
Best Molecular Tools to Investigate Coronavirus Diversity in Mammals: A Comparison

 , , , , and
, , , , and
Abstract
:1. How Diagnostic Failure Can Affect Animal Surveillance
2. Literature Review
3. In Silico Evaluation
4. In Vitro Evaluation
5. Field Evaluation
6. A Look into the Future of Coronavirus Surveillance
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Du, J.; Yang, L.; Ren, X.; Zhang, J.; Dong, J.; Sun, L.; Zhu, Y.; Yang, F.; Zhang, S.; Wu, Z.; et al. Genetic Diversity of Coronaviruses in Miniopterus Fuliginosus Bats. Sci. China Life Sci. 2016, 59, 604–614. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Wang, M.; Lam, C.S.F.; Xu, H.; Guo, R.; Chan, K.; Zheng, B.; et al. Complete Genome Sequence of Bat Coronavirus HKU2 from Chinese Horseshoe Bats Revealed a Much Smaller Spike Gene with a Different Evolutionary Lineage from the Rest of the Genome. Virology 2007, 367, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Herrewegh, A.A.P.M.; Smeenk, I.; Horzinek, M.C.; Rottier, P.J.M.; Groot, R.J.D.E. Feline Coronavirus Type II Strains 79-1683 and 79-1146 Originate from a Double Recombination between Feline Coronavirus Type I and Canine Coronavirus. J. Virol. 1998, 72, 4508–4514. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Lau, S.K.P.; Huang, Y.; Yuen, K.-Y. Coronavirus Diversity, Phylogeny and Interspecies Jumping. Exp. Biol. Med. 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and Evolution of Pathogenic Coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A Pneumonia Outbreak Associated with a New Coronavirus of Probable Bat Origin. Nature 2020. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; Van der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global Patterns in Coronavirus Diversity. Virus Evol. 2017, 3, 1–15. [Google Scholar] [CrossRef]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between Co-Divergence and Cross-Species Transmission in the Evolutionary History of Bat Coronaviruses. Infect. Genet. Evol. 2018, 58. [Google Scholar] [CrossRef] [PubMed]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Thoelen, I.; Wollants, E.; Lemey, P.; Vandamme, A.-M.; Van Ranst, M. Complete Genomic Sequence of Human Coronavirus OC43: Molecular Clock Analysis Suggests a Relatively Recent Zoonotic Coronavirus Transmission Event. J. Virol. 2005, 79, 1595–1604. [Google Scholar] [CrossRef] [Green Version]
- Pyrc, K.; Dijkman, R.; Deng, L.; Jebbink, M.F.; Ross, H.A.; Berkhout, B.; van der Hoek, L. Mosaic Structure of Human Coronavirus NL63, One Thousand Years of Evolution. J. Mol. Biol. 2006, 364, 964–973. [Google Scholar] [CrossRef]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Sabir, J.S.M.; Lam, T.T.Y.; Ahmed, M.M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.M.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-Circulation of Three Camel Coronavirus Species and Recombination of MERS-CoVs in Saudi Arabia. Science 2016, 351, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niederwerder, M.C.; Hesse, R.A. Swine Enteric Coronavirus Disease: A Review of 4 Years with Porcine Epidemic Diarrhoea Virus and Porcine Deltacoronavirus in the United States and Canada. Transbound. Emerg. Dis. 2018, 65, 660–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boileau, M.J.; Kapil, S. Bovine Coronavirus Associated Syndromes. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 123–146. [Google Scholar] [CrossRef] [PubMed]
