Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins



Abstract
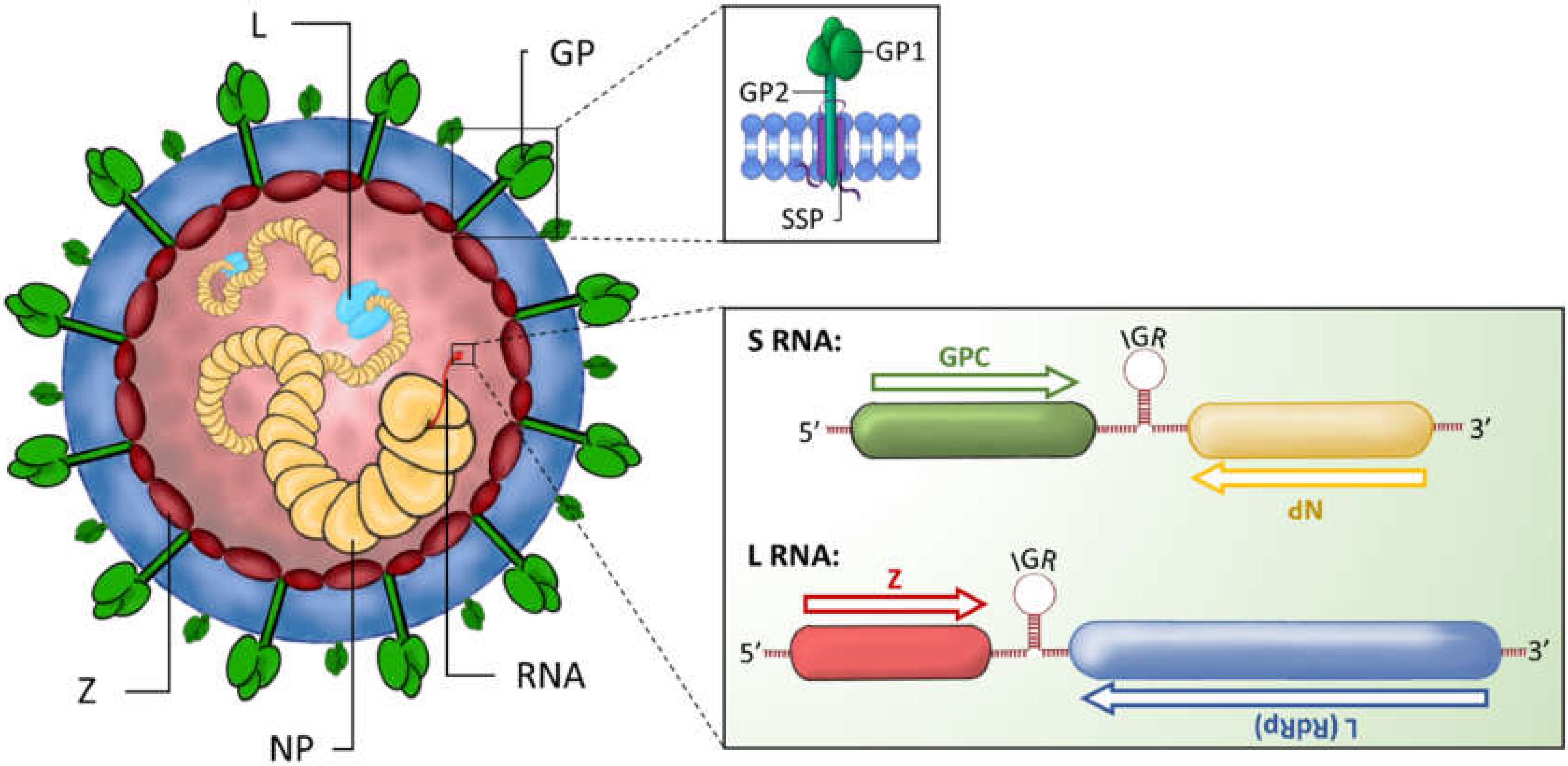
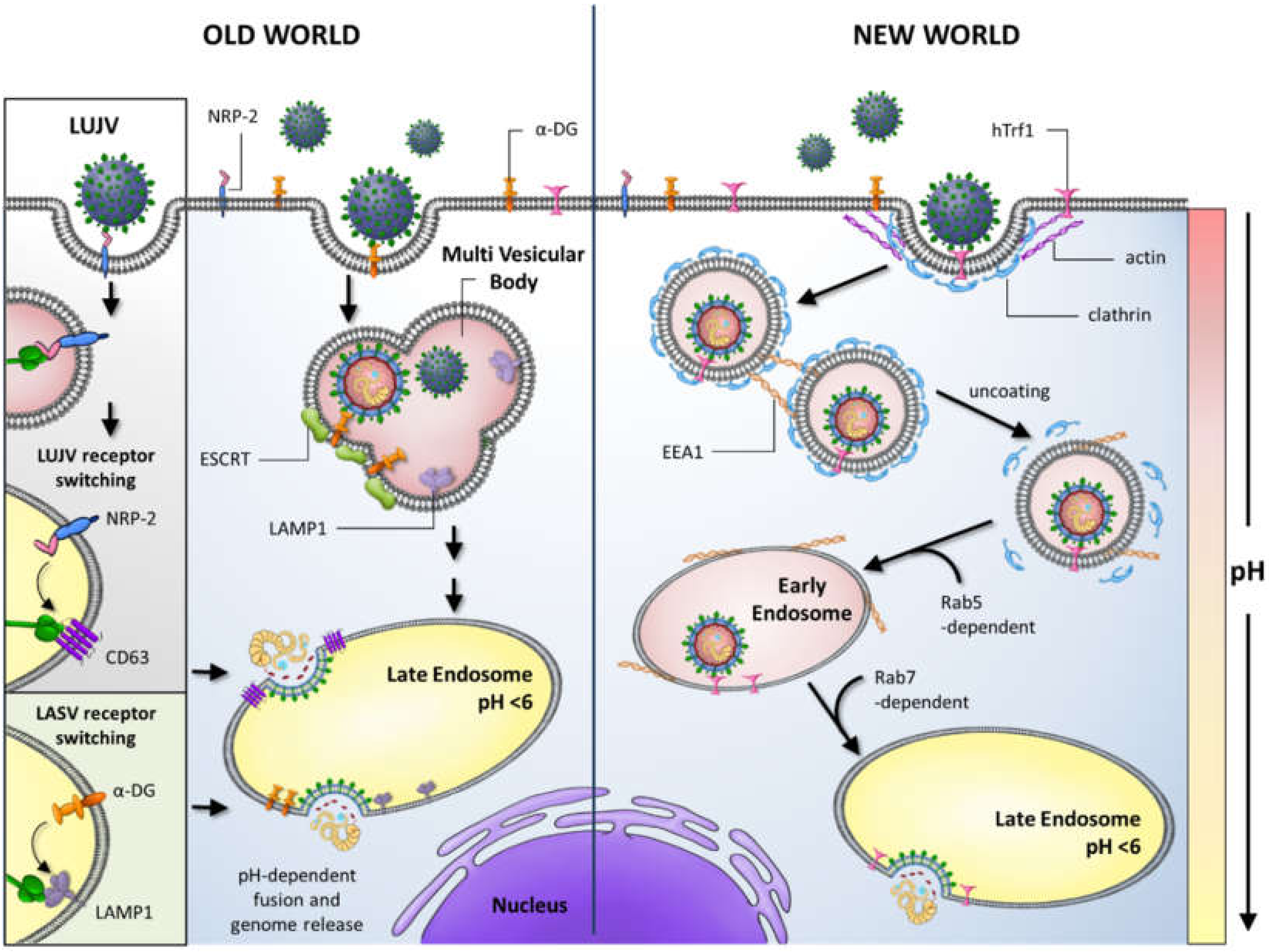
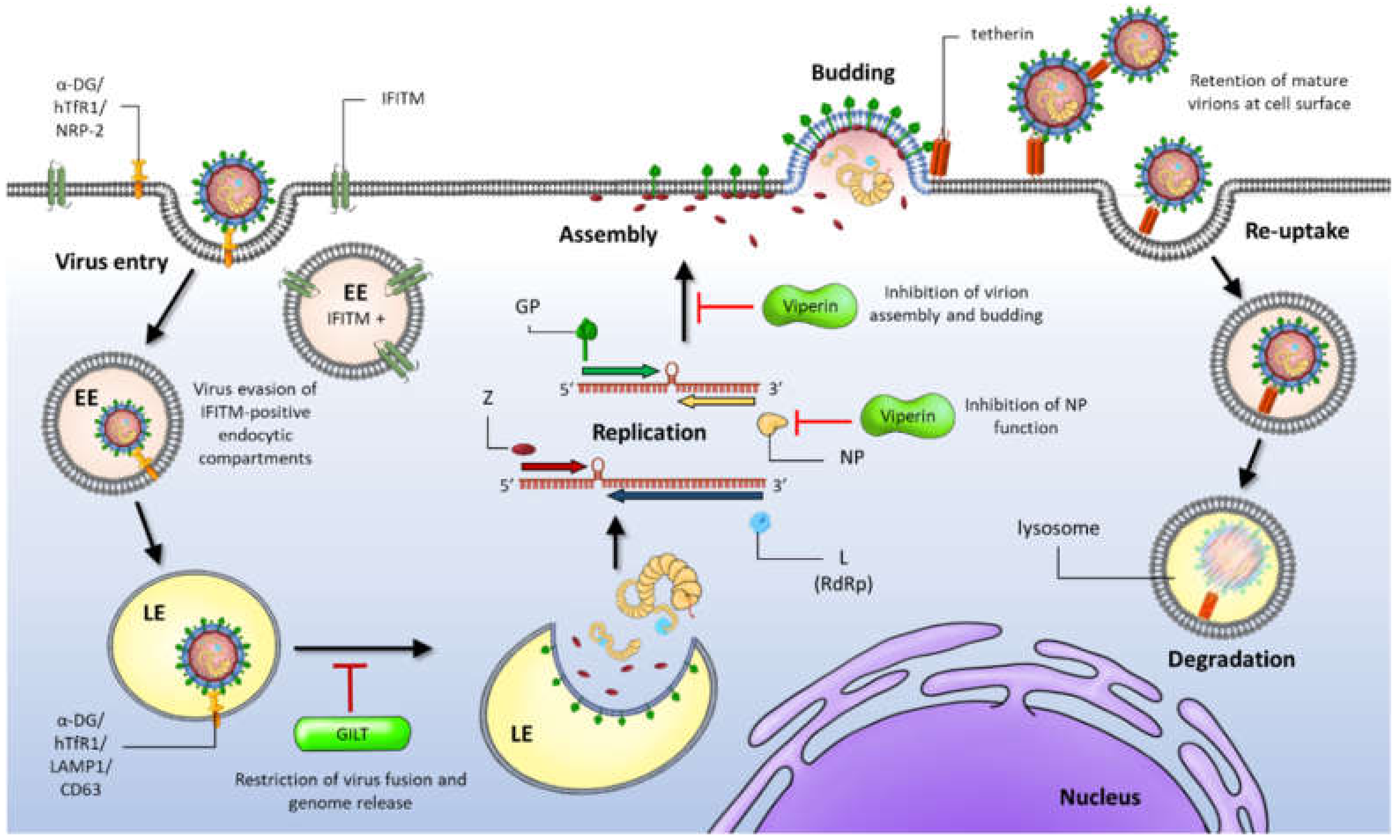
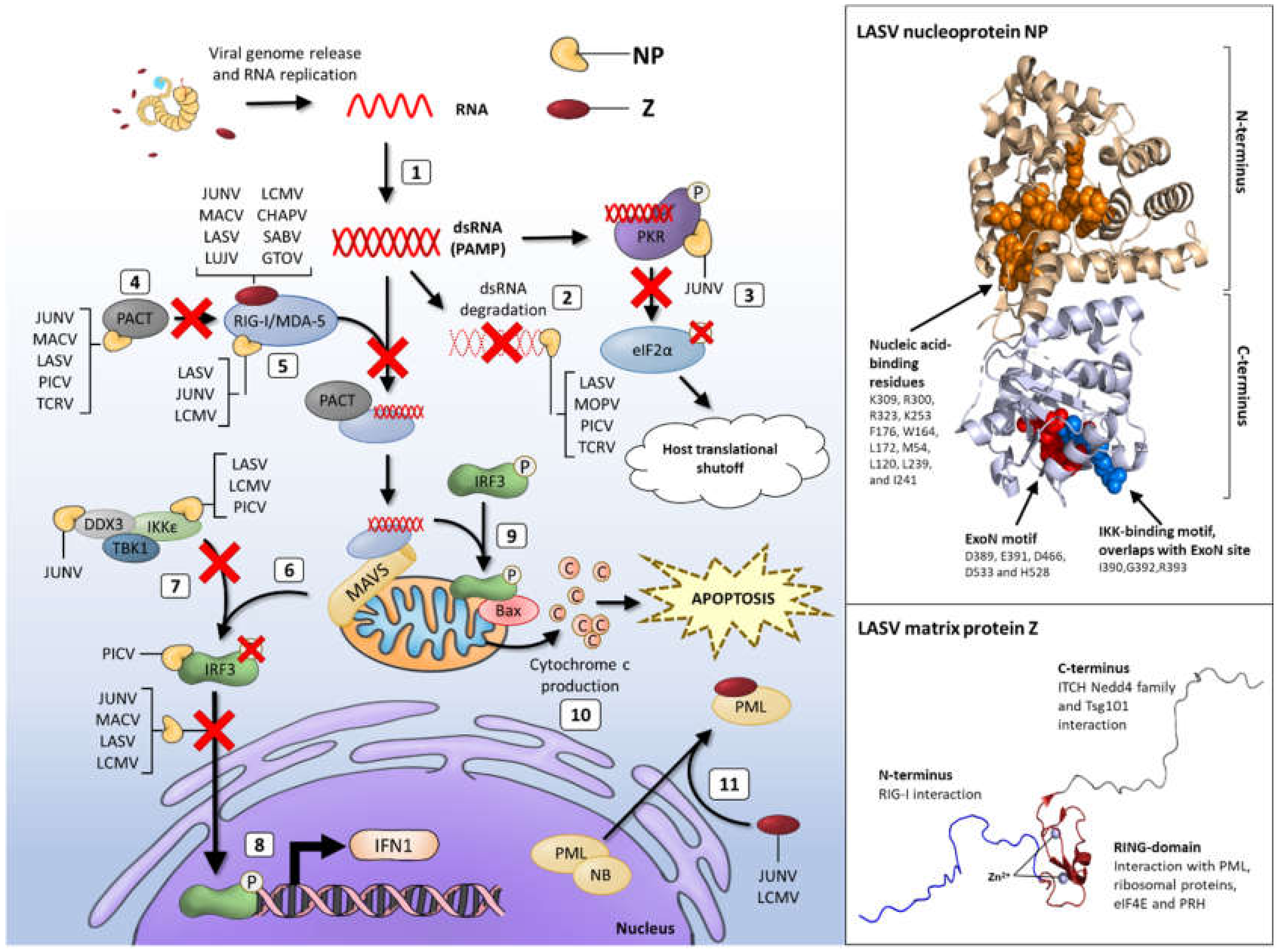
:1. Introduction
2. Host Immune Responses during Arenavirus Infection
3. Immunosuppressive Mechanisms of Mammarenavirus NP Proteins
3.1. NP-Mediated IFN Inhibition and dsRNA Degradation
3.2. NP Interactions with PRRs
3.3. NP-DDX3 Interaction Suppresses IFN1 Induction
3.4. NP-Driven Inhibition of Apoptosis
4. Evasion Strategies of the Arenavirus Matrix Protein, Z
Z Protein-Mediated Inhibition of IFN Responses
5. Development of NP- and Z-Specific Antivirals
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Katze, M.G.; Fornek, J.L.; Palermo, R.E.; Walters, K.A.; Korth, M.J. Innate immune modulation by RNA viruses: Emerging insights from functional genomics. Nat. Rev. Immunol 2008, 8, 644–654. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.Y.; Suthar, M.S. Mechanisms of innate immune evasion in re-emerging RNA viruses. Curr. Opin. Virol. 2015, 12, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J.; Neil, S.J. Host factors involved in retroviral budding and release. Nat. Rev. Microbiol. 2011, 9, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Ilori, E.A.; Furuse, Y.; Ipadeola, O.B.; Dan-Nwafor, C.C.; Abubakar, A.; Womi-Eteng, O.E.; Ogbaini-Emovon, E.; Okogbenin, S.; Unigwe, U.; Ogah, E.; et al. Epidemiologic and Clinical Features of Lassa Fever Outbreak in Nigeria, January 1-May 6, 2018. Emerg. Infect. Dis. 2019, 25, 1066–1074. [Google Scholar] [CrossRef]
- Kafetzopoulou, L.E.; Pullan, S.T.; Lemey, P.; Suchard, M.A.; Ehichioya, D.U.; Pahlmann, M.; Thielebein, A.; Hinzmann, J.; Oestereich, L.; Wozniak, D.M.; et al. Metagenomic sequencing at the epicenter of the Nigeria 2018 Lassa fever outbreak. Science 2019, 363, 74–77. [Google Scholar] [CrossRef] [Green Version]
- Oloniniyi, O.K.; Unigwe, U.S.; Okada, S.; Kimura, M.; Koyano, S.; Miyazaki, Y.; Iroezindu, M.O.; Ajayi, N.A.; Chukwubike, C.M.; Chika-Igwenyi, N.M.; et al. Genetic characterization of Lassa virus strains isolated from 2012 to 2016 in southeastern Nigeria. PLoS Negl. Trop. Dis. 2018, 12, e0006971. [Google Scholar] [CrossRef]
- Siddle, K.J.; Eromon, P.; Barnes, K.G.; Mehta, S.; Oguzie, J.U.; Odia, I.; Schaffner, S.F.; Winnicki, S.M.; Shah, R.R.; Qu, J.; et al. Genomic Analysis of Lassa Virus during an Increase in Cases in Nigeria in 2018. N. Engl. J. Med. 2018, 379, 1745–1753. [Google Scholar] [CrossRef]
- Mantlo, E.; Paessler, S.; Huang, C. Differential Immune Responses to Hemorrhagic Fever-Causing Arenaviruses. Vaccines 2019, 7, 138. [Google Scholar] [CrossRef] [Green Version]
- Garry, C.E.; Garry, R.F. Proteomics Computational Analyses Suggest that the Antennavirus Glycoprotein Complex Includes a Class I Viral Fusion Protein (α-Penetrene) with an Internal Zinc-Binding Domain and a Stable Signal Peptide. Viruses 2019, 11, 750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallam, S.J.; Koma, T.; Maruyama, J.; Paessler, S. Review of Mammarenavirus Biology and Replication. Front. Microbiol. 2018, 9, 1751. [Google Scholar] [CrossRef] [PubMed]
- Hepojoki, J.; Hepojoki, S.; Smura, T.; Szirovicza, L.; Dervas, E.; Prähauser, B.; Nufer, L.; Schraner, E.M.; Vapalahti, O.; Kipar, A.; et al. Characterization of Haartman Institute snake virus-1 (HISV-1) and HISV-like viruses-The representatives of genus Hartmanivirus, family Arenaviridae. PLoS Pathog. 2018, 14, e1007415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinschewer, D.D.; Perez, M.; de la Torre, J.C. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J. Virol. 2005, 79, 4519–4526. [Google Scholar] [CrossRef] [Green Version]
- Burri, D.J.; da Palma, J.R.; Kunz, S.; Pasquato, A. Envelope glycoprotein of arenaviruses. Viruses 2012, 4, 2162–2181. [Google Scholar] [CrossRef]
- Eichler, R.; Lenz, O.; Strecker, T.; Garten, W. Signal peptide of Lassa virus glycoprotein GP-C exhibits an unusual length. FEBS Lett. 2003, 538, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Lenz, O.; ter Meulen, J.; Klenk, H.D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GP-C is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef] [Green Version]
- Buchmeier, M.; Peters, C.J.; De la Torre, C. Arenaviridae: The virus and their replication. Fields Virol. 2007, 2, 1792–1827. [Google Scholar]
- Eschli, B.; Quirin, K.; Wepf, A.; Weber, J.; Zinkernagel, R.; Hengartner, H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J. Virol. 2006, 80, 5897–5907. [Google Scholar] [CrossRef] [Green Version]
- Nunberg, J.H.; York, J. The curious case of arenavirus entry, and its inhibition. Viruses 2012, 4, 83–101. [Google Scholar] [CrossRef] [Green Version]
- Perez, M.; Craven, R.C.; de la Torre, J.C. The small RING finger protein Z drives arenavirus budding: Implications for antiviral strategies. Proc. Natl. Acad. Sci. USA 2003, 100, 12978–12983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvato, M.S. Molecular Biology of the Prototype Arenavirus, Lymphocytic Choriomeningitis Virus. In The Arenaviridae. The Viruses; Salvato, M.S., Ed.; Springer: Boston, MA, USA, 1993. [Google Scholar] [CrossRef]
