Structural Insights into APOBEC3-Mediated Lentiviral Restriction



Abstract
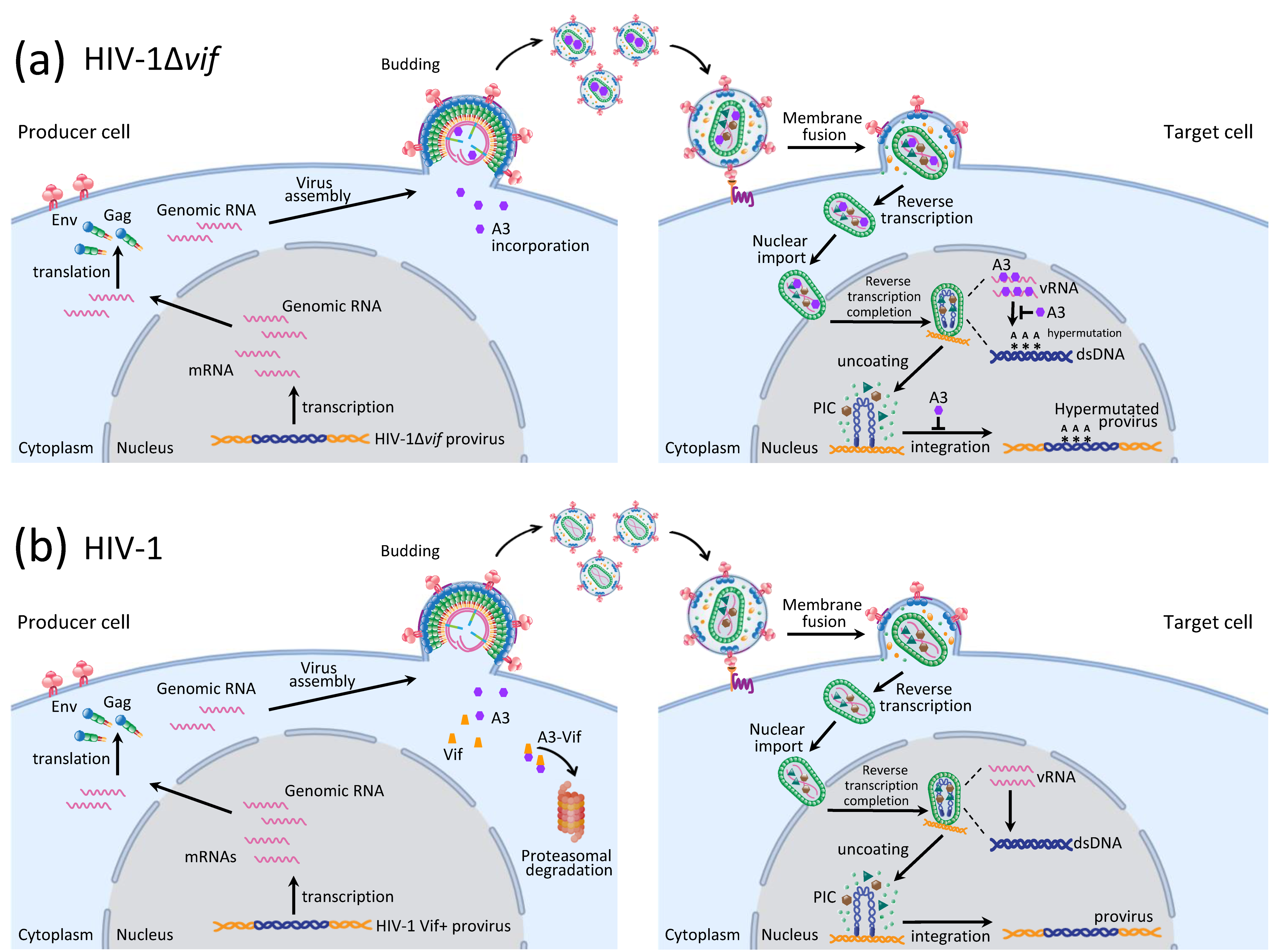
:1. Introduction
2. Overview of A3 Protein and Vif Structures
3. Identification of A3 Residues Critical for A3-Vif Interactions through Mutational Analyses
3.1. A3G Determinants That Interact with Vif
3.2. A3F, A3C, and A3D Determinants That Interact with Vif
3.3. A3H Determinants That Interact with Vif
4. Identification of HIV-1 Vif Residues Critical for Interactions with the CRL5 E3 Ubiquitin Ligase, A3 Proteins, and CBFβ through Mutational Analyses
4.1. HIV-1 Vif Determinants That Interact with CRL5 E3 Ubiquitin Ligase
4.2. HIV-1 Vif Determinants That Interact with A3 Proteins
4.3. HIV-1 Vif Determinants That Interact with CBFβ
5. Structure of the Pentameric Complex of Vif, Cul5, EloB/C, and CBFβ
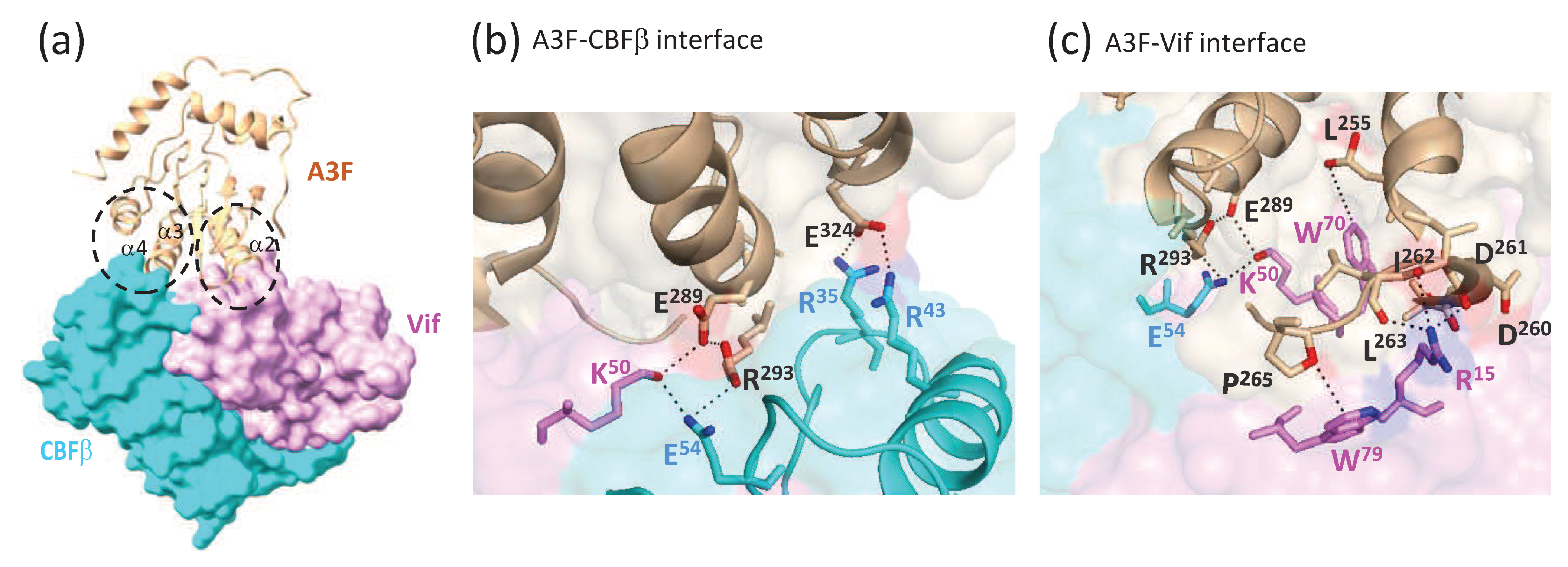
6. Structure of a Vif-CBFβ-A3FCTD Ternary Complex
7. Insights into Substrate Selection, A3 Deamination, and Editing-Site Selection
7.1. ssDNA Substrate Selection
7.2. Deamination and Editing-Site Selection
8. Summary
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stenglein, M.D.; Burns, M.B.; Li, M.; Lengyel, J.; Harris, R.S. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Nat. Struct. Mol. Biol. 2010, 17, 222–229. [Google Scholar] [CrossRef] [PubMed]
- Jarmuz, A.; Chester, A.; Bayliss, J.; Gisbourne, J.; Dunham, I.; Scott, J.; Navaratnam, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, M.; Kinoshita, K.; Fagarasan, S.; Yamada, S.; Shinkai, Y.; Honjo, T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 2000, 102, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Rogozin, I.B.; Iyer, L.M.; Liang, L.; Glazko, G.V.; Liston, V.G.; Pavlov, Y.I.; Aravind, L.; Pancer, Z. Evolution and diversification of lamprey antigen receptors: Evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat. Immunol. 2007, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Habib, G.; Yang, C.Y.; Gu, Z.W.; Lee, B.R.; Weng, S.A.; Silberman, S.R.; Cai, S.J.; Deslypere, J.P.; Rosseneu, M.; et al. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366. [Google Scholar] [CrossRef]
- Powell, L.M.; Wallis, S.C.; Pease, R.J.; Edwards, Y.H.; Knott, T.J.; Scott, J. A novel form of tissue-specific RNA processing produces apolipoprotein-B48 in intestine. Cell 1987, 50, 831–840. [Google Scholar] [CrossRef]
- Liao, W.; Hong, S.H.; Chan, B.H.; Rudolph, F.B.; Clark, S.C.; Chan, L. APOBEC-2, a cardiac- and skeletal muscle-specific member of the cytidine deaminase supergene family. Biochem. Biophys. Res. Commun. 1999, 260, 398–404. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Basu, M.K.; Jordan, I.K.; Pavlov, Y.I.; Koonin, E.V. APOBEC4, a new member of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases predicted by computational analysis. Cell Cycle 2005, 4, 1281–1285. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G.; Thomas, C.J.; Petersen-Mahrt, S.K.; Neuberger, M.S. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Mol. Biol. Evol. 2005, 22, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaRue, R.S.; Andresdottir, V.; Blanchard, Y.; Conticello, S.G.; Derse, D.; Emerman, M.; Greene, W.C.; Jonsson, S.R.; Landau, N.R.; Lochelt, M.; et al. Guidelines for naming nonprimate APOBEC3 genes and proteins. J. Virol. 2009, 83, 494–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, F.; Bollman, B.; Chen, H.; Konig, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary function of the two catalytic domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svarovskaia, E.S.; Xu, H.; Mbisa, J.L.; Barr, R.; Gorelick, R.J.; Ono, A.; Freed, E.O.; Hu, W.S.; Pathak, V.K. Human apolipoprotein B mRNA-editing enzyme-catalytic polypeptide-like 3G (APOBEC3G) is incorporated into HIV-1 virions through interactions with viral and nonviral RNAs. J. Biol. Chem. 2004, 279, 35822–35828. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Tian, C.; Zhang, W.; Luo, K.; Sarkis, P.T.; Yu, L.; Liu, B.; Yu, Y.; Yu, X.F. 7SL RNA mediates virion packaging of the antiviral cytidine deaminase APOBEC3G. J. Virol. 2007, 81, 13112–13124. [Google Scholar] [CrossRef] [Green Version]
- Chelico, L.; Prochnow, C.; Erie, D.A.; Chen, X.S.; Goodman, M.F. Structural model for deoxycytidine deamination mechanisms of the HIV-1 inactivation enzyme APOBEC3G. J. Biol. Chem. 2010, 285, 16195–16205. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Chelico, L. Intensity of deoxycytidine deamination of HIV-1 proviral DNA by the retroviral restriction factor APOBEC3G is mediated by the noncatalytic domain. J. Biol. Chem. 2011, 286, 11415–11426. [Google Scholar] [CrossRef] [Green Version]
- Holden, L.G.; Prochnow, C.; Chang, Y.P.; Bransteitter, R.; Chelico, L.; Sen, U.; Stevens, R.C.; Goodman, M.F.; Chen, X.S. Crystal structure of the anti-viral APOBEC3G catalytic domain and functional implications. Nature 2008, 456, 121–124. [Google Scholar] [CrossRef]
- Nowarski, R.; Britan-Rosich, E.; Shiloach, T.; Kotler, M. Hypermutation by intersegmental transfer of APOBEC3G cytidine deaminase. Nat. Struct. Mol. Biol. 2008, 15, 1059–1066. [Google Scholar] [CrossRef]
- Betts, L.; Xiang, S.; Short, S.A.; Wolfenden, R.; Carter, C.W., Jr. Cytidine deaminase. The 2.3 A crystal structure of an enzyme: Transition-state analog complex. J. Mol. Biol. 1994, 235, 635–656. [Google Scholar] [CrossRef]
- Ireton, G.C.; McDermott, G.; Black, M.E.; Stoddard, B.L. The structure of Escherichia coli cytosine deaminase. J. Mol. Biol. 2002, 315, 687–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navaratnam, N.; Fujino, T.; Bayliss, J.; Jarmuz, A.; How, A.; Richardson, N.; Somasekaram, A.; Bhattacharya, S.; Carter, C.; Scott, J. Escherichia coli cytidine deaminase provides a molecular model for ApoB RNA editing and a mechanism for RNA substrate recognition. J. Mol. Biol. 1998, 275, 695–714. [Google Scholar] [CrossRef] [PubMed]
- Ta, V.T.; Nagaoka, H.; Catalan, N.; Durandy, A.; Fischer, A.; Imai, K.; Nonoyama, S.; Tashiro, J.; Ikegawa, M.; Ito, S.; et al. AID mutant analyses indicate requirement for class-switch-specific cofactors. Nat. Immunol. 2003, 4, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Zaim, J.; Kierzek, A.M. Domain organization of activation-induced cytidine deaminase. Nat. Immunol. 2003, 4, 1153. [Google Scholar] [CrossRef] [PubMed]
