
Molecular Epidemiology of Rabies in Wild Canidae in Tunisia

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Origin of Virus Isolates
2.2. RNA Extraction and Amplification
2.3. DNA Sequencing
2.4. Maximum Likelihood Phylogeny
2.5. Bayesian Phylogeny
2.6. Reservoir Species Analysis
3. Results
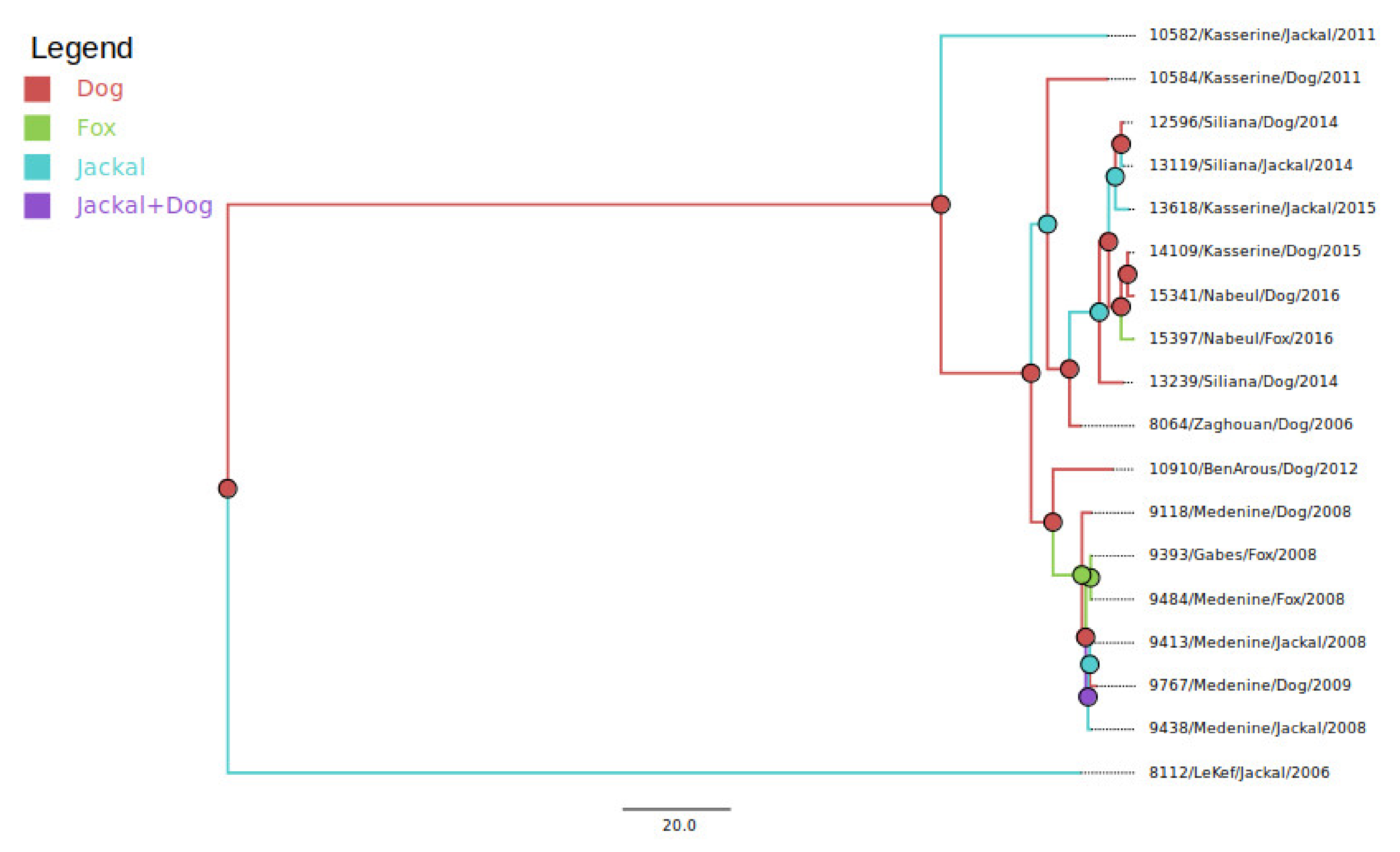
3.1. Virus Taxonomy and Clade Membership
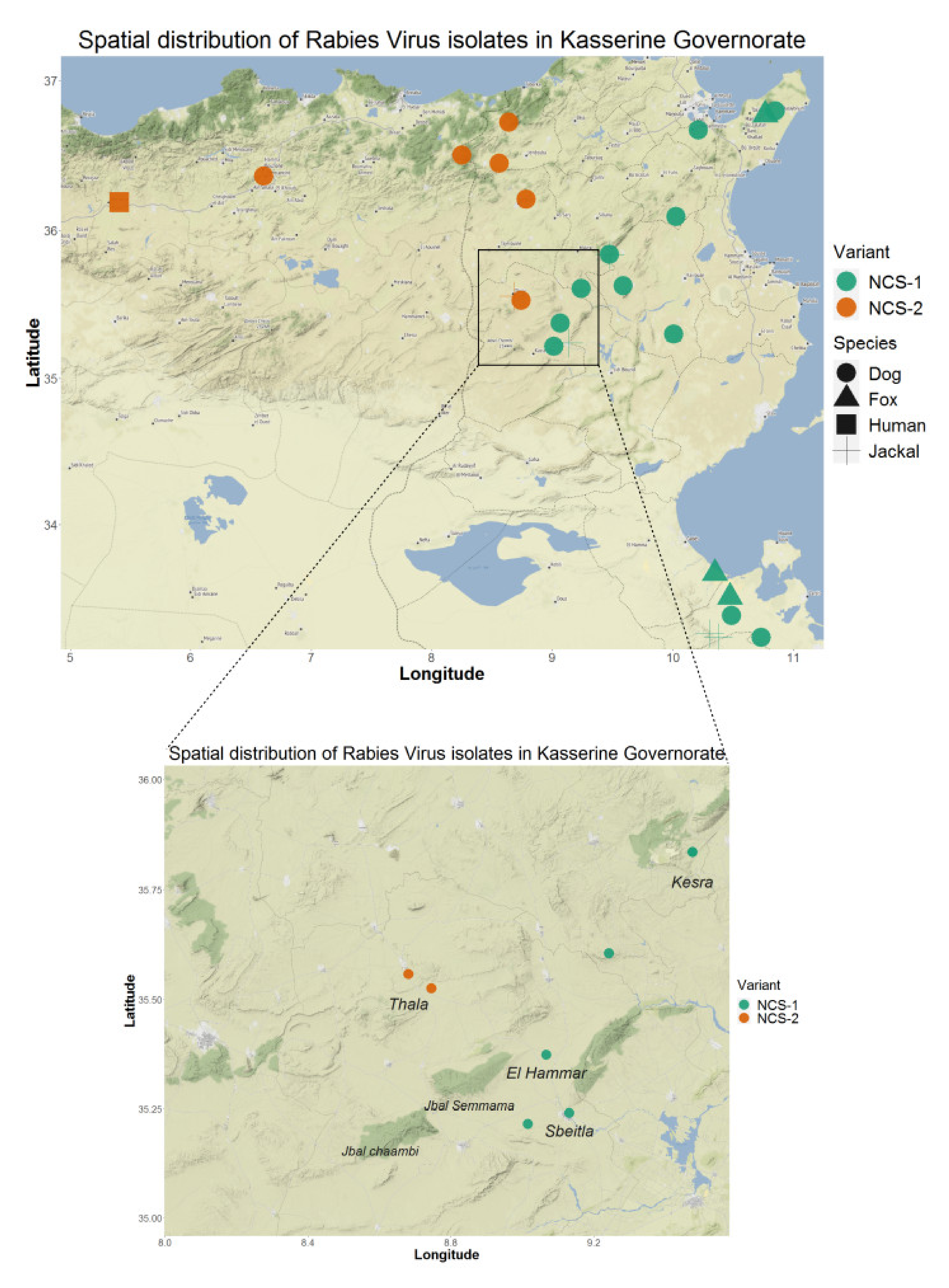
3.2. Comparison of Wild Canidae Virus Isolates with Tunisian Canine Variant Strains