- Strecker, T.; Eichler, R.; Meulen, J.t.; Weissenhorn, W.; Klenk, H.D.; Garten, W.; Lenz, O. Lassa virus Z protein is a matrix protein and sufficient for the release of virus-like particles [corrected]. J. Virol. 2003, 77, 10700–10705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strandin, T.; Hepojoki, J.; Vaheri, A. Cytoplasmic tails of bunyavirus Gn glycoproteins-Could they act as matrix protein surrogates? Virology 2013, 437, 73–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLay, L.; Liang, Y.; Ly, H. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J. Gen. Virol. 2014, 95, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wolff, H.; Lange, J.V.; Webb, P.A. Interrelationships among arenaviruses measured by indirect immunofluorescence. Intervirology 1978, 9, 344–350. [Google Scholar] [CrossRef]
- Albariño, C.G.; Posik, D.M.; Ghiringhelli, P.D.; Lozano, M.E.; Romanowski, V. Arenavirus Phylogeny: A New Insight. Virus Genes 1998, 16, 39–46. [Google Scholar] [CrossRef]
- Bowen, M.D.; Peters, C.J.; Nichol, S.T. The Phylogeny of New World (Tacaribe Complex) Arenaviruses. Virology 1996, 219, 285–290. [Google Scholar] [CrossRef]
- Clegg, J.C. Molecular phylogeny of the arenaviruses. Curr. Top. Microbiol. Immunol. 2002, 262, 1–24. [Google Scholar] [CrossRef]
- Radoshitzky, S.R.; Bào, Y.; Buchmeier, M.J.; Charrel, R.N.; Clawson, A.N.; Clegg, C.S.; DeRisi, J.L.; Emonet, S.; Gonzalez, J.P.; Kuhn, J.H.; et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 2015, 160, 1851–1874. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef] [Green Version]
- Fehling, S.K.; Lennartz, F.; Strecker, T. Multifunctional nature of the arenavirus RING finger protein Z. Viruses 2012, 4, 2973–3011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Manzione, N.; Salas, R.A.; Paredes, H.; Godoy, O.; Rojas, L.; Araoz, F.; Fulhorst, C.F.; Ksiazek, T.G.; Mills, J.N.; Ellis, B.A.; et al. Venezuelan hemorrhagic fever: Clinical and epidemiological studies of 165 cases. Clin. Infect. Dis. 1998, 26, 308–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enria, D.A.; Barrera Oro, J.G. Junin virus vaccines. Curr Top. Microbiol Immunol 2002, 263, 239–261. [Google Scholar] [CrossRef]
- Patterson, M.; Grant, A.; Paessler, S. Epidemiology and pathogenesis of Bolivian hemorrhagic fever. Curr. Opin. Virol. 2014, 5, 82–90. [Google Scholar] [CrossRef] [Green Version]
- Olayemi, A.; Cadar, D.; Magassouba, N.F.; Obadare, A.; Kourouma, F.; Oyeyiola, A.; Fasogbon, S.; Igbokwe, J.; Rieger, T.; Bockholt, S.; et al. New Hosts of The Lassa Virus. Sci. Rep. 2016, 6, 25280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downs, W.G.; Anderson, C.R.; Spence, L.; Aitken, T.H.; Greenhall, A.H. Tacaribe virus, a new agent isolated from Artibeus bats and mosquitoes in Trinidad, West Indies. Am. J. Trop. Med. Hyg 1963, 12, 640–646. [Google Scholar] [CrossRef]
- Salazar-Bravo, J.; Ruedas, L.A.; Yates, T.L. Mammalian reservoirs of arenaviruses. Curr. Top. Microbiol. Immunol. 2002, 262, 25–63. [Google Scholar] [CrossRef]
- Bonwitt, J.; Sáez, A.M.; Lamin, J.; Ansumana, R.; Dawson, M.; Buanie, J.; Lamin, J.; Sondufu, D.; Borchert, M.; Sahr, F.; et al. At Home with Mastomys and Rattus: Human-Rodent Interactions and Potential for Primary Transmission of Lassa Virus in Domestic Spaces. Am. J. Trop. Med. Hyg. 2017, 96, 935–943. [Google Scholar] [CrossRef] [Green Version]
- Dzingirai, V.; Bett, B.; Bukachi, S.; Tweneboah Lawson, E.; Mangwanya, L.; Scoones, I.; Waldman, L.; Wilkinson, A.; Leach, M.; Winnebah, T. Zoonotic diseases: Who gets sick, and why? Explorations from Africa. Crit. Public Health 2016. [Google Scholar] [CrossRef] [Green Version]
- Mazzola, L.T.; Kelly-Cirino, C. Diagnostics for Lassa fever virus: A genetically diverse pathogen found in low-resource settings. BMJ Glob. Health 2019, 4, e001116. [Google Scholar] [CrossRef] [Green Version]
- Ajayi, N.A.; Nwigwe, C.G.; Azuogu, B.N.; Onyire, B.N.; Nwonwu, E.U.; Ogbonnaya, L.U.; Onwe, F.I.; Ekaete, T.; Günther, S.; Ukwaja, K.N. Containing a Lassa fever epidemic in a resource-limited setting: Outbreak description and lessons learned from Abakaliki, Nigeria (January-March 2012). Int. J. Infect. Dis 2013, 17, e1011–e1016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damonte, E.B.; Coto, C.E. Treatment of arenavirus infections: From basic studies to the challenge of antiviral therapy. Adv. Virus Res. 2002, 58, 125–155. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.B.; King, I.J.; Webb, P.A.; Scribner, C.L.; Craven, R.B.; Johnson, K.M.; Elliott, L.H.; Belmont-Williams, R. Lassa fever. Effective therapy with ribavirin. N. Engl. J. Med. 1986, 314, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, J.G.; Grant, D.S.; Schieffelin, J.S.; Boisen, M.L.; Goba, A.; Hartnett, J.N.; Levy, D.C.; Yenni, R.E.; Moses, L.M.; Fullah, M.; et al. Lassa fever in post-conflict sierra leone. PLoS Negl. Trop. Dis. 2014, 8, e2748. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Hum. Vaccin. 2011, 7, 694–700. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. Dose-ranging Study: Safety, Tolerability and Immunogenicity of INO-4500 in Healthy Volunteers in Ghana. Available online: https://clinicaltrials.gov/ct2/show/NCT04093076 (accessed on 7 May 2020).
- ClinicalTrials.gov. A Trial to Evaluate the Optimal Dose of MV-LASV. Available online: https://clinicaltrials.gov/ct2/show/NCT04055454 (accessed on 7 May 2020).