- Xie, K.; Sowden, M.P.; Dance, G.S.; Torelli, A.T.; Smith, H.C.; Wedekind, J.E. The structure of a yeast RNA-editing deaminase provides insight into the fold and function of activation-induced deaminase and APOBEC-1. Proc. Natl. Acad. Sci. USA 2004, 101, 8114–8119. [Google Scholar] [CrossRef] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Adolph, M.B.; Ara, A.; Feng, Y.; Wittkopp, C.J.; Emerman, M.; Fraser, J.S.; Chelico, L. Cytidine deaminase efficiency of the lentiviral viral restriction factor APOBEC3C correlates with dimerization. Nucleic Acids Res. 2017, 45, 3378–3394. [Google Scholar] [CrossRef]
- Wittkopp, C.J.; Adolph, M.B.; Wu, L.I.; Chelico, L.; Emerman, M. A Single Nucleotide Polymorphism in Human APOBEC3C Enhances Restriction of Lentiviruses. PLoS Pathog. 2016, 12, e1005865. [Google Scholar] [CrossRef]
- Lecossier, D.; Bouchonnet, F.; Clavel, F.; Hance, A.J. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science 2003, 300, 1112. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA deamination mediates innate immunity to retroviral infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Desimmie, B.A.; Burdick, R.C.; Izumi, T.; Doi, H.; Shao, W.; Alvord, W.G.; Sato, K.; Koyanagi, Y.; Jones, S.; Wilson, E.; et al. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res. 2016, 44, 7848–7865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Pathak, V.K.; Temin, H.M. Broad spectrum of in vivo forward mutations, hypermutations, and mutational hotspots in a retroviral shuttle vector after a single replication cycle: Substitutions, frameshifts, and hypermutations. Proc. Natl. Acad. Sci. USA 1990, 87, 6019–6023. [Google Scholar] [CrossRef] [Green Version]
- Arias, J.F.; Koyama, T.; Kinomoto, M.; Tokunaga, K. Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbiol. 2012, 3, 275. [Google Scholar] [CrossRef] [Green Version]
- Treger, R.S.; Tokuyama, M.; Dong, H.; Salas-Briceno, K.; Ross, S.R.; Kong, Y.; Iwasaki, A. Human APOBEC3G prevents emergence of infectious endogenous retrovirus in mice. J. Virol. 2019, e00728. [Google Scholar] [CrossRef] [Green Version]
- Nitta, T.; Lee, S.; Ha, D.; Arias, M.; Kozak, C.A.; Fan, H. Moloney murine leukemia virus glyco-gag facilitates xenotropic murine leukemia virus-related virus replication through human APOBEC3-independent mechanisms. Retrovirology 2012, 9, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okeoma, C.M.; Lovsin, N.; Peterlin, B.M.; Ross, S.R. APOBEC3 inhibits mouse mammary tumour virus replication in vivo. Nature 2007, 445, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Okeoma, C.M.; Shen, M.; Ross, S.R. A novel block to mouse mammary tumor virus infection of lymphocytes in B10.BR mice. J. Virol. 2008, 82, 1314–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussil, B.; Sauermann, U.; Motzkus, D.; Stahl-Hennig, C.; Sopper, S. Increased APOBEC3G and APOBEC3F expression is associated with low viral load and prolonged survival in simian immunodeficiency virus infected rhesus monkeys. Retrovirology 2011, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Sasada, A.; Takaori-Kondo, A.; Shirakawa, K.; Kobayashi, M.; Abudu, A.; Hishizawa, M.; Imada, K.; Tanaka, Y.; Uchiyama, T. APOBEC3G targets human T-cell leukemia virus type 1. Retrovirology 2005, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Mahieux, R.; Suspene, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.P. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, H.L.; Cullen, B.R. Inhibition of alpharetrovirus replication by a range of human APOBEC3 proteins. J. Virol. 2007, 81, 13694–13699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lochelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rosler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Lilley, C.E.; Yu, Q.; Lee, D.V.; Chou, J.; Narvaiza, I.; Landau, N.R.; Weitzman, M.D. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr. Biol. 2006, 16, 480–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turelli, P.; Mangeat, B.; Jost, S.; Vianin, S.; Trono, D. Inhibition of hepatitis B virus replication by APOBEC3G. Science 2004, 303, 1829. [Google Scholar] [CrossRef] [PubMed]
- Vartanian, J.P.; Guetard, D.; Henry, M.; Wain-Hobson, S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science 2008, 320, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Nakaya, Y.; Stavrou, S.; Blouch, K.; Tattersall, P.; Ross, S.R. In Vivo Examination of Mouse APOBEC3- and Human APOBEC3A- and APOBEC3G-Mediated Restriction of Parvovirus and Herpesvirus Infection in Mouse Models. J. Virol. 2016, 90, 8005–8012. [Google Scholar] [CrossRef] [Green Version]
- Burdick, R.C.; Li, C.; Munshi, M.; Rawson, J.M.O.; Nagashima, K.; Hu, W.S.; Pathak, V.K. HIV-1 uncoats in the nucleus near sites of integration. Proc. Natl. Acad. Sci. USA 2020, 117, 5486–5493. [Google Scholar] [CrossRef]
- Shirakawa, K.; Takaori-Kondo, A.; Kobayashi, M.; Tomonaga, M.; Izumi, T.; Fukunaga, K.; Sasada, A.; Abudu, A.; Miyauchi, Y.; Akari, H.; et al. Ubiquitination of APOBEC3 proteins by the Vif-Cullin5-ElonginB-ElonginC complex. Virology 2006, 344, 263–266. [Google Scholar] [CrossRef]
- Marin, M.; Rose, K.M.; Kozak, S.L.; Kabat, D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat. Med. 2003, 9, 1398–1403. [Google Scholar] [CrossRef]
- Mehle, A.; Strack, B.; Ancuta, P.; Zhang, C.; McPike, M.; Gabuzda, D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J. Biol. Chem. 2004, 279, 7792–7798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef] [PubMed]
- Stopak, K.; de Noronha, C.; Yonemoto, W.; Greene, W.C. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol. Cell. 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Irwin, D.; Kurosu, T.; Tokunaga, K.; Sata, T.; Peterlin, B.M. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J. Virol. 2004, 78, 6073–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegand, H.L.; Doehle, B.P.; Bogerd, H.P.; Cullen, B.R. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004, 23, 2451–2458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hultquist, J.F.; Lengyel, J.A.; Refsland, E.W.; LaRue, R.S.; Lackey, L.; Brown, W.L.; Harris, R.S. Human and rhesus APOBEC3D, APOBEC3F, APOBEC3G, and APOBEC3H demonstrate a conserved capacity to restrict Vif-deficient HIV-1. J. Virol. 2011, 85, 11220–11234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OhAinle, M.; Kerns, J.A.; Malik, H.S.; Emerman, M. Adaptive evolution and antiviral activity of the conserved mammalian cytidine deaminase APOBEC3H. J. Virol. 2006, 80, 3853–3862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Y.; Wang, X.; Esselman, W.J.; Zheng, Y.H. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 2006, 80, 10522–10533. [Google Scholar] [CrossRef] [Green Version]
