA cut-off | Hollow Fibre. (KrosFlo Research II TFF System) | [216] |
| VSV-G | Chromatography/ Benzonase | UF only (30–40× CF) | Membrane type (n.m.). (100 kDA, 50 cm) | Hollow fibre. (Cytiva) | [244] |
| VSV-G | Chromatography/ Benzonase | UF only (30–40× CF) | Membrane type (n.m.). VivaFlow (100 kDA, 50 cm) | Flat sheet cassette. (Vivaflow, Sartorius) | [244] |
| VSV-G | Chromatography/ Dilution | DF/UF/DF | Membrane type (n.m.). VivaFlow (100 kDA, 50 cm) | Flat sheet cassette. (Vivaflow, Sartorius) | [98] |
| BaEV-R-less | Chromatography/ Dilution | DF/UF/DF | Membrane type (n.m.). VivaFlow50 (100 kDA, 50 cm) | Flat sheet cassette. (Vivaflow, Sartorius) | [98] |
| VSV-G | Chromatography | UF (10× CF). DF (2 DV) | Membrane type (n.m.). Pellicon-2 (500 kDA, 0.1 m) | Flat Sheet Cassette. (Pellicon-2 Mini, Merck Millipore) | [140] |
| VSV-G | Chromatography | UF Only | Membrane type (n.m.). VivaFlow (100 kDA, 50 cm) | Flat sheet cassette. (Vivaflow, Sartorius) | [190] |
| VSV-G | Chromatography | UF Only | Membrane type (n.m.). (100 kDA, 50 cm) | Hollow fibre. (Cytiva) | [96] |
Table 8.
Examples of tangential flow filtration operating conditions in lentiviral vector processing.
Table 8.
Examples of tangential flow filtration operating conditions in lentiviral vector processing.
| Pseudotype | TFF Mode: Permeate Flux Control (Flux, TMP) or TMP Control * | Notes | Ref. |
|---|---|---|---|
| VSV-G | Constant flux (n.m., 0.05–0.1 bar) | 59–74 L/m feed load. DF buffer 50 mM HEPES + 300 mM NaCl (pH 7.5) | [145] |
| VSV-G | Constant flux (n.m., 0.10 bar < TMP < 0.2 bar) | 30–35 L/m feed load during concentration. DF buffer 50 mM HEPES + 300 mM NaCl (pH 7.5) | [145] |
| VSV-G | Constant flux. (10 LMH, 0.06 bar) | 10–70 kg/m feed load. 3 L/h Feed flowrate ** (64% crossflow). DF buffer 50 mM HEPES + 300 mM NaCl (pH 7.5) | [145] |
| VSV-G | Constant flux. (24–100 LMH, 0.2–0.5 bar) | 50–120 kg/m. Typically 60 kg/m feed load. 20–110 L/h Feed flowrate ** (88–90% crossflow). DF buffer 50 mM HEPES + 300 mM NaCl (pH 7.5) | [145] |
| VSV-G | Constant flux (13–14 LMH, 0.1–0.2 bar) | 42–43 kg/m feed load. 350–496 L/h Feed flowrate ** (89% crossflow). 70% recovery, >70% dsDNA clearance. DF buffer 50 mM HEPES + 300 mM NaCl (pH 7.5) | [145] |
| VSV-G | Constant TMP (feed control, <6 psi (0.4 bar)) | DF buffer is a 1L mixture of Dulbecco’s Phosphate Buffered Saline with fetal calf serum (FCS). >97% recovery | [242] |
| VSV-G | Constant TMP (feed control at <9 psi, (0.6 bar)) | >97% Recovery. | [242] |
| VSV-G | Constant TMP (feed control at 7 psi, 0.5 bar) | 40% recovery. Membranes pre-treated with DMEM and FBS. Performed at 4 °C. Feed flowrate, at 40 mL/min | [243] |
| VSV-G | Constant TMP (feed control at 7 psi, 0.5 bar) | Up to 100% recovery depending on buffer composition (pH, salt, sucrose, etc). Membranes pre-treated with DMEM and FBS. Performed at 4 °C. Feed flowrate, at 40 mL/min | [243] |
| GalV-TR | Constant TMP (feed control at 7 psi, 0.5 bar) | Up to 80% recovery depending on buffer composition (pH, salt, sucrose, etc). Membranes pre-treated with DMEM and FBS. Performed at 4 °C. Feed flowrate, at 40 mL/min | [243] |
| VSV-G. | Constant TMP (feed control at 5 psi (0.3 bar), retentate backpressure 0–5 psi) | “Low TMP”. Recoveries are 3% (50 kDA), 21% (100 kDa), and 11% (300kDa) at 0.3 bar TMP | [65] |
| VSV-G | Constant TMP (feed control at 15 psi (1 bar), retentate backpressure 10 psi) | “high TMP” (0.8 bar). Recoveries are 100% (50 kDa), 91% (100 kDa), and 81% (300 kDa). Overall recovery using 50 kDa is 99.6% | [65] |
| GP JenV | Constant TMP (feed control at 5 psi (0.3 bar) | “Low TMP” (0.3bar TMP). 99% recovery | [65] |