3.3. Reservoir Species Analysis
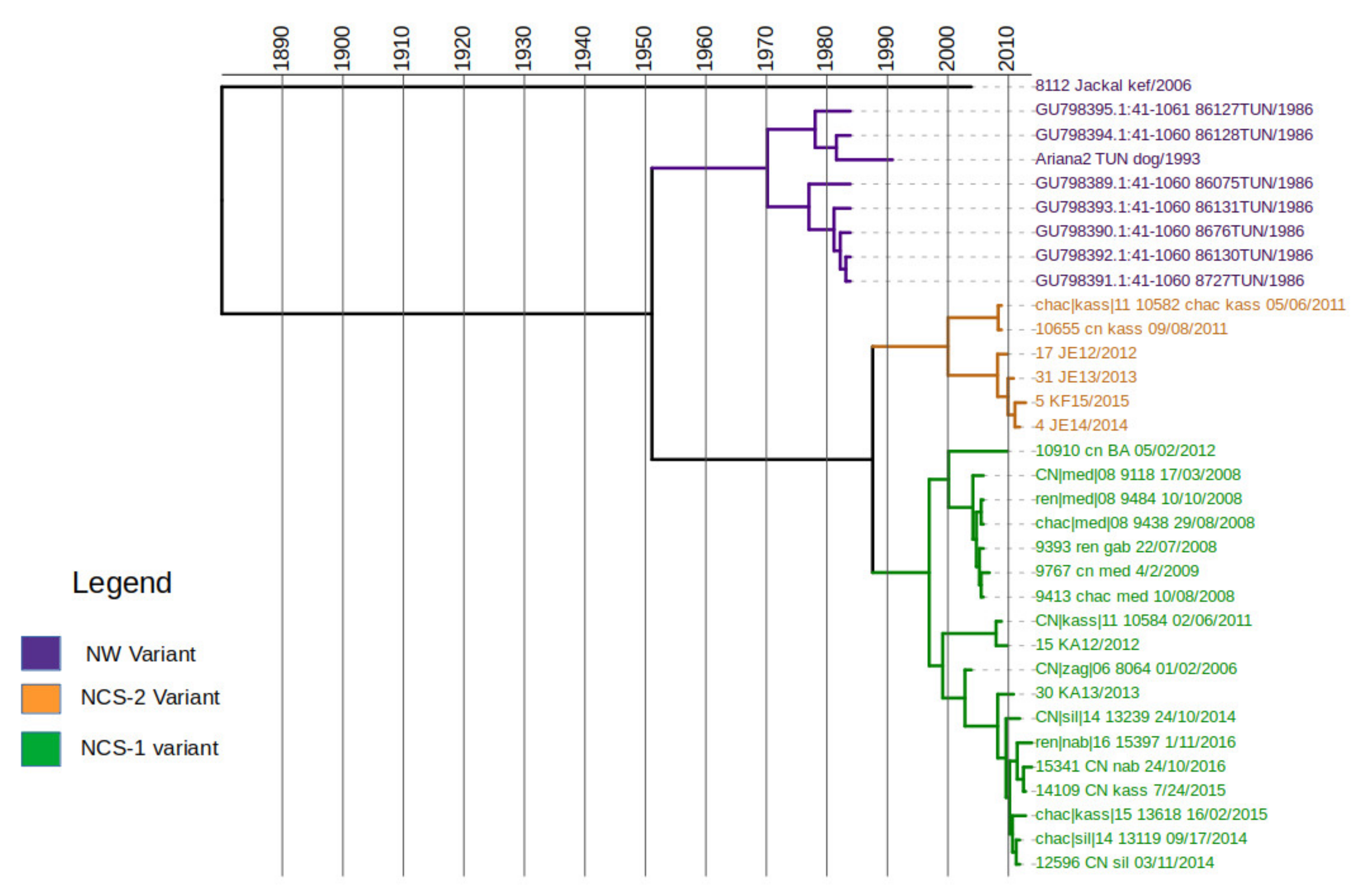
3.4. Time Scale of Rabies Virus Evolution in Tunisia
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bourhy, H.; Kissi, B.; Tordo, N. Molecular Diversity of the Lyssavirus Genus. Virology 1993, 194, 70–81. [Google Scholar] [CrossRef]
- Tordo, N.; Poch, O.; Ermine, A.; Keith, G. Primary Structure of Leader RNA and Nucleoprotein Genes of the Rabies Genome: Segmented Homology with VSV. Nucleic Acids Res. 1986, 14, 2671–2683. [Google Scholar] [CrossRef] [Green Version]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H. Large-Scale Phylogenomic Analysis Reveals the Complex Evolutionary History of Rabies Virus in Multiple Carnivore Hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef] [PubMed]
- International Commitee on Taxonomy of Viruses (ICTV). 2021. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 21 October 2021).