- Refsland, E.W.; Hultquist, J.F.; Harris, R.S. Endogenous origins of HIV-1 G-to-A hypermutation and restriction in the nonpermissive T cell line CEM2n. PLoS Pathog. 2012, 8, e1002800. [Google Scholar] [CrossRef]
- Chaipan, C.; Smith, J.L.; Hu, W.S.; Pathak, V.K. APOBEC3G restricts HIV-1 to a greater extent than APOBEC3F and APOBEC3DE in human primary CD4+ T cells and macrophages. J. Virol. 2013, 87, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doehle, B.P.; Schafer, A.; Wiegand, H.L.; Bogerd, H.P.; Cullen, B.R. Differential sensitivity of murine leukemia virus to APOBEC3-mediated inhibition is governed by virion exclusion. J. Virol. 2005, 79, 8201–8207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Liu, B.; Xiao, Z.; Yu, Y.; Yu, X.; Gorelick, R.; Yu, X.F. Amino-terminal region of the human immunodeficiency virus type 1 nucleocapsid is required for human APOBEC3G packaging. J. Virol. 2004, 78, 11841–11852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zennou, V.; Perez-Caballero, D.; Gottlinger, H.; Bieniasz, P.D. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.A.; Kao, S.; Miyagi, E.; Takeuchi, H.; Goila-Gaur, R.; Opi, S.; Gipson, C.L.; Parslow, T.G.; Ly, H.; Strebel, K. Viral RNA is required for the association of APOBEC3G with human immunodeficiency virus type 1 nucleoprotein complexes. J. Virol. 2005, 79, 5870–5874. [Google Scholar] [CrossRef] [Green Version]
- Alce, T.M.; Popik, W. APOBEC3G is incorporated into virus-like particles by a direct interaction with HIV-1 Gag nucleocapsid protein. J. Biol. Chem. 2004, 279, 34083–34086. [Google Scholar] [CrossRef] [Green Version]
- Schafer, A.; Bogerd, H.P.; Cullen, B.R. Specific packaging of APOBEC3G into HIV-1 virions is mediated by the nucleocapsid domain of the gag polyprotein precursor. Virology 2004, 328, 163–168. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Chertova, E.; Chen, J.; Ott, D.E.; Roser, J.D.; Hu, W.S.; Pathak, V.K. Stoichiometry of the antiviral protein APOBEC3G in HIV-1 virions. Virology 2007, 360, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Suspene, R.; Sommer, P.; Henry, M.; Ferris, S.; Guetard, D.; Pochet, S.; Chester, A.; Navaratnam, N.; Wain-Hobson, S.; Vartanian, J.P. APOBEC3G is a single-stranded DNA cytidine deaminase and functions independently of HIV reverse transcriptase. Nucleic Acids Res. 2004, 32, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [Green Version]
- Yu, Q.; Konig, R.; Pillai, S.; Chiles, K.; Kearney, M.; Palmer, S.; Richman, D.; Coffin, J.M.; Landau, N.R. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat. Struct. Mol. Biol. 2004, 11, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Sheehy, A.M.; Craig, H.M.; Malim, M.H.; Neuberger, M.S. DNA deamination: Not just a trigger for antibody diversification but also a mechanism for defense against retroviruses. Nat. Immunol. 2003, 4, 641–643. [Google Scholar] [CrossRef] [PubMed]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G contributes to HIV-1 variation through sublethal mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hache, G.; Mansky, L.M.; Harris, R.S. Human APOBEC3 proteins, retrovirus restriction, and HIV drug resistance. AIDS Rev. 2006, 8, 148–157. [Google Scholar]
- Deforche, K.; Camacho, R.; Laethem, K.V.; Shapiro, B.; Moreau, Y.; Rambaut, A.; Vandamme, A.M.; Lemey, P. Estimating the relative contribution of dNTP pool imbalance and APOBEC3G/3F editing to HIV evolution in vivo. J. Comput. Biol. 2007, 14, 1105–1114. [Google Scholar] [CrossRef] [Green Version]
- Wood, N.; Bhattacharya, T.; Keele, B.F.; Giorgi, E.; Liu, M.; Gaschen, B.; Daniels, M.; Ferrari, G.; Haynes, B.F.; McMichael, A.; et al. HIV evolution in early infection: Selection pressures. PLoS Pathog. 2009, 5, e1000414. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.Y.; Bhattacharya, T.; Kunstman, K.; Swantek, P.; Koning, F.A.; Malim, M.H.; Wolinsky, S.M. Human APOBEC3G-mediated editing can promote HIV-1 sequence diversification and accelerate adaptation to selective pressure. J. Virol. 2010, 84, 10402–10405. [Google Scholar] [CrossRef] [Green Version]
- Neogi, U.; Shet, A.; Sahoo, P.N.; Bontell, I.; Ekstrand, M.L.; Banerjea, A.C.; Sonnerborg, A. Human APOBEC3G-mediated hypermutation is associated with antiretroviral therapy failure in HIV-1 subtype C-infected individuals. J. Int. AIDS Soc. 2013, 16, 18472. [Google Scholar] [CrossRef]
- Mulder, L.C.; Harari, A.; Simon, V. Cytidine deamination induced HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 2008, 105, 5501–5506. [Google Scholar] [CrossRef] [Green Version]
- Jern, P.; Russell, R.A.; Pathak, V.K.; Coffin, J.M. Likely role of APOBEC3G-mediated G-to-A mutations in HIV-1 evolution and drug resistance. PLoS Pathog. 2009, 5, e1000367. [Google Scholar] [CrossRef]
- McCallum, M.; Oliveira, M.; Ibanescu, R.I.; Kramer, V.G.; Moisi, D.; Asahchop, E.L.; Brenner, B.G.; Harrigan, P.R.; Xu, H.; Wainberg, M.A. Basis for early and preferential selection of the E138K mutation in HIV-1 reverse transcriptase. Antimicrob. Agents Chemother. 2013, 57, 4681–4688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.M.; Fahrny, A.; Jayaprakash, A.; Gers-Huber, G.; Dillon-White, M.; Audige, A.; Mulder, L.C.F.; Sachidanandam, R.; Speck, R.F.; Simon, V. Impact of Suboptimal APOBEC3G Neutralization on the Emergence of HIV Drug Resistance in Humanized Mice. J. Virol. 2020, 94, e01543. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.E.; Deforche, K.; Chang, C.H.; Wee, E.; Kramer, B.; Welch, J.J.; Gerstoft, J.; Fugger, L.; McMichael, A.; Rambaut, A.; et al. APOBEC3G-induced hypermutation of human immunodeficiency virus type-1 is typically a discrete “all or nothing” phenomenon. PLoS Genet. 2012, 8, e1002550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delviks-Frankenberry, K.A.; Nikolaitchik, O.A.; Burdick, R.C.; Gorelick, R.J.; Keele, B.F.; Hu, W.S.; Pathak, V.K. Minimal Contribution of APOBEC3-Induced G-to-A Hypermutation to HIV-1 Recombination and Genetic Variation. PLoS Pathog. 2016, 12, e1005646. [Google Scholar] [CrossRef]
- Ebrahimi, D.; Anwar, F.; Davenport, M.P. APOBEC3 has not left an evolutionary footprint on the HIV-1 genome. J. Virol. 2011, 85, 9139–9146. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.A.; Moore, M.D.; Hu, W.S.; Pathak, V.K. APOBEC3G induces a hypermutation gradient: Purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology 2009, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Chan, D.S.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Newman, E.N.; Holmes, R.K.; Craig, H.M.; Klein, K.C.; Lingappa, J.R.; Malim, M.H.; Sheehy, A.M. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr. Biol. 2005, 15, 166–170. [Google Scholar] [CrossRef] [Green Version]