- Mateo, M.; Reynard, S.; Carnec, X.; Journeaux, A.; Baillet, N.; Schaeffer, J.; Picard, C.; Legras-Lachuer, C.; Allan, R.; Perthame, E.; et al. Vaccines inducing immunity to Lassa virus glycoprotein and nucleoprotein protect macaques after a single shot. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Hulseberg, C.E.; Fénéant, L.; Szymańska, K.M.; White, J.M. Lamp1 Increases the Efficiency of Lassa Virus Infection by Promoting Fusion in Less Acidic Endosomal Compartments. mBio 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Jae, L.T.; Raaben, M.; Herbert, A.S.; Kuehne, A.I.; Wirchnianski, A.S.; Soh, T.K.; Stubbs, S.H.; Janssen, H.; Damme, M.; Saftig, P.; et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 2014, 344, 1506–1510. [Google Scholar] [CrossRef] [Green Version]
- Raaben, M.; Jae, L.T.; Herbert, A.S.; Kuehne, A.I.; Stubbs, S.H.; Chou, Y.Y.; Blomen, V.A.; Kirchhausen, T.; Dye, J.M.; Brummelkamp, T.R.; et al. NRP2 and CD63 Are Host Factors for Lujo Virus Cell Entry. Cell Host Microbe 2017, 22, 688.e5–696.e5. [Google Scholar] [CrossRef] [Green Version]
- Abraham, J.; Kwong, J.A.; Albariño, C.G.; Lu, J.G.; Radoshitzky, S.R.; Salazar-Bravo, J.; Farzan, M.; Spiropoulou, C.F.; Choe, H. Host-Species Transferrin Receptor 1 Orthologs Are Cellular Receptors for Nonpathogenic New World Clade B Arenaviruses. PLoS Pathog. 2009, 5, e1000358. [Google Scholar] [CrossRef] [Green Version]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Sanchez, A.B.; Nguyen, N.T.; de la Torre, J.C.; Kunz, S. Different mechanisms of cell entry by human-pathogenic Old World and New World arenaviruses. J. Virol. 2008, 82, 7677–7687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Simone, C.; Zandonatti, M.A.; Buchmeier, M.J. Acidic pH triggers LCMV membrane fusion activity and conformational change in the glycoprotein spike. Virology 1994, 198, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Beachboard, D.C.; Horner, S.M. Innate immune evasion strategies of DNA and RNA viruses. Curr. Opin. Microbiol. 2016, 32, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Scutigliani, E.M.; Kikkert, M. Interaction of the innate immune system with positive-strand RNA virus replication organelles. Cytokine Growth Factor Rev. 2017, 37, 17–27. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Jensen, S.; Thomsen, A.R. Sensing of RNA viruses: A review of innate immune receptors involved in recognizing RNA virus invasion. J. Virol. 2012, 86, 2900–2910. [Google Scholar] [CrossRef] [Green Version]
- Suprunenko, T.; Hofer, M.J. Complexities of Type I Interferon Biology: Lessons from LCMV. Viruses 2019, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Meyer, B.; Ly, H. Inhibition of Innate Immune Responses Is Key to Pathogenesis by Arenaviruses. J. Virol. 2016, 90, 3810–3818. [Google Scholar] [CrossRef] [Green Version]
- Zuniga, E.I.; Liou, L.Y.; Mack, L.; Mendoza, M.; Oldstone, M.B. Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 2008, 4, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.E.; Ly, H. Hemorrhagic Fever-Causing Arenaviruses: Lethal Pathogens and Potent Immune Suppressors. Front. Immunol. 2019, 10, 372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pannetier, D.; Reynard, S.; Russier, M.; Journeaux, A.; Tordo, N.; Deubel, V.; Baize, S. Human dendritic cells infected with the nonpathogenic Mopeia virus induce stronger T-cell responses than those infected with Lassa virus. J. Virol. 2011, 85, 8293–8306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaeffer, J.; Reynard, S.; Carnec, X.; Pietrosemoli, N.; Dillies, M.A.; Baize, S. Non-Pathogenic Mopeia Virus Induces More Robust Activation of Plasmacytoid Dendritic Cells than Lassa Virus. Viruses 2019, 11, 172. [Google Scholar] [CrossRef] [Green Version]
- Bieniasz, P.D. Intrinsic immunity: A front-line defense against viral attack. Nat. Immunol. 2004, 5, 1109–1115. [Google Scholar] [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and interferons: Who’s interfering with whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef]
- Foster, T.L.; Wilson, H.; Iyer, S.S.; Coss, K.; Doores, K.; Smith, S.; Kellam, P.; Finzi, A.; Borrow, P.; Hahn, B.H.; et al. Resistance of Transmitted Founder HIV-1 to IFITM-Mediated Restriction. Cell Host Microbe 2016, 20, 429–442. [Google Scholar] [CrossRef] [Green Version]
- Goujon, C.; Malim, M.H. Characterization of the alpha interferon-induced postentry block to HIV-1 infection in primary human macrophages and T cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [Green Version]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, B.; Phan, U.T.; Geuze, H.J.; Cresswell, P. Enzymatic reduction of disulfide bonds in lysosomes: Characterization of a Gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc. Natl. Acad. Sci. USA 2000, 97, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luster, A.D.; Weinshank, R.L.; Feinman, R.; Ravetch, J.V. Molecular and biochemical characterization of a novel gamma-interferon-inducible protein. J. Biol. Chem. 1988, 263, 12036–12043. [Google Scholar]
- Singh, R.; Cresswell, P. Defective cross-presentation of viral antigens in GILT-free mice. Science 2010, 328, 1394–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, L.C.; Cresswell, P. Expanding roles for GILT in immunity. Curr. Opin. Immunol. 2013, 25, 103–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Hou, Z.; Jiang, D.; Zheng, M.; Li, G.; Zhang, Y.; Li, R.; Lin, H.; Chang, J.; Zeng, H.; et al. GILT restricts the cellular entry mediated by the envelope glycoproteins of SARS-CoV, Ebola virus and Lassa fever virus. Emerg. Microbes Infect. 2019, 8, 1511–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mudhasani, R.; Tran, J.P.; Retterer, C.; Radoshitzky, S.R.; Kota, K.P.; Altamura, L.A.; Smith, J.M.; Packard, B.Z.; Kuhn, J.H.; Costantino, J.; et al. IFITM-2 and IFITM-3 but not IFITM-1 restrict Rift Valley fever virus. J. Virol. 2013, 87, 8451–8464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, G.; Schwartz, O.; Compton, A.A. More than meets the I: The diverse antiviral and cellular functions of interferon-induced transmembrane proteins. Retrovirology 2017, 14, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef] [Green Version]