- Mollentze, N.; Biek, R.; Streicker, D.G. The Role of Viral Evolution in Rabies Host Shifts and Emergence. Curr. Opin. Virol. 2014, 8, 68–72. [Google Scholar] [CrossRef] [Green Version]
- Rupprecht, C.E.; Hanlon, C.A.; Hemachudha, T. Rabies Re-Examined. Lancet Infect. Dis. 2002, 2, 327–343. [Google Scholar] [CrossRef]
- Bourhy, H.; Reynes, J.-M.; Dunham, E.J.; Dacheux, L.; Larrous, F.; Huong, V.T.Q.; Xu, G.; Yan, J.; Miranda, M.E.G.; Holmes, E.C. The Origin and Phylogeography of Dog Rabies Virus. J. Gen. Virol. 2008, 89, 2673–2681. [Google Scholar] [CrossRef] [PubMed]
- Fooks, A.R.; Banyard, A.C.; Horton, D.L.; Johnson, N.; McElhinney, L.M.; Jackson, A.C. Current Status of Rabies and Prospects for Elimination. Lancet 2014, 384, 1389–1399. [Google Scholar] [CrossRef]
- Blancou, J. Ecology and Epidemiology of Fox Rabies. Rev. Infect. Dis. 1988, 10, S606–S609. [Google Scholar] [CrossRef]
- Oliveira, R.N.; Freire, C.C.; Iamarino, A.; Zanotto, P.M.; Pessoa, R.; Sanabani, S.S.; de Souza, S.P.; Castilho, J.G.; Batista, H.B.C.R.; Carnieli, P., Jr.; et al. Rabies Virus Diversification in Aerial and Terrestrial Mammals. Genet. Mol. Biol. 2020, 43, e20190370. [Google Scholar] [CrossRef] [PubMed]
- Blancou, J.; Aubert, M.F. Transmission of rabies virus: Importance of the species barrier. Bull. Acad. Natl. Med. 1997, 181, 301–311; discussion 311–312. [Google Scholar] [PubMed]
- Amouri, I.K.; Kharmachi, H.; Djebbi, A.; Saadi, M.; Hogga, N.; Zakour, L.B.; Ghram, A. Molecular Characterization of Rabies Virus Isolated from Dogs in Tunisia: Evidence of Two Phylogenetic Variants. Virus Res. 2011, 158, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Hogga, N. Etude Moléculaire des Isolats de Virus Rabique Chez les Animaux Domestiques; Mémoire de Master de Microbiologie; Faculté des Sciences de Tunis: Tunis, Tunisia, 2007. [Google Scholar]
- Lamine, H. Caractérisation Moléculaire des Isolats du Virus Rabique à Partir des Cas de Rage Humaine en Tunisie; Mémoire de fin d’études; Institut de Biotechnologie de Monastir: Monastir, Tunisia, 2007. [Google Scholar]
- Talbi, C.; Lemey, P.; Suchard, M.A.; Abdelatif, E.; Elharrak, M.; Jalal, N.; Faouzi, A.; Echevarría, J.E.; Morón, S.V.; Rambaut, A.; et al. Phylodynamics and Human-Mediated Dispersal of a Zoonotic Virus. PLoS Pathog. 2010, 6, e1001166. [Google Scholar] [CrossRef] [Green Version]
- Available online: http://www.rage.tn/upload/1516305067.pdf (accessed on 30 September 2021).