- Adolph, M.B.; Webb, J.; Chelico, L. Retroviral restriction factor APOBEC3G delays the initiation of DNA synthesis by HIV-1 reverse transcriptase. PLoS ONE 2013, 8, e64196. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.N.; Verma, M.; Kim, E.Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [Green Version]
- Li, X.Y.; Guo, F.; Zhang, L.; Kleiman, L.; Cen, S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J. Biol. Chem. 2007, 282, 32065–32074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, R.K.; Koning, F.A.; Bishop, K.N.; Malim, M.H. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J. Biol. Chem. 2007, 282, 2587–2595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbisa, J.L.; Barr, R.; Thomas, J.A.; Vandegraaff, N.; Dorweiler, I.J.; Svarovskaia, E.S.; Brown, W.L.; Mansky, L.M.; Gorelick, R.J.; Harris, R.S.; et al. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J. Virol. 2007, 81, 7099–7110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mbisa, J.L.; Bu, W.; Pathak, V.K. APOBEC3F and APOBEC3G inhibit HIV-1 DNA integration by different mechanisms. J. Virol. 2010, 84, 5250–5259. [Google Scholar] [CrossRef] [Green Version]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep sequencing of HIV-1 reverse transcripts reveals the multifaceted antiviral functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef]
- Luo, K.; Wang, T.; Liu, B.; Tian, C.; Xiao, Z.; Kappes, J.; Yu, X.F. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J. Virol. 2007, 81, 7238–7248. [Google Scholar] [CrossRef] [Green Version]
- Dang, Y.; Siew, L.M.; Wang, X.; Han, Y.; Lampen, R.; Zheng, Y.H. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J. Biol. Chem. 2008, 283, 11606–11614. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.B.; Komor, A.C.; Levy, J.M.; Packer, M.S.; Zhao, K.T.; Liu, D.R. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol. 2017, 35, 371–376. [Google Scholar] [CrossRef]
- McGrath, E.; Shin, H.; Zhang, L.; Phue, J.N.; Wu, W.W.; Shen, R.F.; Jang, Y.Y.; Revollo, J.; Ye, Z. Targeting specificity of APOBEC-based cytosine base editor in human iPSCs determined by whole genome sequencing. Nat. Commun. 2019, 10, 5353. [Google Scholar] [CrossRef]
- Zong, Y.; Song, Q.; Li, C.; Jin, S.; Zhang, D.; Wang, Y.; Qiu, J.L.; Gao, C. Efficient C-to-T base editing in plants using a fusion of nCas9 and human APOBEC3A. Nat. Biotechnol. 2018, 36, 950–953. [Google Scholar] [CrossRef]
- Chen, K.M.; Harjes, E.; Gross, P.J.; Fahmy, A.; Lu, Y.; Shindo, K.; Harris, R.S.; Matsuo, H. Structure of the DNA deaminase domain of the HIV-1 restriction factor APOBEC3G. Nature 2008, 452, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, S.; Ode, H.; Iwatani, Y. Structural Features of Antiviral APOBEC3 Proteins are Linked to Their Functional Activities. Front. Microbiol. 2011, 2, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byeon, I.J.; Ahn, J.; Mitra, M.; Byeon, C.H.; Hercik, K.; Hritz, J.; Charlton, L.M.; Levin, J.G.; Gronenborn, A.M. NMR structure of human restriction factor APOBEC3A reveals substrate binding and enzyme specificity. Nat. Commun. 2013, 4, 1890. [Google Scholar] [CrossRef] [PubMed]
- Shandilya, S.M.; Nalam, M.N.; Nalivaika, E.A.; Gross, P.J.; Valesano, J.C.; Shindo, K.; Li, M.; Munson, M.; Royer, W.E.; Harjes, E.; et al. Crystal structure of the APOBEC3G catalytic domain reveals potential oligomerization interfaces. Structure 2010, 18, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, M.F.; Shandilya, S.M.; Albin, J.S.; Kouno, T.; Anderson, B.D.; McDougle, R.M.; Carpenter, M.A.; Rathore, A.; Evans, L.; Davis, A.N.; et al. Crystal structure of the DNA cytosine deaminase APOBEC3F: The catalytically active and HIV-1 Vif-binding domain. Structure 2013, 21, 1042–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitamura, S.; Ode, H.; Nakashima, M.; Imahashi, M.; Naganawa, Y.; Kurosawa, T.; Yokomaku, Y.; Yamane, T.; Watanabe, N.; Suzuki, A.; et al. The APOBEC3C crystal structure and the interface for HIV-1 Vif binding. Nat. Struct. Mol. Biol. 2012, 19, 1005–1010. [Google Scholar] [CrossRef]
- Bohn, J.A.; Thummar, K.; York, A.; Raymond, A.; Brown, W.C.; Bieniasz, P.D.; Hatziioannou, T.; Smith, J.L. APOBEC3H structure reveals an unusual mechanism of interaction with duplex RNA. Nat. Commun. 2017, 8, 1021. [Google Scholar] [CrossRef] [Green Version]
- Shaban, N.M.; Shi, K.; Lauer, K.V.; Carpenter, M.A.; Richards, C.M.; Salamango, D.; Wang, J.; Lopresti, M.W.; Banerjee, S.; Levin-Klein, R.; et al. The Antiviral and Cancer Genomic DNA Deaminase APOBEC3H Is Regulated by an RNA-Mediated Dimerization Mechanism. Mol. Cell. 2018, 69, 75–86.e9. [Google Scholar] [CrossRef] [Green Version]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, O.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Kouno, T.; Silvas, T.V.; Hilbert, B.J.; Shandilya, S.M.D.; Bohn, M.F.; Kelch, B.A.; Royer, W.E.; Somasundaran, M.; Kurt Yilmaz, N.; Matsuo, H.; et al. Crystal structure of APOBEC3A bound to single-stranded DNA reveals structural basis for cytidine deamination and specificity. Nat. Commun. 2017, 8, 15024. [Google Scholar] [CrossRef] [Green Version]
- Byeon, I.J.; Byeon, C.H.; Wu, T.; Mitra, M.; Singer, D.; Levin, J.G.; Gronenborn, A.M. Nuclear Magnetic Resonance Structure of the APOBEC3B Catalytic Domain: Structural Basis for Substrate Binding and DNA Deaminase Activity. Biochemistry 2016, 55, 2944–2959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, A.; Myint, W.; Kanai, T.; Delviks-Frankenberry, K.; Sierra Rodriguez, C.; Pathak, V.K.; Schiffer, C.A.; Matsuo, H. Crystal structure of the catalytic domain of HIV-1 restriction factor APOBEC3G in complex with ssDNA. Nat. Commun. 2018, 9, 2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Li, S.X.; Yang, H.; Chen, X.S. Crystal structures of APOBEC3G N-domain alone and its complex with DNA. Nat. Commun. 2016, 7, 12193. [Google Scholar] [CrossRef]