| GP JenV. | Constant TMP (feed control at 15 psi (1 bar), retentate backpressure 10 psi) | “high TMP” (0.8 bar). 98% recovery. Overall recovery using TFF is higher than overall recovery using ultracentrifugation | [65] |
| RDPro | Constant TMP = 1 psig (feed control at 20–30 mL/min) | 20 mM Tris (pH 7.4) | [216] |
| VSV-G | Constant TMP (feed flow at 75 mL/min, 0.3 bar TMP) | Benzonase addition and 1:1 dilution immediately performed after elution (i.e., before TFF). Measured permeate flux is 30–35 LMH | [244] |
| VSV-G | Constant TMP (feed control at 1–1.5 bar) | Benzonase addition and 1:1 dilution immediately performed after elution (i.e., before TFF). Measured permeate flow is 5 mL/min | [244] |
| VSV-G | n.m. | From suspension cells. 40–70% overall recovery. 50-fold concentration from harvest | [98] |
| BaEV-R-less | n.m. | From suspension cells. 30–36% overall recovery. 50-fold concentration from harvest | [98] |
| VSV-G | n.m. | [140] | |
| VSV-G | Constant TMP (feed control at 1 bar) | Feed flowrate at 7 mL/min. 72% recovery. Membranes pre-treated with human serum albumin (HSA) | [190] |
| VSV-G | n.m. | Overall yield: 16–24% | [96] |
n.m., not mentioned in reference; * pressure control—feed control is a form of Transmembrane Pressure (TMP) control if the difference between inlet and outlet (retentate) pressure is maintained; ** calculated from data in the reference.
Table 9.
Known challenges in LV Production.
| No. | Challenge * | Solution ** |
|---|---|---|
| 1 | Reliance on transient transfection methods and lack of suitable stable cell line | Development of stable packaging cell line (e.g., WinPac by Sanber et al. [7] in [270]) |
| 2 | Use of adherent cell in LV production | Development of suspension cultures McCarron et al. [270] particularly ([98,129,146,170]) |
| 3 | Low recovery and loss of vector functionality | The fundamental study and understanding of LV bioprocessing and rational bioprocess development will be crucial to address this. |
| 4 | Lack of international standards for LV vector products | This is an industry-wide challenge. Product-specifications and process-specifications may vary depending on application. Review on a case-by-case basis ([270]) |
| 5 | Difference in purity and potency requirements for in vivo and ex vivo applications | As in No. 4 |
| 6 | Greater batch-to-batch variability (e.g., ratio of total vs functional vector) | Ensure measurement of both total and functional vectors as well as assessments following cell transduction (e.g., proviral DNA titres, RNA, etc.) ([270]). |
| 7 | Inter-assay variation of LV titering between laboratories and operators | This is a challenge which needs to be addressed at company-level. Implementing automated, analytical assays and comparing to international standards such as WHO LV standard [255]. Training in established protocols and maintaining personnel expertise. |
| 8 | Challenges in testing for replication-competent lentivirus | Development of more sensitive and rapid PCR-based techniques (McCarron et al. [270]). |
* Summarised from McCarron et al. [270]. ** Text in italics are the current authors comments.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Perry, C.; Rayat, A.C.M.E. Lentiviral Vector Bioprocessing. Viruses 2021, 13, 268. https://doi.org/10.3390/v13020268
AMA Style
Perry C, Rayat ACME. Lentiviral Vector Bioprocessing. Viruses. 2021; 13(2):268. https://doi.org/10.3390/v13020268
Chicago/Turabian StylePerry, Christopher, and Andrea C. M. E. Rayat. 2021. "Lentiviral Vector Bioprocessing" Viruses 13, no. 2: 268. https://doi.org/10.3390/v13020268
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.