- Spence, J.S.; He, R.; Hoffmann, H.H.; Das, T.; Thinon, E.; Rice, C.M.; Peng, T.; Chandran, K.; Hang, H.C. IFITM3 directly engages and shuttles incoming virus particles to lysosomes. Nat. Chem. Biol. 2019, 15, 259–268. [Google Scholar] [CrossRef]
- Suddala, K.C.; Lee, C.C.; Meraner, P.; Marin, M.; Markosyan, R.M.; Desai, T.M.; Cohen, F.S.; Brass, A.L.; Melikyan, G.B. Interferon-induced transmembrane protein 3 blocks fusion of sensitive but not resistant viruses by partitioning into virus-carrying endosomes. PLoS Pathog. 2019, 15, e1007532. [Google Scholar] [CrossRef] [Green Version]
- Peña Cárcamo, J.R.; Morell, M.L.; Vázquez, C.A.; Vatansever, S.; Upadhyay, A.S.; Överby, A.K.; Cordo, S.M.; García, C.C. The interplay between viperin antiviral activity, lipid droplets and Junín mammarenavirus multiplication. Virology 2018, 514, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, F.; Black, S.G.; Murphy, L.; Griffiths, D.J.; Neil, S.J.; Spencer, T.E.; Palmarini, M. Interplay between Ovine Bone Marrow Stromal Cell Antigen 2/Tetherin and Endogenous Retroviruses. J. Virol. 2010, 84, 4415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Tortorec, A.; Willey, S.; Neil, S.J.D. Antiviral inhibition of enveloped virus release by tetherin/BST-2: Action and counteraction. Viruses 2011, 3, 520–540. [Google Scholar] [CrossRef] [PubMed]
- Perez-Caballero, D.; Zang, T.; Ebrahimi, A.; McNatt, M.W.; Gregory, D.A.; Johnson, M.C.; Bieniasz, P.D. Tetherin inhibits HIV-1 release by directly tethering virions to cells. Cell 2009, 139, 499–511. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Radoshitzky, S.R.; Dong, L.; Chi, X.; Clester, J.C.; Retterer, C.; Spurgers, K.; Kuhn, J.H.; Sandwick, S.; Ruthel, G.; Kota, K.; et al. Infectious Lassa virus, but not filoviruses, is restricted by BST-2/tetherin. J. Virol. 2010, 84, 10569–10580. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Noda, T.; Urata, S.; Kawaoka, Y.; Yasuda, J. Inhibition of Lassa and Marburg virus production by tetherin. J. Virol. 2009, 83, 2382–2385. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, T.; Sakurai, A.; Yasuda, J. Dimerization of tetherin is not essential for its antiviral activity against Lassa and Marburg viruses. PLoS ONE 2009, 4, e6934. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, V.R.; Urata, S.; Sakaguchi, M.; Yasuda, J. Human BST-2/tetherin inhibits Junin virus release from host cells and its inhibition is partially counteracted by viral nucleoprotein. J. Gen. Virol. 2020. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Eastman, S.W.; Jouvenet, N.; Bieniasz, P.D. HIV-1 Vpu Promotes Release and Prevents Endocytosis of Nascent Retrovirus Particles from the Plasma Membrane. PLOS Pathog. 2006, 2, e39. [Google Scholar] [CrossRef]
- Rollason, R.; Korolchuk, V.; Hamilton, C.; Schu, P.; Banting, G. Clathrin-mediated endocytosis of a lipid-raft-associated protein is mediated through a dual tyrosine motif. J. Cell Sci. 2007, 120, 3850. [Google Scholar] [CrossRef] [Green Version]
- Göttlinger, H.G. Virus kept on a leash. Nature 2008, 451, 407–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groseth, A.; Wolff, S.; Strecker, T.; Hoenen, T.; Becker, S. Efficient budding of the tacaribe virus matrix protein z requires the nucleoprotein. J. Virol. 2010, 84, 3603–3611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3’ to 5’ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef] [Green Version]
- Hastie, K.M.; Liu, T.; Li, S.; King, L.B.; Ngo, N.; Zandonatti, M.A.; Woods, V.L., Jr.; de la Torre, J.C.; Saphire, E.O. Crystal structure of the Lassa virus nucleoprotein-RNA complex reveals a gating mechanism for RNA binding. Proc. Natl. Acad. Sci. USA 2011, 108, 19365–19370. [Google Scholar] [CrossRef] [Green Version]
- Levingston Macleod, J.M.; D’Antuono, A.; Loureiro, M.E.; Casabona, J.C.; Gomez, G.A.; Lopez, N. Identification of two functional domains within the arenavirus nucleoprotein. J. Virol. 2011, 85, 2012–2023. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Riaño, E.; Cheng, B.Y.; de la Torre, J.C.; Martínez-Sobrido, L. Self-association of lymphocytic choriomeningitis virus nucleoprotein is mediated by its N-terminal region and is not required for its anti-interferon function. J. Virol. 2012, 86, 3307–3317. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sobrido, L.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; de la Torre, J.C. Differential Inhibition of Type I Interferon Induction by Arenavirus Nucleoproteins. J. Virol. 2007, 81, 12696. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sobrido, L.; Emonet, S.; Giannakas, P.; Cubitt, B.; García-Sastre, A.; de la Torre, J.C. Identification of amino acid residues critical for the anti-interferon activity of the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2009, 83, 11330–11340. [Google Scholar] [CrossRef] [Green Version]