- Bohn, M.F.; Shandilya, S.M.D.; Silvas, T.V.; Nalivaika, E.A.; Kouno, T.; Kelch, B.A.; Ryder, S.P.; Kurt-Yilmaz, N.; Somasundaran, M.; Schiffer, C.A. The ssDNA Mutator APOBEC3A Is Regulated by Cooperative Dimerization. Structure 2015, 23, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Carpenter, M.A.; Kurahashi, K.; Harris, R.S.; Aihara, H. Crystal Structure of the DNA Deaminase APOBEC3B Catalytic Domain. J. Biol. Chem. 2015, 290, 28120–28130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, K.; Demir, O.; Carpenter, M.A.; Wagner, J.; Kurahashi, K.; Harris, R.S.; Amaro, R.E.; Aihara, H. Conformational Switch Regulates the DNA Cytosine Deaminase Activity of Human APOBEC3B. Sci. Rep. 2017, 7, 17415. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Yang, H.; Arutiunian, V.; Fang, Y.; Besse, G.; Morimoto, C.; Zirkle, B.; Chen, X.S. Structural determinants of APOBEC3B non-catalytic domain for molecular assembly and catalytic regulation. Nucleic Acids Res. 2017, 45, 7540, Erratum in 2017, 45, 7494–7506. [Google Scholar] [CrossRef]
- Shaban, N.M.; Shi, K.; Li, M.; Aihara, H.; Harris, R.S. 1.92 Angstrom Zinc-Free APOBEC3F Catalytic Domain Crystal Structure. J. Mol. Biol. 2016, 428, 2307–2316. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, M.; Ode, H.; Kawamura, T.; Kitamura, S.; Naganawa, Y.; Awazu, H.; Tsuzuki, S.; Matsuoka, K.; Nemoto, M.; Hachiya, A.; et al. Structural Insights into HIV-1 Vif-APOBEC3F Interaction. J. Virol. 2016, 90, 1034–1047. [Google Scholar] [CrossRef] [Green Version]
- Siu, K.K.; Sultana, A.; Azimi, F.C.; Lee, J.E. Structural determinants of HIV-1 Vif susceptibility and DNA binding in APOBEC3F. Nat. Commun. 2013, 4, 2593. [Google Scholar] [CrossRef]
- Fang, Y.; Xiao, X.; Li, S.X.; Wolfe, A.; Chen, X.S. Molecular Interactions of a DNA Modifying Enzyme APOBEC3F Catalytic Domain with a Single-Stranded DNA. J. Mol. Biol. 2018, 430, 87–101. [Google Scholar] [CrossRef]
- Cheng, C.; Zhang, T.; Wang, C.; Lan, W.; Ding, J.; Cao, C. Crystal Structure of Cytidine Deaminase Human APOBEC3F Chimeric Catalytic Domain in Complex with DNA. Chin. J. Chem. 2018, 36, 1241–1248. [Google Scholar] [CrossRef]
- Hu, Y.; Desimmie, B.A.; Nguyen, H.C.; Ziegler, S.J.; Cheng, T.C.; Chen, J.; Wang, J.; Wang, H.; Zhang, K.; Pathak, V.K.; et al. Structural basis of antagonism of human APOBEC3F by HIV-1 Vif. Nat. Struct. Mol. Biol. 2019, 26, 1176–1183. [Google Scholar] [CrossRef]
- Yan, X.; Lan, W.; Wang, C.; Cao, C. Structural Investigations on the Interactions between Cytidine Deaminase Human APOBEC3G and DNA. Chem. Asian J. 2019, 14, 2235–2241. [Google Scholar] [CrossRef]
- Ziegler, S.J.; Liu, C.; Landau, M.; Buzovetsky, O.; Desimmie, B.A.; Zhao, Q.; Sasaki, T.; Burdick, R.C.; Pathak, V.K.; Anderson, K.S.; et al. Insights into DNA substrate selection by APOBEC3G from structural, biochemical, and functional studies. PLoS ONE 2018, 13, e0195048. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Zhang, T.; Xu, Z.; Liu, S.; Zhao, B.; Lan, W.; Wang, C.; Ding, J.; Cao, C. Crystal structure of DNA cytidine deaminase ABOBEC3G catalytic deamination domain suggests a binding mode of full-length enzyme to single-stranded DNA. J. Biol. Chem. 2015, 290, 4010–4021. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Shandilya, S.M.; Carpenter, M.A.; Rathore, A.; Brown, W.L.; Perkins, A.L.; Harki, D.A.; Solberg, J.; Hook, D.J.; Pandey, K.K.; et al. First-in-class small molecule inhibitors of the single-strand DNA cytosine deaminase APOBEC3G. ACS Chem. Biol. 2012, 7, 506–517. [Google Scholar] [CrossRef] [Green Version]
- Harjes, E.; Gross, P.J.; Chen, K.M.; Lu, Y.; Shindo, K.; Nowarski, R.; Gross, J.D.; Kotler, M.; Harris, R.S.; Matsuo, H. An extended structure of the APOBEC3G catalytic domain suggests a unique holoenzyme model. J. Mol. Biol. 2009, 389, 819–832. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, A.; Nagata, T.; Matsugami, A.; Habu, Y.; Sugiyama, R.; Hayashi, F.; Kobayashi, N.; Yokoyama, S.; Takaku, H.; Katahira, M. Structure, interaction and real-time monitoring of the enzymatic reaction of wild-type APOBEC3G. EMBO J. 2009, 28, 440–451. [Google Scholar] [CrossRef] [Green Version]
- Kouno, T.; Luengas, E.M.; Shigematsu, M.; Shandilya, S.M.; Zhang, J.; Chen, L.; Hara, M.; Schiffer, C.A.; Harris, R.S.; Matsuo, H. Structure of the Vif-binding domain of the antiviral enzyme APOBEC3G. Nat. Struct. Mol. Biol. 2015, 22, 485–491. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Ito, F.; Wolfe, A.D.; Li, S.; Mohammadzadeh, N.; Love, R.P.; Yan, M.; Zirkle, B.; Gaba, A.; Chelico, L.; et al. Understanding the structural basis of HIV-1 restriction by the full length double-domain APOBEC3G. Nat. Commun. 2020, 11, 632. [Google Scholar] [CrossRef] [Green Version]
- Ito, F.; Yang, H.; Xiao, X.; Li, S.X.; Wolfe, A.; Zirkle, B.; Arutiunian, V.; Chen, X.S. Understanding the Structure, Multimerization, Subcellular Localization and mC Selectivity of a Genomic Mutator and Anti-HIV Factor APOBEC3H. Sci. Rep. 2018, 8, 3763. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, T.; Nagae, T.; Ode, H.; Awazu, H.; Kurosawa, T.; Hamano, A.; Matsuoka, K.; Hachiya, A.; Imahashi, M.; Yokomaku, Y.; et al. Structural basis of chimpanzee APOBEC3H dimerization stabilized by double-stranded RNA. Nucleic Acids Res. 2018, 46, 10368–10379. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Dong, L.; Qiu, X.; Wang, Y.; Zhang, B.; Liu, H.; Yu, Y.; Zang, Y.; Yang, M.; Huang, Z. Structural basis for hijacking CBF-beta and CUL5 E3 ligase complex by HIV-1 Vif. Nature 2014, 505, 229–233. [Google Scholar] [CrossRef]
- Lu, Z.; Bergeron, J.R.; Atkinson, R.A.; Schaller, T.; Veselkov, D.A.; Oregioni, A.; Yang, Y.; Matthews, S.J.; Malim, M.H.; Sanderson, M.R. Insight into the HIV-1 Vif SOCS-box-ElonginBC interaction. Open Biol. 2013, 3, 130100. [Google Scholar] [CrossRef] [Green Version]
- Stanley, B.J.; Ehrlich, E.S.; Short, L.; Yu, Y.; Xiao, Z.; Yu, X.F.; Xiong, Y. Structural insight into the human immunodeficiency virus Vif SOCS box and its role in human E3 ubiquitin ligase assembly. J. Virol. 2008, 82, 8656–8663. [Google Scholar] [CrossRef] [Green Version]
- Binning, J.M.; Chesarino, N.M.; Emerman, M.; Gross, J.D. Structural Basis for a Species-Specific Determinant of an SIV Vif Protein toward Hominid APOBEC3G Antagonism. Cell Host Microbe 2019, 26, 739–747.e4. [Google Scholar] [CrossRef]
- Prochnow, C.; Bransteitter, R.; Klein, M.G.; Goodman, M.F.; Chen, X.S. The APOBEC-2 crystal structure and functional implications for the deaminase AID. Nature 2007, 445, 447–451. [Google Scholar] [CrossRef]
- Aydin, H.; Taylor, M.W.; Lee, J.E. Structure-guided analysis of the human APOBEC3-HIV restrictome. Structure 2014, 22, 668–684. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Bogerd, H.P.; Doehle, B.P.; Wiegand, H.L.; Cullen, B.R. A single amino acid difference in the host APOBEC3G protein controls the primate species specificity of HIV type 1 virion infectivity factor. Proc. Natl. Acad. Sci. USA 2004, 101, 3770–3774. [Google Scholar] [CrossRef] [Green Version]