- Malbon, A.J.; Fonfara, S.; Meli, M.L.; Hahn, S.; Egberink, H.; Kipar, A. Feline Infectious Peritonitis as a Systemic Inflammatory Disease: Contribution of Liver and Heart to the Pathogenesis. Viruses 2019, 11, 1144. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Hasoksuz, M.; Spiro, D.; Halpin, R.; Wang, S.; Vlasova, A.; Janies, D.; Jones, L.R.; Ghedin, E.; Saif, L.J. Quasispecies of Bovine Enteric and Respiratory Coronaviruses Based on Complete Genome Sequences and Genetic Changes after Tissue Culture Adaptation. Virology 2007, 363, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.M.; Drosten, C. Ecology, Evolution and Classification of Bat Coronaviruses in the Aftermath of SARS. Antivir. Res. 2014, 101, 45–56. [Google Scholar] [CrossRef]
- Olival, K.J.; Hosseini, P.R.; Zambrana-Torrelio, C.; Ross, N.; Bogich, T.L.; Daszak, P. Host and Viral Traits Predict Zoonotic Spillover from Mammals. Nature 2017, 546, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Daszak, P.; Olival, K.J.; Li, H. A Strategy to Prevent Future Epidemics Similar to the 2019-NCoV Outbreak. Biosaf. Health 2020, 10–12. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Sh, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal Swine Acute Diarrhoea Syndrome Caused by an HKU2-Related Coronavirus of Bat Origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Haagmans, B.L.; Müller, M.A.; Gutierrez, C.; Godeke, G.J.; Meyer, B.; Muth, D.; Raj, V.S.; Smits-De Vries, L.; Corman, V.M.; et al. Middle East Respiratory Syndrome Coronavirus Neutralising Serum Antibodies in Dromedary Camels: A Comparative Serological Study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Leopardi, S.; Terregino, C.; De Benedictis, P. Silent Circulation of Coronaviruses in Pigs. Vet. Rec. 2020, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.; Chen, W.; Chen, J.P. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis Javanica). Viruses 2019, 11, 979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Si, H.-R.; Zhu, Y.; Yang, X.-L.; Anderson, D.E.; Shi, Z.-L.; Wang, L.-F.; Zhou, P. Discovery of Bat Coronaviruses through Surveillance and Probe Capture-Based Next-Generation Sequencing. mSphere 2020, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1805–1820. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and Re-Emerging Coronaviruses in Pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Marthaler, D.; Jiang, Y.; Collins, J.; Rossow, K. Porcine Deltacoronavirus from the United States. Genome Announc. 2014, 2, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Mihindukulasuriya, K.A.; Wu, G.; St Leger, J.; Nordhausen, R.W.; Wang, D. Identification of a Novel Coronavirus from a Beluga Whale by Using a Panviral Microarray. J. Virol. 2008, 82, 5084–5088. [Google Scholar] [CrossRef] [Green Version]
- Wille, M.; Holmes, E.C. Wild Birds as Reservoirs for Diverse and Abundant Gamma- and Deltacoronaviruses. FEMS Microbiol. Rev. 2020, 1–14. [Google Scholar] [CrossRef]
- Hu, H.; Jung, K.; Wang, Q.; Saif, L.J.; Vlasova, A.N. Development of a One-Step RT-PCR Assay for Detection of Pancoronaviruses (α-, β-, γ-, and δ-Coronaviruses) Using Newly Designed Degenerate Primers for Porcine and Avian ‘fecal Samples. J. Virol. Methods 2018, 256, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Geldenhuys, M.; Mortlock, M.; Epstein, J.H.; Pawęska, J.T.; Weyer, J.; Markotter, W. Overview of Bat and Wildlife Coronavirus Surveillance in Africa: A Framework for Global Investigations. Viruses 2021, 13, 936. [Google Scholar] [CrossRef] [PubMed]