- Horton, D.L.; McElhinney, L.M.; Freuling, C.M.; Marston, D.A.; Banyard, A.C.; Goharrriz, H.; Wise, E.; Breed, A.C.; Saturday, G.; Kolodziejek, J.; et al. Complex Epidemiology of a Zoonotic Disease in a Culturally Diverse Region: Phylogeography of Rabies Virus in the Middle East. PLoS Negl. Trop. Dis. 2015, 9, e0003569. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.oie.int/fr/ce-que-nous-faisons/normes/codes-et-manuels/acces-en-ligne-au-manuel-terrestre/ (accessed on 26 October 2021).
- World Health Organization. WHO Expert Consultation on Rabies: Third Report; WHO Technical Report Series; World Health Organization: Geneva, Switzerland, 2018; ISBN 978-92-4-121021-8. [Google Scholar]
- Nadin-Davis, S.A. Polymerase Chain Reaction Protocols for Rabies Virus Discrimination. J. Virol. Methods 1998, 75, 1–8. [Google Scholar] [CrossRef]
- Wang, L.; Wu, H.; Tao, X.; Li, H.; Rayner, S.; Liang, G.; Tang, Q. Genetic and Evolutionary Characterization of RABVs from China Using the Phosphoprotein Gene. Virol. J. 2013, 10, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 12 January 2021).
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; Maio, N.D.; et al. BEAST 2.5: An Advanced Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouckaert, R.R.; Drummond, A.J. BModelTest: Bayesian Phylogenetic Site Model Averaging and Model Comparison. BMC Evol. Biol. 2017, 17, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Kahle, D.; Wickham, H. Ggmap: Spatial Visualization with Ggplot2. R J. 2013, 5, 144–161. [Google Scholar] [CrossRef] [Green Version]
- Bonnaud, E.M.; Troupin, C.; Dacheux, L.; Holmes, E.C.; Monchatre-Leroy, E.; Tanguy, M.; Bouchier, C.; Cliquet, F.; Barrat, J.; Bourhy, H. Comparison of Intra- and Inter-Host Genetic Diversity in Rabies Virus during Experimental Cross-Species Transmission. PLoS Pathog. 2019, 15, e1007799. [Google Scholar] [CrossRef] [Green Version]
- Omodo, M.; Ar Gouilh, M.; Mwiine, F.N.; Okurut, A.R.A.; Nantima, N.; Namatovu, A.; Nakanjako, M.F.; Isingoma, E.; Arinaitwe, E.; Esau, M.; et al. Rabies in Uganda: Rabies Knowledge, Attitude and Practice and Molecular Characterization of Circulating Virus Strains. BMC Infect. Dis. 2020, 20, 200. [Google Scholar] [CrossRef]
- Bourhy, H.; Kissi, B.; Audry, L.; Smreczak, M.; Sadkowska-Todys, M.; Kulonen, K.; Tordo, N.; Zmudzinski, J.F.; Holmes, E.C. Ecology and Evolution of Rabies Virus in Europe. J. Gen. Virol. 1999, 80 Pt 10, 2545–2557. [Google Scholar] [CrossRef] [PubMed]
- Bouslama, Z.; Belkhiria, J.A.; Turki, I.; Kharmachi, H. Spatio-Temporal Evolution of Canine Rabies in Tunisia, 2011–2016. Prev. Vet. Med. 2020, 185, 105195. [Google Scholar] [CrossRef]
- Brunker, K.; Lemey, P.; Marston, D.A.; Fooks, A.R.; Lugelo, A.; Ngeleja, C.; Hampson, K.; Biek, R. Landscape Attributes Governing Local Transmission of an Endemic Zoonosis: Rabies Virus in Domestic Dogs. Mol. Ecol. 2018, 27, 773–788. [Google Scholar] [CrossRef] [Green Version]
- Saito, M.; Oshitani, H.; Orbina, J.R.C.; Tohma, K.; de Guzman, A.S.; Kamigaki, T.; Demetria, C.S.; Manalo, D.L.; Noguchi, A.; Inoue, S.; et al. Genetic Diversity and Geographic Distribution of Genetically Distinct Rabies Viruses in the Philippines. PLoS Negl. Trop. Dis. 2013, 7, e2144. [Google Scholar] [CrossRef]