- Mangeat, B.; Turelli, P.; Liao, S.; Trono, D. A single amino acid determinant governs the species-specific sensitivity of APOBEC3G to Vif action. J. Biol. Chem. 2004, 279, 14481–14483. [Google Scholar] [CrossRef] [Green Version]
- Schrofelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Svarovskaia, E.S.; Barr, R.; Zhang, Y.; Khan, M.A.; Strebel, K.; Pathak, V.K. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. USA 2004, 101, 5652–5657. [Google Scholar] [CrossRef] [Green Version]
- Huthoff, H.; Malim, M.H. Identification of amino acid residues in APOBEC3G required for regulation by human immunodeficiency virus type 1 Vif and Virion encapsidation. J. Virol. 2007, 81, 3807–3815. [Google Scholar] [CrossRef] [Green Version]
- Letko, M.; Booiman, T.; Kootstra, N.; Simon, V.; Ooms, M. Identification of the HIV-1 Vif and Human APOBEC3G Protein Interface. Cell Rep. 2015, 13, 1789–1799. [Google Scholar] [CrossRef] [Green Version]
- Compton, A.A.; Hirsch, V.M.; Emerman, M. The host restriction factor APOBEC3G and retroviral Vif protein coevolve due to ongoing genetic conflict. Cell Host Microbe 2012, 11, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Sawyer, S.L.; Emerman, M.; Malik, H.S. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004, 2, E275. [Google Scholar] [CrossRef]
- Hache, G.; Liddament, M.T.; Harris, R.S. The retroviral hypermutation specificity of APOBEC3F and APOBEC3G is governed by the C-terminal DNA cytosine deaminase domain. J. Biol. Chem. 2005, 280, 10920–10924. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.A.; Smith, J.; Barr, R.; Bhattacharyya, D.; Pathak, V.K. Distinct domains within APOBEC3G and APOBEC3F interact with separate regions of human immunodeficiency virus type 1 Vif. J. Virol. 2009, 83, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.L.; Pathak, V.K. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J. Virol. 2010, 84, 12599–12608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albin, J.S.; LaRue, R.S.; Weaver, J.A.; Brown, W.L.; Shindo, K.; Harjes, E.; Matsuo, H.; Harris, R.S. A single amino acid in human APOBEC3F alters susceptibility to HIV-1 Vif. J. Biol. Chem. 2010, 285, 40785–40792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harari, A.; Ooms, M.; Mulder, L.C.; Simon, V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J. Virol. 2009, 83, 295–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OhAinle, M.; Kerns, J.A.; Li, M.M.; Malik, H.S.; Emerman, M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe 2008, 4, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Sarkis, P.T.; Wang, T.; Tian, C.; Yu, X.F. Sole copy of Z2-type human cytidine deaminase APOBEC3H has inhibitory activity against retrotransposons and HIV-1. FASEB J. 2009, 23, 279–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Abudu, A.; Son, S.; Dang, Y.; Venta, P.J.; Zheng, Y.H. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type 1 activity. J. Virol. 2011, 85, 3142–3152. [Google Scholar] [CrossRef] [Green Version]
- Li, M.M.; Wu, L.I.; Emerman, M. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J. Virol. 2010, 84, 88–95. [Google Scholar] [CrossRef] [Green Version]
- Zhen, A.; Wang, T.; Zhao, K.; Xiong, Y.; Yu, X.F. A single amino acid difference in human APOBEC3H variants determines HIV-1 Vif sensitivity. J. Virol. 2010, 84, 1902–1911. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, M.; Tsuzuki, S.; Awazu, H.; Hamano, A.; Okada, A.; Ode, H.; Maejima, M.; Hachiya, A.; Yokomaku, Y.; Watanabe, N.; et al. Mapping Region of Human Restriction Factor APOBEC3H Critical for Interaction with HIV-1 Vif. J. Mol. Biol. 2017, 429, 1262–1276. [Google Scholar] [CrossRef]
- Ooms, M.; Letko, M.; Simon, V. The Structural Interface between HIV-1 Vif and Human APOBEC3H. J. Virol. 2017, 91, e02289. [Google Scholar] [CrossRef] [Green Version]
- Conticello, S.G.; Harris, R.S.; Neuberger, M.S. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr. Biol. 2003, 13, 2009–2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Gonda, M.A. Conservation of amino-acid sequence motifs in lentivirus Vif proteins. Virus Genes 1992, 6, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Okumura, F.; Matsuzaki, M.; Nakatsukasa, K.; Kamura, T. The Role of Elongin BC-Containing Ubiquitin Ligases. Front. Oncol. 2012, 2, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, K.; Xiao, Z.; Ehrlich, E.; Yu, Y.; Liu, B.; Zheng, S.; Yu, X.F. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc. Natl. Acad. Sci. USA 2005, 102, 11444–11449. [Google Scholar] [CrossRef] [Green Version]
- Mehle, A.; Thomas, E.R.; Rajendran, K.S.; Gabuzda, D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J. Biol. Chem. 2006, 281, 17259–17265. [Google Scholar] [CrossRef] [Green Version]
- Paul, I.; Cui, J.; Maynard, E.L. Zinc binding to the HCCH motif of HIV-1 virion infectivity factor induces a conformational change that mediates protein-protein interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 18475–18480. [Google Scholar] [CrossRef] [Green Version]
- Evans, S.L.; Schon, A.; Gao, Q.; Han, X.; Zhou, X.; Freire, E.; Yu, X.F. HIV-1 Vif N-terminal motif is required for recruitment of Cul5 to suppress APOBEC3. Retrovirology 2014, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Gu, Q.; Zhang, Z.; Gertzen, C.G.W.; Haussinger, D.; Gohlke, H.; Munk, C. Identification of a Conserved Interface of Human Immunodeficiency Virus Type 1 and Feline Immunodeficiency Virus Vifs with Cullin 5. J. Virol. 2018, 92, e01697. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.A.; Pathak, V.K. Identification of two distinct human immunodeficiency virus type 1 Vif determinants critical for interactions with human APOBEC3G and APOBEC3F. J. Virol. 2007, 81, 8201–8210. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Takeuchi, J.S.; Misawa, N.; Izumi, T.; Kobayashi, T.; Kimura, Y.; Iwami, S.; Takaori-Kondo, A.; Hu, W.S.; Aihara, K.; et al. APOBEC3D and APOBEC3F potently promote HIV-1 diversification and evolution in humanized mouse model. PLoS Pathog. 2014, 10, e1004453. [Google Scholar] [CrossRef] [Green Version]
- Dang, Y.; Wang, X.; York, I.A.; Zheng, Y.H. Identification of a critical T(Q/D/E)x5ADx2(I/L) motif from primate lentivirus Vif proteins that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 2010, 84, 8561–8570. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; He, Z.; Wang, T.; Xu, R.; Yu, X.F. A patch of positively charged amino acids surrounding the human immunodeficiency virus type 1 Vif SLVx4Yx9Y motif influences its interaction with APOBEC3G. J. Virol. 2009, 83, 8674–8682. [Google Scholar] [CrossRef] [Green Version]