- Woo, P.C.Y.; Lau, S.K.P.; Chu, C.; Chan, K.; Tsoi, H.; Huang, Y.; Wong, B.H.L.; Poon, R.W.S.; Cai, J.J.; Luk, W.; et al. Characterization and Complete Genome Sequence of a Novel Coronavirus, Coronavirus HKU1, from Patients with Pneumonia. J. Virol. 2005, 79, 884–895. [Google Scholar] [CrossRef] [Green Version]
- Poon, L.L.M.; Chu, D.K.W.; Chan, K.H.; Wong, O.K.; Ellis, T.M.; Leung, Y.H.C.; Lau, S.K.P.; Woo, P.C.Y.; Suen, K.Y.; Yuen, K.Y.; et al. Identification of a Novel Coronavirus in Bats. J. Virol. 2005, 79, 2001–2009. [Google Scholar] [CrossRef] [Green Version]
- De Souza Luna, L.K.; Heiser, V.; Regamey, N.; Panning, M.; Drexler, J.F.; Mulangu, S.; Poon, L.; Baumgarte, S.; Haijema, B.J.; Kaiser, L.; et al. Generic Detection of Coronaviruses and Differentiation at the Prototype Strain Level by Reverse Transcription-PCR and Nonfluorescent Low-Density Microarray. J. Clin. Microbiol. 2007, 45, 1049–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, D.K.W.; Leung, C.Y.H.; Gilbert, M.; Joyner, P.H.; Ng, E.M.; Tse, T.M.; Guan, Y.; Peiris, J.S.M.; Poon, L.L.M. Avian Coronavirus in Wild Aquatic Birds. J. Virol. 2011, 85, 12815–12820. [Google Scholar] [CrossRef] [Green Version]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a Severe Acute Respiratory Syndrome Coronavirus-like Virus in a Leaf-Nosed Bat in Nigeria. mBio 2010, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, Q.-Y.; Wang, K.-C.; Liu, S.; Hou, G.-Y.; Jiang, W.-M.; Wang, S.-C.; Li, J.-P.; Yu, J.-M.; Chen, J.-M. Genomic Analysis and Surveillance of the Coronavirus Dominant in Ducks in China. PLoS ONE 2015, 10, e0129256. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of Seven Novel Mammalian and Avian Coronaviruses in the Genus Deltacoronavirus Supports Bat Coronaviruses as the Gene Source of Alphacoronavirus and Betacoronavirus and Avian Coronaviruses as the Gene Source of Gammacoronavirus and Deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [Green Version]
- Jordan, B.J.; Hilt, D.A.; Poulson, R.; Stallknecht, D.E.; Jackwood, M.W. Identification of Avian Coronavirus in Wild Aquatic Birds of the Central and Eastern USA. J. Wildl. Dis. 2015, 51, 218–221. [Google Scholar] [CrossRef]
- Jonassen, C.M.; Kofstad, T.; Larsen, I.-L.; Løvland, A.; Handeland, K.; Follestad, A.; Lillehaug, A. Molecular Identification and Characterization of Novel Coronaviruses Infecting Graylag Geese (Anser Anser), Feral Pigeons (Columbia Livia) and Mallards (Anas Platyrhynchos). J. Gen. Virol. 2005, 86, 1597–1607. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Poon, L.L.M.; Chan, K.H.; Chen, H.; Guan, Y.; Yuen, K.Y.; Peiris, J.S.M. Coronaviruses in Bent-Winged Bats (Miniopterus Spp.). J. Gen. Virol. 2006, 87, 2461–2466. [Google Scholar] [CrossRef] [PubMed]
- Holbrook, M.G.; Anthony, S.J.; Navarrete-Macias, I.; Bestebroer, T.; Munster, V.J.; van Doremalen, N. Updated and Validated Pan-Coronavirus PCR Assay to Detect All Coronavirus Genera. Viruses 2021, 13, 599. [Google Scholar] [CrossRef]
- Xiu, L.; Binder, R.A.; Alarja, N.A.; Kochek, K.; Coleman, K.K.; Than, S.T.; Bailey, E.S.; Bui, V.N.; Toh, T.-H.; Erdman, D.D.; et al. A RT-PCR Assay for the Detection of Coronaviruses from Four Genera. J. Clin. Virol. 2020, 128, 104391. [Google Scholar] [CrossRef] [PubMed]
- ICTV Coronaviridae Virus Taxonomy: 2020 Release. Available online: https://talk.ictvonline.org/ictv-reports/ictv_9th_report/positive-sense-rna-viruses-2011/w/posrna_viruses/222/coronaviridae (accessed on 1 June 2021).