- Tohma, K.; Saito, M.; Kamigaki, T.; Tuason, L.T.; Demetria, C.S.; Orbina, J.R.C.; Manalo, D.L.; Miranda, M.E.; Noguchi, A.; Inoue, S.; et al. Phylogeographic Analysis of Rabies Viruses in the Philippines. Infect. Genet. Evol. 2014, 23, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.E.; Small, C. Hypsographic Demography: The Distribution of Human Population by Altitude. PNAS 1998, 95, 14009–14014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maurer, G. Agriculture in the Rif and Tell Mountains of North Africa. Mt. Res. Dev. 1992, 12, 337–347. [Google Scholar] [CrossRef]
- Holmes, E.C.; Woelk, C.H.; Kassis, R.; Bourhy, H. Genetic Constraints and the Adaptive Evolution of Rabies Virus in Nature. Virology 2002, 292, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Isolate | Animal Species | Collection Date | Location (Governorate) | Location (Delegation) | Latitude | Longitude | Nucleoprotein Sequence Accession Number | Phosphoprotein Sequence Accession Number |
|---|---|---|---|---|---|---|---|---|
| 15841 | Fox | 10 April 2017 | Nabeul | Nabeul | 36.4911995 | 10.6626997 | OK275685 | - |
| 15397 | Fox | 1 November 2016 | Nabeul | El Mida | 36.773047 | 10.7656 | OK27568 | OK27568 |
| 11284 | Fox | 17 November 2012 | Ben Arous | Mhamdia | 36.647136 | 10.066214 | OK275688 | - |
| 9484 | Fox | 10 October 2008 | Medenine | Sidi Makhlouf | 33.499538 | 10.473707 | OK27569 | OK275673 |
| 9393 | Fox | 22 July 2008 | Gabes | Mareth | 33.663899 | 10.348131 | OK275692 | OK275670 |
| 9413 | Jackal | 10 August 2008 | Medenine | Beni Khdache | 33.436471 | 10.206275 | OK275691 | OK275671 |
| 13618 | Jackal | 16 February 2015 | Kasserine | Sbeitla | 35.241052 | 9.131288 | OK275687 | OK275681 |
| 13119 | Jackal | 17 September 2014 | Siliana | Kesra | 35.8359985 | 9.4758101 | OK275693 | OK275679 |
| 10582 | Jackal | 5 June 2011 | Kasserine | Thala | 35.557643 | 8.680733 | OK275689 | OK275675 |
| 9438 | Jackal | 29 August 2008 | Medenine | Beni Khdache | 33.436471 | 10.206275 | OK275690 | OK275672 |
| 8112 | Jackal | 23 February 2006 | El Kef | Dahmani | 36.021820 | 8.906841 | OK275695 | OK275668 |
| 12596 | Dog | 3 November 2014 | Siliana | Kesra | 35.8359985 | 9.4758101 | - | OK275678 |
| 14109 | Dog | 24 July 2015 | Kasserine | Sbeitla | 35.2153015 | 9.0153503 | - | OK275682 |
| 10910 | Dog | 2 May 2012 | Ben Arous | Fouchana | 36.6683998 | 10.2124996 | - | OK275677 |
| 15341 | Dog | 24 October 2016 | Nabeul | Menzel Temime | 36.796965 | 10.853034 | - | OK275683 |
| 13239 | Dog | 24 October 2014 | Siliana | Rouhia | 35.7809982 | 9.0813599 | - | OK275680 |
| 10584 | Dog | 2 June 2011 | Kasserine | El Hammar | 35.3737984 | 9.0671101 | - | OK275676 |
| 9118 | Dog | 17 Mars 2008 | Medenine | Ben Guerdane | 33.2186012 | 10.7330999 | - | OK275669 |
| 8064 | Dog | 1 November 2006 | Zaghouan | Nadhour | 36.0915985 | 10.0277004 | - | OK275667 |
| 9767 | Dog | 2 April 2009 | Medenine | Medenine Nord | 33.3704987 | 10.4882002 | - | OK275674 |
| 10792 | Dog | 31 January 2011 | Jendouba | Ouled Mliz | 36.4488983 | 8.5616598 | - | OK275665 |
| 11747 | Dog | 11 June 2013 | Jendouba | Sloul | 36.7193985 | 8.6416702 | - | OK275664 |
| 12900 | Dog | 13 June 1014 | Jendouba | Ouechtata | 36.5028000 | 8.2497797 | - | OK275663 |
| 13961 | Dog | 5 June 2015 | El Kef | Eddir | 36.2057991 | 8.7846699 | - | OK275666 |
| Primer Name | Primer Sequence | Sense | Target Gene | Reference |
|---|---|---|---|---|
| Rab N1 | AACACCTCTACAATGGATGCCGACAA | Forward | Nucleoprotein | Nadin Davis S, 1998 |
| Rab N5 | GGATTGAC (AG) AAGATCTTGCTCAT | Reverse | Nucleoprotein | Nadin Davis S, 1998 |
| Rab Nfor | TTGT(AG) GA (TC) CAATATGAGTACAA | Forward | Nucleoprotein | Nadin Davis S, 1998 |