- Dang, Y.; Wang, X.; Zhou, T.; York, I.A.; Zheng, Y.H. Identification of a novel WxSLVK motif in the N terminus of human immunodeficiency virus and simian immunodeficiency virus Vif that is critical for APOBEC3G and APOBEC3F neutralization. J. Virol. 2009, 83, 8544–8552. [Google Scholar] [CrossRef] [Green Version]
- Ooms, M.; Letko, M.; Binka, M.; Simon, V. The resistance of human APOBEC3H to HIV-1 NL4-3 molecular clone is determined by a single amino acid in Vif. PLoS ONE 2013, 8, e57744. [Google Scholar] [CrossRef]
- Binka, M.; Ooms, M.; Steward, M.; Simon, V. The activity spectrum of Vif from multiple HIV-1 subtypes against APOBEC3G, APOBEC3F, and APOBEC3H. J. Virol. 2012, 86, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Jager, S.; Kim, D.Y.; Hultquist, J.F.; Shindo, K.; LaRue, R.S.; Kwon, E.; Li, M.; Anderson, B.D.; Yen, L.; Stanley, D.; et al. Vif hijacks CBF-beta to degrade APOBEC3G and promote HIV-1 infection. Nature 2011, 481, 371–375. [Google Scholar] [CrossRef]
- Zhang, W.; Du, J.; Evans, S.L.; Yu, Y.; Yu, X.F. T-cell differentiation factor CBF-beta regulates HIV-1 Vif-mediated evasion of host restriction. Nature 2011, 481, 376–379. [Google Scholar] [CrossRef]
- Wang, Q.; Stacy, T.; Miller, J.D.; Lewis, A.F.; Gu, T.L.; Huang, X.; Bushweller, J.H.; Bories, J.C.; Alt, F.W.; Ryan, G.; et al. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 1996, 87, 697–708. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Shigesada, K.; Ito, K.; Wee, H.J.; Yokomizo, T.; Ito, Y. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. EMBO J. 2001, 20, 723–733. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.; Littman, D.R.; Taniuchi, I. RUNX proteins in transcription factor networks that regulate T-cell lineage choice. Nat. Rev. Immunol. 2009, 9, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Taniuchi, I.; Osato, M.; Egawa, T.; Sunshine, M.J.; Bae, S.C.; Komori, T.; Ito, Y.; Littman, D.R. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell 2002, 111, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.Y.; Kwon, E.; Hartley, P.D.; Crosby, D.C.; Mann, S.; Krogan, N.J.; Gross, J.D. CBFbeta stabilizes HIV Vif to counteract APOBEC3 at the expense of RUNX1 target gene expression. Mol. Cell. 2013, 49, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Shindo, K.; Nagata, K.; Yoshinaga, N.; Shirakawa, K.; Kobayashi, M.; Takaori-Kondo, A. Core Binding Factor beta Protects HIV, Type 1 Accessory Protein Viral Infectivity Factor from MDM2-mediated Degradation. J. Biol. Chem. 2016, 291, 24892–24899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyagi, E.; Kao, S.; Yedavalli, V.; Strebel, K. CBFbeta enhances de novo protein biosynthesis of its binding partners HIV-1 Vif and RUNX1 and potentiates the Vif-induced degradation of APOBEC3G. J. Virol. 2014, 88, 4839–4852. [Google Scholar] [CrossRef] [Green Version]
- Anderson, B.D.; Harris, R.S. Transcriptional regulation of APOBEC3 antiviral immunity through the CBF-beta/RUNX axis. Sci. Adv. 2015, 1, e1500296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fribourgh, J.L.; Nguyen, H.C.; Wolfe, L.S.; Dewitt, D.C.; Zhang, W.; Yu, X.F.; Rhoades, E.; Xiong, Y. Core binding factor beta plays a critical role by facilitating the assembly of the Vif-cullin 5 E3 ubiquitin ligase. J. Virol. 2014, 88, 3309–3319. [Google Scholar] [CrossRef] [Green Version]
- Matsui, Y.; Shindo, K.; Nagata, K.; Io, K.; Tada, K.; Iwai, F.; Kobayashi, M.; Kadowaki, N.; Harris, R.S.; Takaori-Kondo, A. Defining HIV-1 Vif residues that interact with CBFbeta by site-directed mutagenesis. Virology 2014, 449, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Han, X.; Zhao, K.; Du, J.; Evans, S.L.; Wang, H.; Li, P.; Zheng, W.; Rui, Y.; Kang, J.; et al. Dispersed and conserved hydrophobic residues of HIV-1 Vif are essential for CBFbeta recruitment and A3G suppression. J. Virol. 2014, 88, 2555–2563. [Google Scholar] [CrossRef] [Green Version]
- Desimmie, B.A.; Smith, J.L.; Matsuo, H.; Hu, W.S.; Pathak, V.K. Identification of a tripartite interaction between the N-terminus of HIV-1 Vif and CBFbeta that is critical for Vif function. Retrovirology 2017, 14, 19. [Google Scholar] [CrossRef] [Green Version]
- Hultquist, J.F.; McDougle, R.M.; Anderson, B.D.; Harris, R.S. HIV type 1 viral infectivity factor and the RUNX transcription factors interact with core binding factor beta on genetically distinct surfaces. AIDS Res. Hum. Retroviruses 2012, 28, 1543–1551. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Kwak, M.J.; Ku, B.; Suh, H.Y.; Joo, K.; Lee, J.; Jung, J.U.; Oh, B.H. Structural basis of intersubunit recognition in elongin BC-cullin 5-SOCS box ubiquitin-protein ligase complexes. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, Y.; Davis, R.W.; York, I.A.; Zheng, Y.H. Identification of 81LGxGxxIxW89 and 171EDRW174 domains from human immunodeficiency virus type 1 Vif that regulate APOBEC3G and APOBEC3F neutralizing activity. J. Virol. 2010, 84, 5741–5750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Liu, B.; Liu, X.; Li, Z.; Yu, X.F.; Zhang, W. Identification of HIV-1 Vif regions required for CBF-beta interaction and APOBEC3 suppression. PLoS ONE 2014, 9, e95738. [Google Scholar] [CrossRef]
- Chelico, L.; Pham, P.; Calabrese, P.; Goodman, M.F. APOBEC3G DNA deaminase acts processively 3′ --> 5′ on single-stranded DNA. Nat. Struct. Mol. Biol. 2006, 13, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Chelico, L.; Pham, P.; Goodman, M.F. Mechanisms of APOBEC3G-catalyzed processive deamination of deoxycytidine on single-stranded DNA. Nat. Struct. Mol. Biol. 2009, 16, 454–456. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Takeuchi, H.; Strebel, K.; Levin, J.G. Biochemical activities of highly purified, catalytically active human APOBEC3G: Correlation with antiviral effect. J. Virol. 2006, 80, 5992–6002. [Google Scholar] [CrossRef] [Green Version]
- Salter, J.D.; Smith, H.C. Modeling the Embrace of a Mutator: APOBEC Selection of Nucleic Acid Ligands. Trends Biochem. Sci. 2018, 43, 606–622. [Google Scholar] [CrossRef] [Green Version]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, R.S.; Lovsin, N.; Tanuri, A.; Peterlin, B.M. Vpr.A3A chimera inhibits HIV replication. J. Biol. Chem. 2008, 283, 2518–2525. [Google Scholar] [CrossRef] [Green Version]
- Doehle, B.P.; Schafer, A.; Cullen, B.R. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology 2005, 339, 281–288. [Google Scholar] [CrossRef] [Green Version]