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.M.; Schutten, M. The Effect of Primer-Template Mismatches on the Detection and Quantification of Nucleic Acids Using the 5′ Nuclease Assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Molenkamp, R. Detection of 2019 Novel Coronavirus ( 2019-NCoV ) By real-time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, D.A.; Joffrin, L.; Lebarbenchon, C.; Mavingui, P. Analysis of Partial Sequences of the RNA-Dependent RNA Polymerase Gene as a Tool for Genus and Subgenus Classification of Coronaviruses. J. Gen. Virol. 2020, 101, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.-F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny.Fr: Robust Phylogenetic Analysis for the Non-Specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v3: An Online Tool for the Display and Annotation of Phylogenetic and Other Trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Lee, C. Porcine Epidemic Diarrhea Virus: An Emerging and Re-Emerging Epizootic Swine Virus. Virol. J. 2015, 12, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, S.K.P.; Woo, P.C.Y.; Li, K.S.M.; Huang, Y.; Tsoi, H.-W.; Wong, B.H.L.; Wong, S.S.Y.; Leung, S.-Y.; Chan, K.-H.; Yuen, K.-Y. Severe Acute Respiratory Syndrome Coronavirus-like Virus in Chinese Horseshoe Bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Lin, X.D.; Zhang, H.L.; Wang, M.R.; Guan, X.Q.; Holmes, E.C.; Zhang, Y.Z. Extensive Genetic Diversity and Host Range of Rodent-Borne Coronaviruses. Virus Evol. 2020, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Streicker, B.D.G.; Gilbert, A.T. Contextualizing Bats as Viral