| Rab Nrev | CCGGCTCAAACATTCTTCTTA | Reverse | Nucleoprotein | Nadin Davis S, 1998 |
| P for | GAACCATCCCAAAYATG | Forward | Phosphoprotein | Wang L. et al., 2013 [21] |
| P rev | CTATCTTGCGCAGAAARTTCAT | Reverse | Phosphoprotein | Wang L. et al., 2013 |
| Isolate | GenBank Accession Number | Host Species | Location (Country) | Reference |
|---|---|---|---|---|
| ALG/08-153 | GU798549.1 | Dog | Algeria | (Talbi et al., 2010) |
| ALG/08-200 | GU798548.1 | Human | Algeria | (Talbi et al., 2010) |
| 8676TUN/1986 | GU798390.1 | Human | Tunisia | (Talbi et al., 2010) |
| 8727TUN/1986 | GU798391.1 | Human | Tunisia | (Talbi et al., 2010) |
| 86075TUN/1986 | GU798389.1 | Human | Tunisia | (Talbi et al., 2010) |
| 86130TUN/1986 | GU798392.1 | Human | Tunisia | (Talbi et al., 2010) |
| 86131TUN/1986 | GU798393.1 | Human | Tunisia | (Talbi et al., 2010) |
| 86127TUN/1986 | GU798395.1 | Human | Tunisia | (Talbi et al., 2010) |
| 86128TUN/1986 | GU798394.1 | Human | Tunisia | (Talbi et al., 2010) |
| Ariana2 | MK981888.1 | Dog | Tunisia | [31] (Bonnaud et al., 2019) |
| DogTN/Bj01 | EU643525.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Bz98 | EU643527.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Bz99 | EU643528.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Jd01 | EU643534.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Jd96 | EU643535.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Jd98 | EU643536.1 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Ar98 | EU643555 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Gb96 | EU643532 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Gb98 | EU643533 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Jd99 | EU643537 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Kf96 | EU643540 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Kr02 | EU643538 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Kr03 | EU643553 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Kr98 | EU643539 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Ks00 | EU643541 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Ks98 | EU643543 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Md95 | EU643545 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Md99 | EU643546 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Nb00 | EU643548 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Ta02 | EU643550 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Tn03 | EU643554 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Tz96 | EU643551 | Dog | Tunisia | (Amouri et al., 2011) |
| DogTN/Zg00 | EU643552 | Dog | Tunisia | (Amouri et al., 2011) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bouslama, Z.; Kharmachi, H.; Basdouri, N.; Ben Salem, J.; Ben Maiez, S.; Handous, M.; Saadi, M.; Ghram, A.; Turki, I. Molecular Epidemiology of Rabies in Wild Canidae in Tunisia. Viruses 2021, 13, 2473. https://doi.org/10.3390/v13122473
Bouslama Z, Kharmachi H, Basdouri N, Ben Salem J, Ben Maiez S, Handous M, Saadi M, Ghram A, Turki I. Molecular Epidemiology of Rabies in Wild Canidae in Tunisia. Viruses. 2021; 13(12):2473. https://doi.org/10.3390/v13122473
Chicago/Turabian StyleBouslama, Zied, Habib Kharmachi, Nourhene Basdouri, Jihen Ben Salem, Samia Ben Maiez, Mariem Handous, Mohamed Saadi, Abdeljalil Ghram, and Imed Turki. 2021. "Molecular Epidemiology of Rabies in Wild Canidae in Tunisia" Viruses 13, no. 12: 2473. https://doi.org/10.3390/v13122473