- Harmon, B.; Kozina, C.; Maar, D.; Carpenter, T.S.; Branda, C.S.; Negrete, O.A.; Carson, B.D. Identification of critical amino acids within the nucleoprotein of Tacaribe virus important for anti-interferon activity. J. Biol. Chem. 2013, 288, 8702–8711. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef] [Green Version]
- Carnec, X.; Mateo, M.; Page, A.; Reynard, S.; Hortion, J.; Picard, C.; Yekwa, E.; Barrot, L.; Barron, S.; Vallve, A.; et al. A Vaccine Platform against Arenaviruses Based on a Recombinant Hyperattenuated Mopeia Virus Expressing Heterologous Glycoproteins. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Q.; Shao, J.; Lan, S.; Zhou, Y.; Xing, J.; Dong, C.; Liang, Y.; Ly, H. In vitro and in vivo characterizations of pichinde viral nucleoprotein exoribonuclease functions. J. Virol. 2015, 89, 6595–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, B.R.; Hastie, K.M.; Saphire, E.O. Structure of the LCMV nucleoprotein provides a template for understanding arenavirus replication and immunosuppression. Acta Crystallogr. D. Biol. Crystallogr. 2014, 70, 1764–1769. [Google Scholar] [CrossRef] [PubMed]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa Virus Nucleoprotein Mutants Generated by Reverse Genetics Induce a Robust Type I Interferon Response in Human Dendritic Cells and Macrophages. J. Virol. 2011, 85, 12093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateer, E.J.; Maruyama, J.; Card, G.E.; Paessler, S.; Huang, C. Lassa Virus, but Not Highly Pathogenic New World Arenaviruses, Restricts Immunostimulatory Double-Stranded RNA Accumulation during Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Mateer, E.J.; Paessler, S.; Huang, C. Visualization of Double-Stranded RNA Colocalizing With Pattern Recognition Receptors in Arenavirus Infected Cells. Front. Cell Infect. Microbiol. 2018, 8, 251. [Google Scholar] [CrossRef]
- Weber, F.; Wagner, V.; Rasmussen, S.B.; Hartmann, R.; Paludan, S.R. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 2006, 80, 5059–5064. [Google Scholar] [CrossRef] [Green Version]
- Son, K.N.; Liang, Z.; Lipton, H.L. Double-Stranded RNA Is Detected by Immunofluorescence Analysis in RNA and DNA Virus Infections, Including Those by Negative-Stranded RNA Viruses. J. Virol. 2015, 89, 9383–9392. [Google Scholar] [CrossRef] [Green Version]
- King, B.R.; Hershkowitz, D.; Eisenhauer, P.L.; Weir, M.E.; Ziegler, C.M.; Russo, J.; Bruce, E.A.; Ballif, B.A.; Botten, J. A Map of the Arenavirus Nucleoprotein-Host Protein Interactome Reveals that Junín Virus Selectively Impairs the Antiviral Activity of Double-Stranded RNA-Activated Protein Kinase (PKR). J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Kolokoltsova, O.A.; Mateer, E.J.; Koma, T.; Paessler, S. Highly Pathogenic New World Arenavirus Infection Activates the Pattern Recognition Receptor Protein Kinase R without Attenuating Virus Replication in Human Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pythoud, C.; Rodrigo, W.W.S.I.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [Green Version]
- Pythoud, C.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Lymphocytic Choriomeningitis Virus Differentially Affects the Virus-Induced Type I Interferon Response and Mitochondrial Apoptosis Mediated by RIG-I/MAVS. J. Virol. 2015, 89, 6240–6250. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, W.W.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of nuclear factor kappa B. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.; Huang, Q.; Liu, X.; Di, D.; Liang, Y.; Ly, H. Arenaviral Nucleoproteins Suppress PACT-Induced Augmentation of RIG-I Function To Inhibit Type I Interferon Production. J. Virol. 2018, 92, e00418–e00482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Li, L.; Liu, X.; Dong, S.; Wang, W.; Huo, T.; Guo, Y.; Rao, Z.; Yang, C. Crystal structure of Junin virus nucleoprotein. J. Gen. Virol. 2013, 94, 2175–2183. [Google Scholar] [CrossRef] [Green Version]
- Yekwa, E.; Aphibanthammakit, C.; Carnec, X.; Picard, C.; Canard, B.; Baize, S.; Ferron, F. Arenaviridae exoribonuclease presents genomic RNA edition capacity. bioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Shao, J.; Liang, Y.; Ly, H. Assays to Demonstrate the Roles of Arenaviral Nucleoproteins (NPs) in Viral RNA Synthesis and in Suppressing Type I Interferon. Methods Mol. Biol. 2018, 1604, 189–200. [Google Scholar] [CrossRef]
- Ortiz-Riaño, E.; Cheng, B.Y.; de la Torre, J.C.; Martínez-Sobrido, L. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J. Virol. 2011, 85, 13038–13048. [Google Scholar] [CrossRef] [Green Version]
- Reynard, S.; Russier, M.; Fizet, A.; Carnec, X.; Baize, S. Exonuclease domain of the Lassa virus nucleoprotein is critical to avoid RIG-I signaling and to inhibit the innate immune response. J. Virol. 2014, 88, 13923–13927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loo, Y.M.; Gale, M., Jr. Immune signaling by RIG-I-like receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Tanaka, N.; Hata, N.; Oda, E.; Taniguchi, T. Involvement of the IRF family transcription factor IRF-3 in virus-induced activation of the IFN-beta gene. FEBS Lett. 1998, 425, 112–116. [Google Scholar] [CrossRef] [Green Version]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virol. 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kok, K.H.; Lui, P.Y.; Ng, M.H.; Siu, K.L.; Au, S.W.; Jin, D.Y. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe 2011, 9, 299–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef]