Reservoirs. Science 2020, 370, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Zhou, X.; Peng, Q.; Chen, Y.; Zhang, F.; Huang, T.; Zhang, T.; Li, A.; Huang, D.; Wu, Q.; et al. Newly Emerged Porcine Deltacoronavirus Associated With Diarrhoea in Swine in China: Identification, Prevalence and Full-Length Genome Sequence Analysis. Transbound Emerg. Dis. 2015, 62, 575–580. [Google Scholar] [CrossRef]
- Decaro, N.; Elia, G.; Campolo, M.; Desario, C.; Mari, V.; Radogna, A.; Colaianni, M.L.; Cirone, F.; Tempesta, M.; Buonavoglia, C. Detection of Bovine Coronavirus Using a TaqMan-Based Real-Time RT-PCR Assay. J. Virol. Methods 2008, 151, 167–171. [Google Scholar] [CrossRef]
- Chan, W.S.; Au, C.H.; Lam, H.Y.; Wang, C.L.N.; Ho, D.N.-Y.; Lam, Y.M.; Chu, D.K.W.; Poon, L.L.M.; Chan, T.L.; Zee, J.S.-T.; et al. Evaluation on the Use of Nanopore Sequencing for Direct Characterization of Coronaviruses from Respiratory Specimens, and a Study on Emerging Missense Mutations in Partial RdRP Gene of SARS-CoV-2. Virol. J. 2020, 17, 183. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host | N | Targeted animal species | Sampling strategy * | Matrix | |||||||
| Single species | multi species | L | E | P | S | field stabilizer | feces/ anal swab | oral swab | organs | ||
| Bats | 71 | 13 (18) | 58 (82) | 55 (77) | 7 (10) | 4 (6) | 15 (21) | 39 (55) | 61 (86) | 30 (42) | 19 (27) |
| Rodents | 11 | 0 | 11 (100) | 7 (64) | 1 (9) | 1 (9) | 5 (45) | 8 (73) | 7 (64) | 4 (36) | 7 (64) |
| Other wild mammals | 12 | 5 (41) | 8 (67) | 10 (83) | 0 | 2 (17) | 2 (17) | 10 (83) | 9 (75) | 7 (58) | 4 (33) |