- Shao, J.; Liang, Y.; Ly, H. Roles of Arenavirus Z Protein in Mediating Virion Budding, Viral Transcription-Inhibition and Interferon-Beta Suppression. Methods Mol. Biol. 2018, 1604, 217–227. [Google Scholar] [CrossRef]
- Vidya, M.K.; Kumar, V.G.; Sejian, V.; Bagath, M.; Krishnan, G.; Bhatta, R. Toll-like receptors: Significance, ligands, signaling pathways, and functions in mammals. Int. Rev. Immunol. 2018, 37, 20–36. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, C.D.; Lavanya, M.; Wang, E.; Ross, S.R. Junín Virus Infects Mouse Cells and Induces Innate Immune Responses. J. Virol. 2011, 85, 11058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuevas, C.D.; Ross, S.R. Toll-like receptor 2-mediated innate immune responses against Junín virus in mice lead to antiviral adaptive immune responses during systemic infection and do not affect viral replication in the brain. J. Virol. 2014, 88, 7703–7714. [Google Scholar] [CrossRef] [Green Version]
- Hayes, M.W.; Carrion, R., Jr.; Nunneley, J.; Medvedev, A.E.; Salvato, M.S.; Lukashevich, I.S. Pathogenic Old World arenaviruses inhibit TLR2/Mal-dependent proinflammatory cytokines in vitro. J. Virol. 2012, 86, 7216–7226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.N.; Burke, S.; Montoya, M.; Borrow, P. Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J. Immunol. 2009, 182, 7178–7189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macal, M.; Lewis, G.M.; Kunz, S.; Flavell, R.; Harker, J.A.; Zúñiga, E.I. Plasmacytoid dendritic cells are productively infected and activated through TLR-7 early after arenavirus infection. Cell Host Microbe 2012, 11, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Walsh, K.B.; Teijaro, J.R.; Zuniga, E.I.; Welch, M.J.; Fremgen, D.M.; Blackburn, S.D.; von Tiehl, K.F.; Wherry, E.J.; Flavell, R.A.; Oldstone, M.B. Toll-like receptor 7 is required for effective adaptive immune responses that prevent persistent virus infection. Cell Host Microbe 2012, 11, 643–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, M.J. PKR--a protein kinase regulated by double-stranded RNA. Int. J. Biochem. Cell Biol. 1997, 29, 945–949. [Google Scholar] [CrossRef]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhoads, R.E. Regulation of eukaryotic protein synthesis by initiation factors. J. Biol. Chem. 1993, 268, 3017–3020. [Google Scholar]
- Kumar, A.; Haque, J.; Lacoste, J.; Hiscott, J.; Williams, B.R. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. USA 1994, 91, 6288–6292. [Google Scholar] [CrossRef] [Green Version]
- Schulz, O.; Pichlmair, A.; Rehwinkel, J.; Rogers, N.C.; Scheuner, D.; Kato, H.; Takeuchi, O.; Akira, S.; Kaufman, R.J.; Reis e Sousa, C. Protein kinase R contributes to immunity against specific viruses by regulating interferon mRNA integrity. Cell Host Microbe 2010, 7, 354–361. [Google Scholar] [CrossRef] [Green Version]
- Khamina, K.; Lercher, A.; Caldera, M.; Schliehe, C.; Vilagos, B.; Sahin, M.; Kosack, L.; Bhattacharya, A.; Májek, P.; Stukalov, A.; et al. Characterization of host proteins interacting with the lymphocytic choriomeningitis virus L protein. PLoS Pathog. 2017, 13, e1006758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, M.E.; Zorzetto-Fernandes, A.L.; Radoshitzky, S.; Chi, X.; Dallari, S.; Marooki, N.; Lèger, P.; Foscaldi, S.; Harjono, V.; Sharma, S.; et al. DDX3 suppresses type I interferons and favors viral replication during Arenavirus infection. PLoS Pathog. 2018, 14, e1007125. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Fullam, A.; Brennan, R.; Schröder, M. Human DEAD box helicase 3 couples IκB kinase ε to interferon regulatory factor 3 activation. Mol. Cell Biol. 2013, 33, 2004–2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010, 40, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef]
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008, 27, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.C.; Sun, H.S.; Wang, S.W.; Tarn, W.Y. DDX3 functions in antiviral innate immunity through translational control of PACT. FEBS J. 2016, 283, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Becker, S.; Groseth, A. Cleavage of the Junin virus nucleoprotein serves a decoy function to inhibit the induction of apoptosis during infection. J. Virol. 2013, 87, 224–233. [Google Scholar] [CrossRef] [Green Version]
- Wolff, S.; Groseth, A.; Meyer, B.; Jackson, D.; Strecker, T.; Kaufmann, A.; Becker, S. The New World arenavirus Tacaribe virus induces caspase-dependent apoptosis in infected cells. J. Gen. Virol. 2016, 97, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Kolokoltsova, O.A.; Grant, A.M.; Huang, C.; Smith, J.K.; Poussard, A.L.; Tian, B.; Brasier, A.R.; Peters, C.J.; Tseng, C.T.; de la Torre, J.C.; et al. RIG-I enhanced interferon independent apoptosis upon Junin virus infection. PLoS ONE 2014, 9, e99610. [Google Scholar] [CrossRef] [PubMed]
- Moreno, H.; Möller, R.; Fedeli, C.; Gerold, G.; Kunz, S. Comparison of the Innate Immune Responses to Pathogenic and Nonpathogenic Clade B New World Arenaviruses. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, S.; Marques, J.T.; Yamashita, M.; Peters, K.L.; Smith, K.; Desai, A.; Williams, B.R.G.; Sen, G.C. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010, 29, 1762–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chattopadhyay, S.; Yamashita, M.; Zhang, Y.; Sen, G.C. The IRF-3/Bax-Mediated Apoptotic Pathway, Activated by Viral Cytoplasmic RNA and DNA, Inhibits Virus Replication. J. Virol. 2011, 85, 3708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, B.; Groseth, A. Apoptosis during arenavirus infection: Mechanisms and evasion strategies. Microbes Infect. 2018, 20, 65–80. [Google Scholar] [CrossRef] [PubMed]
- Clegg, J.C.; Lloyd, G. Structural and cell-associated proteins of Lassa virus. J. Gen. Virol 1983, 64, 1127–1136. [Google Scholar] [CrossRef]
- Harnish, D.G.; Leung, W.C.; Rawls, W.E. Characterization of polypeptides immunoprecipitable from Pichinde virus-infected BHK-21 cells. J. Virol. 1981, 38, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Young, P.R.; Chanas, A.C.; Lee, S.R.; Gould, E.A.; Howard, C.R. Localization of an arenavirus protein in the nuclei of infected cells. J. Gen. Virol. 1987, 68 (Pt. 9), 2465–2470. [Google Scholar] [CrossRef]
- Hastie, K.M.; Zandonatti, M.; Liu, T.; Li, S.; Woods, V.L., Jr.; Saphire, E.O. Crystal Structure of the Oligomeric Form of Lassa Virus Matrix Protein Z. J. Virol. 2016, 90, 4556–4562. [Google Scholar] [CrossRef] [Green Version]
- Salvato, M.S.; Shimomaye, E.M. The completed sequence of lymphocytic choriomeningitis virus reveals a unique RNA structure and a gene for a zinc finger protein. Virology 1989, 173, 1–10. [Google Scholar] [CrossRef]
- Volpon, L.; Osborne, M.J.; Borden, K.L. NMR assignment of the arenaviral protein Z from Lassa fever virus. Biomol. NMR Assign. 2008, 2, 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volpon, L.; Osborne, M.J.; Capul, A.A.; de la Torre, J.C.; Borden, K.L. Structural characterization of the Z RING-eIF4E complex reveals a distinct mode of control for eIF4E. Proc. Natl. Acad. Sci. USA 2010, 107, 5441–5446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, C.M.; Eisenhauer, P.; Bruce, E.A.; Weir, M.E.; King, B.R.; Klaus, J.P.; Krementsov, D.N.; Shirley, D.J.; Ballif, B.A.; Botten, J. The Lymphocytic Choriomeningitis Virus Matrix Protein PPXY Late Domain Drives the Production of Defective Interfering Particles. PLoS Pathog. 2016, 12, e1005501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strecker, T.; Maisa, A.; Daffis, S.; Eichler, R.; Lenz, O.; Garten, W. The role of myristoylation in the membrane association of the Lassa virus matrix protein Z. J. Virol. 2006, 3, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borden, K.L.; Campbell Dwyer, E.J.; Salvato, M.S. An arenavirus RING (zinc-binding) protein binds the oncoprotein promyelocyte leukemia protein (PML) and relocates PML nuclear bodies to the cytoplasm. J. Virol. 1998, 72, 758–766. [Google Scholar] [CrossRef] [Green Version]
- Borden, K.L.; Campbelldwyer, E.J.; Carlile, G.W.; Djavani, M.; Salvato, M.S. Two RING finger proteins, the oncoprotein PML and the arenavirus Z protein, colocalize with the nuclear fraction of the ribosomal P proteins. J. Virol. 1998, 72, 3819–3826. [Google Scholar] [CrossRef] [Green Version]
- Regad, T.; Chelbi-Alix, M.K. Role and fate of PML nuclear bodies in response to interferon and viral infections. Oncogene 2001, 20, 7274–7286. [Google Scholar] [CrossRef] [Green Version]
- Campbell Dwyer, E.J.; Lai, H.; MacDonald, R.C.; Salvato, M.S.; Borden, K.L. The lymphocytic choriomeningitis virus RING protein Z associates with eukaryotic initiation factor 4E and selectively represses translation in a RING-dependent manner. J. Virol. 2000, 74, 3293–3300. [Google Scholar] [CrossRef] [Green Version]
- Kentsis, A.; Dwyer, E.C.; Perez, J.M.; Sharma, M.; Chen, A.; Pan, Z.Q.; Borden, K.L. The RING domains of the promyelocytic leukemia protein PML and the arenaviral protein Z repress translation by directly inhibiting translation initiation factor eIF4E. J. Mol. Biol. 2001, 312, 609–623. [Google Scholar] [CrossRef]
- Djavani, M.; Topisirovic, I.; Zapata, J.C.; Sadowska, M.; Yang, Y.; Rodas, J.; Lukashevich, I.S.; Bogue, C.W.; Pauza, C.D.; Borden, K.L.; et al. The proline-rich homeodomain (PRH/HEX) protein is down-regulated in liver during infection with lymphocytic choriomeningitis virus. J. Virol. 2005, 79, 2461–2473. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Yasuda, J. Molecular mechanism of arenavirus assembly and budding. Viruses 2012, 4, 2049–2079. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Fehling, S.K.; Noda, T.; Maisner, A.; Lamp, B.; Conzelmann, K.K.; Kawaoka, Y.; Klenk, H.D.; Garten, W.; Strecker, T. The microtubule motor protein KIF13A is involved in intracellular trafficking of the Lassa virus matrix protein Z. Cell Microbiol. 2013, 15, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Shtanko, O.; Watanabe, S.; Jasenosky, L.D.; Watanabe, T.; Kawaoka, Y. ALIX/AIP1 is required for NP incorporation into Mopeia virus Z-induced virus-like particles. J. Virol. 2011, 85, 3631–3641. [Google Scholar] [CrossRef] [Green Version]
- Urata, S.; Noda, T.; Kawaoka, Y.; Yokosawa, H.; Yasuda, J. Cellular factors required for Lassa virus budding. J. Virol. 2006, 80, 4191–4195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, S.; Yasuda, J.; de la Torre, J.C. The z protein of the new world arenavirus tacaribe virus has bona fide budding activity that does not depend on known late domain motifs. J. Virol. 2009, 83, 12651–12655. [Google Scholar] [CrossRef] [Green Version]