| Domestic mammals | 13 | 10 (77) | 3 (23) | 11 (85) | 0 | 3 (23) | 0 | 1 (7.7) | 8 (62) | 6 (46) | 2 (15) |
| Birds | 7 | 0 | 7 (100) | 7 (100) | 0 | 0 | 1 (14) | 6 (86) | 7 (100) | 4 (57) | 1 (14) |
| Total | 100 | 27 | 74 | 77 | 8 | 9 | 21 | 54 | 79 | 41 | 28 |
| Host | N | Woo et al./Poon et al. [34,35] | De Souza Luna et al. [36] | Chu et al. 2011 [37] | Quan et al. [38] | Others (< 3 papers) |
| Bats | 71 | 19 (27%) | 22 (31%) | 2 (3%) | 5 (7%) | 29 (41%) |
| Rodents | 11 | 4 (36%) | 4 (36%) | 0 | 3 (27%) | 4 (36%) |
| Other wild mammals | 12 | 5 (42%) | 3 (25%) | 1 (8%) | 3 (25%) | 4 (33%) |
| Domestic mammals | 13 | 2 (15%) | 2 (15%) | 4 (31%) | 0 | 5 (38%) |
| Birds | 7 | 1 (14%) | 0 | 1 (14%) | 0 | 5 (71%) |
| Total | 100 | 26% | 26% | 8% | 6% | 42% |
| Identification of α-cov | 18 (69%) | 19 (73%) | 3 (38%) | 3 (50%) | 25 (60%) | |
| Identification of β-cov | 20 (17%) | 21 (81%) | 6 (75%) | 6 (100%) | 23 (55%) | |
| Identification of γ-cov | 1 (13%) | 3 (7%) | ||||
| Identification of δ-cov | 1 (13%) | 3 (7%) |
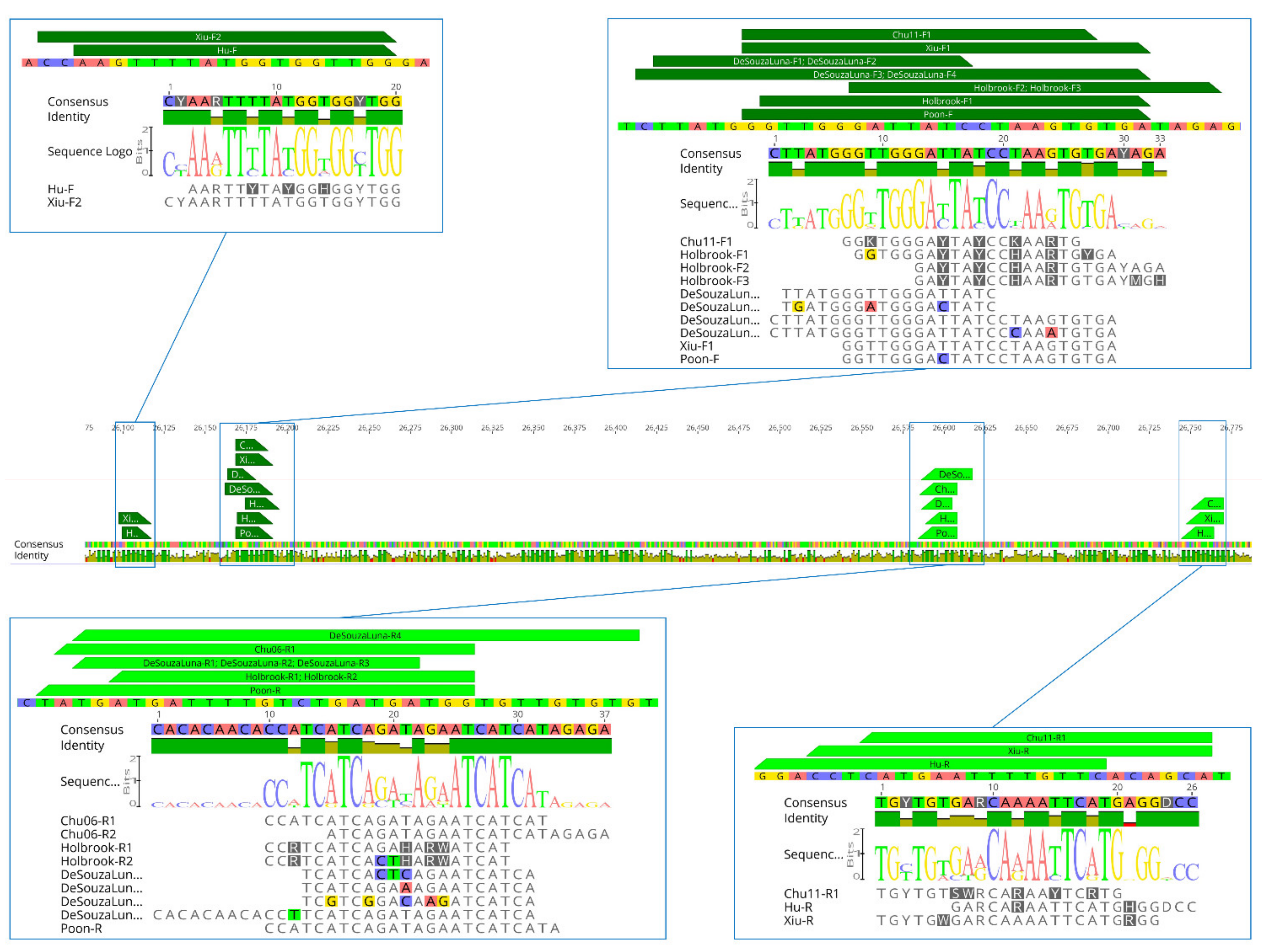
| Ref. | PCR format | Primer | S/As | Sequence (5′→3′) | Deg. * | Number of Mismatches | |||
|---|---|---|---|---|---|---|---|---|---|
| α | β | γ | δ | ||||||
| [44] | Two-step RT- | Holbrook-F1 | S | GGTGGGAYTAYCCHAARTGYGA | 48 | 0–2 | 1–4 | 0–1 | 0–1 |
| Holbrook-R1 | As | CCRTCATCAGAHARWATCAT | 24 | 0–3 | 0–3 | 1–3 | 0–1 | ||
| Holbrook-R2 | As | CCRTCATCACTHARWATCAT | 24 | 0–5 | 0–5 | 1–4 | 2–3 | ||
| Hemi–nested | Holbrook-F2 | S | GAYTAYCCHAARTGTGAYAGA | 48 | 0–3 | 0–3 | 0–1 | 1–2 | |
| Holbrook-F3 | S | GAYTAYCCHAARTGTGAYMGH | 288 | 0–1 | 0–4 | 0–1 | 0–1 | ||
| [45] | One-step RT- | Xiu-F1 | S | CYAARTTTTATGGTGGYTGG | 8 | 0–2 | 0–4 | 0–2 | 0–1 |
| Xiu-R | As | TGYTGWGARCAAAATTCATGRGG | 16 | 0–3 | 0–3 | 0–2 | 0–2 | ||
| Hemi-nested | Xiu-F2 | S | GGTTGGGATTATCCTAAGTGTGA | None | 1–4 | 0–4 | 0–3 | 0–3 | |
| [32] | One-step RT- | Hu-F | S | AARTTYTAYGGHGGYTGG | 48 | 0–1 | 0–1 | 0–1 | 0 |
| Hu-R | As | GARCARAATTCATGHGGDCC | 36 | 0–1 | 0–1 | 0–2 | 0–1 | ||
| [37] | Two-step RT- | Chu11-F1 | S | GGKTGGGAYTAYCCKAARTG | 32 | 0–2 | 0–1 | 0–1 | 0–2 |
| Chu11-R1 | As | TGYTGTSWRCARAAYTCRTG | 128 | 0–1 | 0–1 | 0–1 | 0–1 | ||
| Nested | Chu11-F2 | S | Identical to Poon-F | ||||||
| Chu11-R2 | As | Identical to Chu06-R1 | |||||||
| [43] | Two-step RT- | Chu06-F1 | S | Identical to Poon-F | |||||
| Chu06-R1 | As | CCATCATCAGATAGAATCATCAT | None | 1–6 | 2–5 | 4–8 | 2–5 | ||
| Chu06-F2 | S | Identical to Poon-F | |||||||
| Chu06-R2 | As | ATCAGATAGAATCATCATAGAGA | None | 1–7 | 2–8 | 5–11 | 5–9 | ||
| [36] | One-step RT- | DeSouzaLuna-F1 | S | TTATGGGTTGGGATTATC | None | 0–4 | 0–3 | 0–3 | 3–6 |
| DeSouzaLuna-F2 | S | TGATGGGATGGGACTATC | None | 0–4 | 1–4 | 1–3 | 4–7 | ||
| DeSouzaLuna-R1 | As | TCATCACTCAGAATCATCA | None | 2–7 | 1–6 | 4–7 | 2–7 | ||
| DeSouzaLuna-R2 | As | TCATCAGAAAGAATCATCA | None | 0–5 | 0–5 | 3–4 | 1–5 | ||
| DeSouzaLuna-R3 | As | TCGTCGGACAAGATCATCA | None | 1–7 | 3–7 | 1–5 | 3–5 | ||
| Nested | DeSouzaLuna-F3 | S | CTTATGGGTTGGGATTATCCTAAGTGTGA | None | 1–6 | 0–5 | 0–3 | 3–7 | |
| DeSouzaLuna-F4 | S | CTTATGGGTTGGGATTATCCCAAATGTGA | None | 0–7 | 1–5 | 1–5 | 5–9 | ||
| DeSouzaLuna-R4 | As | CACACAACACCTTCATCAGATAGAATCATCA | None | 2–9 | 4–7 | 5–8 | 4–8 | ||