- Baillet, N.; Krieger, S.; Carnec, X.; Mateo, M.; Journeaux, A.; Merabet, O.; Caro, V.; Tangy, F.; Vidalain, P.-O.; Baize, S. E3 Ligase ITCH Interacts with the Z Matrix Protein of Lassa and Mopeia Viruses and Is Required for the Release of Infectious Particles. Viruses 2019, 12, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, C.M.; Dang, L.; Eisenhauer, P.; Kelly, J.A.; King, B.R.; Klaus, J.P.; Manuelyan, I.; Mattice, E.B.; Shirley, D.J.; Weir, M.E.; et al. NEDD4 family ubiquitin ligases associate with LCMV Z’s PPXY domain and are required for virus budding, but not via direct ubiquitination of Z. PLoS Pathog. 2019, 15, e1008100. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.M.; Eisenhauer, P.; Manuelyan, I.; Weir, M.E.; Bruce, E.A.; Ballif, B.A.; Botten, J. Host-Driven Phosphorylation Appears to Regulate the Budding Activity of the Lassa Virus Matrix Protein. Pathogens 2018, 7, 97. [Google Scholar] [CrossRef] [Green Version]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-I and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Ly, H.; Liang, Y. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-I-like receptor-dependent interferon production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.; Chai, Z.; Ly, H.; Liang, Y. Differential Inhibition of Macrophage Activation by Lymphocytic Choriomeningitis Virus and Pichinde Virus Is Mediated by the Z Protein N-Terminal Domain. J. Virol. 2015, 89, 12513–12517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, U.; Steinhoff, U.; Reis, L.F.; Hemmi, S.; Pavlovic, J.; Zinkernagel, R.M.; Aguet, M. Functional role of type I and type II interferons in antiviral defense. Science 1994, 264, 1918–1921. [Google Scholar] [CrossRef]
- Schaeffer, J.; Carnec, X.; Reynard, S.; Mateo, M.; Picard, C.; Pietrosemoli, N.; Dillies, M.-A.; Baize, S. Lassa virus activates myeloid dendritic cells but suppresses their ability to stimulate T cells. PLOS Pathog. 2018, 14, e1007430. [Google Scholar] [CrossRef] [PubMed]
- Mendenhall, M.; Russell, A.; Juelich, T.; Messina, E.L.; Smee, D.F.; Freiberg, A.N.; Holbrook, M.R.; Furuta, Y.; de la Torre, J.C.; Nunberg, J.H.; et al. T-705 (favipiravir) inhibition of arenavirus replication in cell culture. Antimicrob. Agents Chemother. 2011, 55, 782–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.M.; Rojek, J.M.; Spiropoulou, C.F.; Gundersen, A.T.; Jin, W.; Shaginian, A.; York, J.; Nunberg, J.H.; Boger, D.L.; Oldstone, M.B.; et al. Unique small molecule entry inhibitors of hemorrhagic fever arenaviruses. J. Biol. Chem. 2008, 283, 18734–18742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLay, L.; Ansari, A.; Liang, Y.; Ly, H. Targeting virulence mechanisms for the prevention and therapy of arenaviral hemorrhagic fever. Antiviral. Res. 2013, 97, 81–92. [Google Scholar] [CrossRef] [Green Version]
- Torriani, G.; Trofimenko, E.; Mayor, J.; Fedeli, C.; Moreno, H.; Michel, S.; Heulot, M.; Chevalier, N.; Zimmer, G.; Shrestha, N.; et al. Identification of Clotrimazole Derivatives as Specific Inhibitors of Arenavirus Fusion. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- García, C.C.; Djavani, M.; Topisirovic, I.; Borden, K.L.; Salvato, M.S.; Damonte, E.B. Arenavirus Z protein as an antiviral target: Virus inactivation and protein oligomerization by zinc finger-reactive compounds. J. Gen. Virol. 2006, 87, 1217–1228. [Google Scholar] [CrossRef]
- García, C.C.; Topisirovic, I.; Djavani, M.; Borden, K.L.; Damonte, E.B.; Salvato, M.S. An antiviral disulfide compound blocks interaction between arenavirus Z protein and cellular promyelocytic leukemia protein. Biochem Biophys Res. Commun 2010, 393, 625–630. [Google Scholar] [CrossRef] [Green Version]
- Cordo, S.M.; Candurra, N.A.; Damonte, E.B. Myristic acid analogs are inhibitors of Junin virus replication. Microbes. Infect. 1999, 1, 609–614. [Google Scholar] [CrossRef]
- Miranda, P.O.; Cubitt, B.; Jacob, N.T.; Janda, K.D.; de la Torre, J.C. Mining a Kröhnke Pyridine Library for Anti-Arenavirus Activity. ACS Infect. Dis. 2018, 4, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Zapata, J.C.; Salvato, M.S. Arenavirus variations due to host-specific adaptation. Viruses 2013, 5, 241–278. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| dsRNA Binding | dsRNA Degradation | Inhibits Nuclear Translocation of IRF-3 through Interaction with RIG-I and MDA-5 | Targets Kinase Domain of IKKε and Blocks Its Activity | Inhibition of PACT-Mediated Augmentation of RIG-I Signalling | Inhibits NF-κB Transcriptional Activation | Activation of PKR | Refs | |
|---|---|---|---|---|---|---|---|---|
| Pathogenic | ||||||||
| LCMV | √ | n.d. | √ | √ | n.d. | √ | √ | [102,103,108,116,117,118,119] |
| LASV | √ | √ | √ | √ | √ | √ | × | [96,99,102,103,105,110,115,117,119,120] |
| JUNV | × | × | √ | √ | √ | √ | √ | [92,102,110,111,114,115,117,119,120,121] |
| MACV | n.d. | × | √ | n.d. | √ | √ | √ | [102,110,111,115,119,120] |
| Non-Pathogenic | ||||||||
| MOPV | √ | √ | n.d. | n.d. | n.d. | n.d. | n.d. | [122] |
| PICV | √ | √ | √ | √ | √ | √ | n.d. | [102,107,119,120,123] |
| TCRV | √ | √ | × | n.d. | √ | × | n.d. | [102,105,119,120] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stott, R.J.; Strecker, T.; Foster, T.L. Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins. Viruses 2020, 12, 784. https://doi.org/10.3390/v12070784
Stott RJ, Strecker T, Foster TL. Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins. Viruses. 2020; 12(7):784. https://doi.org/10.3390/v12070784
Chicago/Turabian StyleStott, Robert J., Thomas Strecker, and Toshana L. Foster. 2020. "Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins" Viruses 12, no. 7: 784. https://doi.org/10.3390/v12070784