| [34] [35] | Two-step RT- | Poon-F | S | GGTTGGGACTATCCTAAGTGTGA | None | 0–4 | 0–3 | 1–2 | 1–4 |
| Poon-R | As | CCATCATCAGATAGAATCATCATA | None | 1–6 | 3–5 | 4–8 | 3–6 | ||
| This study | One step RT- | Identical to Hu [32] | |||||||
| Nested | Identical to Poon-F and Chu06-R1 [35,43] | ||||||||
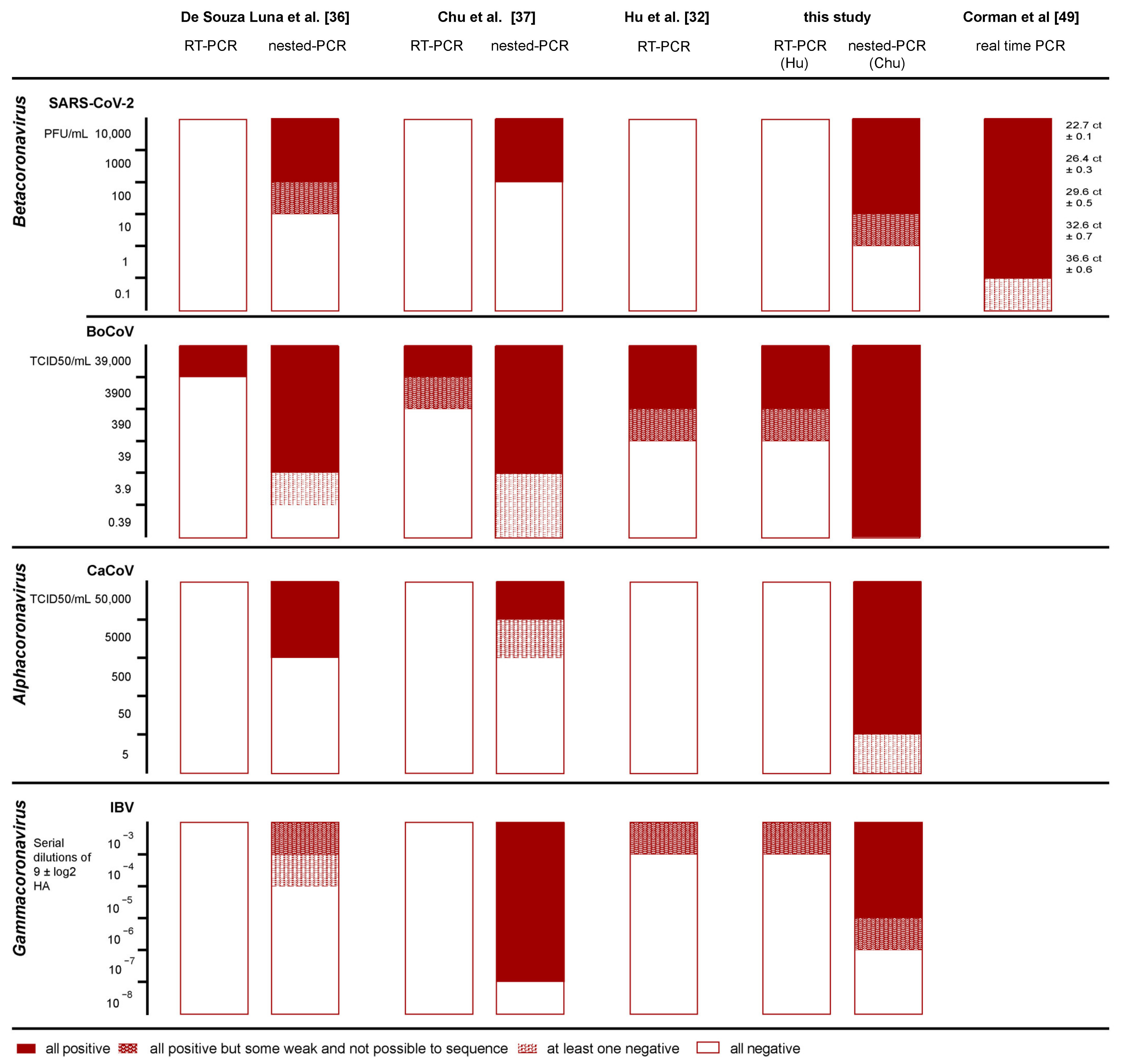
| Genus | Species | Strain | Material | Titer |
|---|---|---|---|---|
| α | Alphacoronavirus 1—Canine CoV | Jan 71 (ATCC VR-809) | cell culture supernatant | 5.0 × 107 TCID50/mL |
| β | Betacoronavirus 1—Bovine CoV | Mebus | cell culture supernatant | 3.9 × 106 TCID50/mL |
| β | SARS-CoV-2 | IZSVe20VIR1935 | cell culture supernatant | 1.0 × 105 PFU/mL |
| γ | Infectious Bronchitis Virus | D1466 | egg allantoic fluid | 9 ± 1 log2 HA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Drzewnioková, P.; Festa, F.; Panzarin, V.; Lelli, D.; Moreno, A.; Zecchin, B.; De Benedictis, P.; Leopardi, S. Best Molecular Tools to Investigate Coronavirus Diversity in Mammals: A Comparison. Viruses 2021, 13, 1975. https://doi.org/10.3390/v13101975
Drzewnioková P, Festa F, Panzarin V, Lelli D, Moreno A, Zecchin B, De Benedictis P, Leopardi S. Best Molecular Tools to Investigate Coronavirus Diversity in Mammals: A Comparison. Viruses. 2021; 13(10):1975. https://doi.org/10.3390/v13101975
Chicago/Turabian StyleDrzewnioková, Petra, Francesca Festa, Valentina Panzarin, Davide Lelli, Ana Moreno, Barbara Zecchin, Paola De Benedictis, and Stefania Leopardi. 2021. "Best Molecular Tools to Investigate Coronavirus Diversity in Mammals: A Comparison" Viruses 13, no. 10: 1975. https://doi.org/10.3390/v13101975