- Langlois, M.A.; Beale, R.C.; Conticello, S.G.; Neuberger, M.S. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005, 33, 1913–1923. [Google Scholar] [CrossRef] [Green Version]
- Liddament, M.T.; Brown, W.L.; Schumacher, A.J.; Harris, R.S. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr. Biol. 2004, 14, 1385–1391. [Google Scholar] [CrossRef] [Green Version]
- Salter, J.D.; Bennett, R.P.; Smith, H.C. The APOBEC Protein Family: United by Structure, Divergent in Function. Trends Biochem. Sci. 2016, 41, 578–594. [Google Scholar] [CrossRef] [Green Version]
- Wedekind, J.E.; Gillilan, R.; Janda, A.; Krucinska, J.; Salter, J.D.; Bennett, R.P.; Raina, J.; Smith, H.C. Nanostructures of APOBEC3G support a hierarchical assembly model of high molecular mass ribonucleoprotein particles from dimeric subunits. J. Biol. Chem. 2006, 281, 38122–38126. [Google Scholar] [CrossRef] [Green Version]
- Qiao, Q.; Wang, L.; Meng, F.L.; Hwang, J.K.; Alt, F.W.; Wu, H. AID Recognizes Structured DNA for Class Switch Recombination. Mol. Cell. 2017, 67, 361–373.e4. [Google Scholar] [CrossRef]
- Furukawa, A.; Nagata, T.; Matsugami, A.; Habu, Y.; Sugiyama, R.; Hayashi, F.; Kobayashi, N.; Yokoyama, S.; Takaku, H.; Katahira, M. Structure and real-time monitoring of the enzymatic reaction of APOBEC3G which is involved in anti-HIV activity. Nucleic Acids Symp. Ser. (Oxf.) 2009, 53, 87–88. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Silvas, T.V.; Leidner, F.; Nalivaika, E.A.; Matsuo, H.; Kurt Yilmaz, N.; Schiffer, C.A. Structural Analysis of the Active Site and DNA Binding of Human Cytidine Deaminase APOBEC3B. J. Chem. Theory Comput. 2019, 15, 637–647. [Google Scholar] [CrossRef] [Green Version]
- Losey, H.C.; Ruthenburg, A.J.; Verdine, G.L. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 2006, 13, 153–159. [Google Scholar] [CrossRef]
- Polevoda, B.; Joseph, R.; Friedman, A.E.; Bennett, R.P.; Greiner, R.; de Zoysa, T.; Stewart, R.A.; Smith, H.C. DNA mutagenic activity and capacity for HIV-1 restriction of the cytidine deaminase APOBEC3G depend on whether DNA or RNA binds to tyrosine 315. J. Biol. Chem. 2017, 292, 8642–8656. [Google Scholar] [CrossRef] [Green Version]
- Friew, Y.N.; Boyko, V.; Hu, W.S.; Pathak, V.K. Intracellular interactions between APOBEC3G, RNA, and HIV-1 Gag: APOBEC3G multimerization is dependent on its association with RNA. Retrovirology 2009, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Holtz, C.M.; Sadler, H.A.; Mansky, L.M. APOBEC3G cytosine deamination hotspots are defined by both sequence context and single-stranded DNA secondary structure. Nucleic Acids Res. 2013, 41, 6139–6148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beale, R.C.; Petersen-Mahrt, S.K.; Watt, I.N.; Harris, R.S.; Rada, C.; Neuberger, M.S. Comparison of the differential context-dependence of DNA deamination by APOBEC enzymes: Correlation with mutation spectra in vivo. J. Mol. Biol. 2004, 337, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.; Bransteitter, R.; Petruska, J.; Goodman, M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003, 424, 103–107. [Google Scholar] [CrossRef]
- Silvas, T.V.; Hou, S.; Myint, W.; Nalivaika, E.; Somasundaran, M.; Kelch, B.A.; Matsuo, H.; Kurt Yilmaz, N.; Schiffer, C.A. Substrate sequence selectivity of APOBEC3A implicates intra-DNA interactions. Sci. Rep. 2018, 8, 7511. [Google Scholar] [CrossRef] [Green Version]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Diaz, M. Cutting edge: DGYW/WRCH is a better predictor of mutability at G:C bases in Ig hypermutation than the widely accepted RGYW/WRCY motif and probably reflects a two-step activation-induced cytidine deaminase-triggered process. J. Immunol. 2004, 172, 3382–3384. [Google Scholar] [CrossRef] [Green Version]
- Ara, A.; Love, R.P.; Follack, T.B.; Ahmed, K.A.; Adolph, M.B.; Chelico, L. Mechanism of Enhanced HIV Restriction by Virion Coencapsidated Cytidine Deaminases APOBEC3F and APOBEC3G. J. Virol. 2017, 91, e02230. [Google Scholar] [CrossRef] [Green Version]
- Rathore, A.; Carpenter, M.A.; Demir, O.; Ikeda, T.; Li, M.; Shaban, N.M.; Law, E.K.; Anokhin, D.; Brown, W.L.; Amaro, R.E.; et al. The local dinucleotide preference of APOBEC3G can be altered from 5′-CC to 5′-TC by a single amino acid substitution. J. Mol. Biol. 2013, 425, 4442–4454. [Google Scholar] [CrossRef] [Green Version]
- Marx, A.; Galilee, M.; Alian, A. Zinc enhancement of cytidine deaminase activity highlights a potential allosteric role of loop-3 in regulating APOBEC3 enzymes. Sci. Rep. 2015, 5, 18191. [Google Scholar] [CrossRef]
- Kohli, R.M.; Abrams, S.R.; Gajula, K.S.; Maul, R.W.; Gearhart, P.J.; Stivers, J.T. A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. J. Biol. Chem. 2009, 284, 22898–22904. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, M.A.; Rajagurubandara, E.; Wijesinghe, P.; Bhagwat, A.S. Determinants of sequence-specificity within human AID and APOBEC3G. DNA Repair (Amst.) 2010, 9, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Chen, G.; Niewiadomska, A.M.; Xu, R.; Yu, X.F. Distinct determinants in HIV-1 Vif and human APOBEC3 proteins are required for the suppression of diverse host anti-viral proteins. PLoS ONE 2008, 3, e3963. [Google Scholar] [CrossRef]
- Chen, Q.; Xiao, X.; Wolfe, A.; Chen, X.S. The in vitro Biochemical Characterization of an HIV-1 Restriction Factor APOBEC3F: Importance of Loop 7 on Both CD1 and CD2 for DNA Binding and Deamination. J. Mol. Biol. 2016, 428, 2661–2670. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A3 Protein and Vif Structures | PDB 1 ID | Resolution (Å) 2 | Modeled DNA or RNA Substrate Sequence | Vif-Binding Interface | Ref. No. |
|---|---|---|---|---|---|
| A3A | 2M65 | NMR | [103] | ||
| 4XXO | 2.84 | [114] | |||
| 5KEG | 2.2 | 5′-TTCTT-3′ | [110] | ||
| 5SWW | 3.15 | 5′-ATCGGG-3′ | [109] | ||
| A3BCTD | 5TD5 3 | 1.72 | 5′-TTCAT-3′ | [109] | |
| 2NBQ | NMR | [111] | |||
| 5CQD | 2.08 | [115] | |||
| 5CQH | 1.73 | dCMP | [115] | ||
| 5CQI | 1.68 | [115] | |||
| 5CQK | 1.88 | [115] | |||
| 5SXG | 1.93 | [116] | |||
| 5SXH | 1.78 | [116] | |||
| A3BNTD | 5TKM | 1.9 | [117] | ||
| A3C | 3VM8 | 3.0 | yes | [106] | |
| 3VOW | 2.15 | yes | [106] | ||
| A3FCTD | 5HX5 | 2.33 | [118] | ||
| 3WUS | 2.54 | yes | [119] | ||
| 4J4J | 3.1 | yes | [120] | ||
| 5HX4 | 1.92 | [118] | |||
| 5W2M | 3.7 | 5′-TTTTTTTTTT-3′ | [121] | ||
| 5ZVA | 2.3 | 5′-ATTTTCAACT-3′ | [122] | ||
| 5ZVB | 2.0 | 5′-ATTTTCAAT-3′ | [122] | ||
| 4IUO | 2.75 | yes | [105] | ||
| A3FCTD-Vif-CBFβ | 6NIL | 3.9, cryoEM | yes | [123] | |
| A3GCTD | 6K3J | NMR | 5′-ATTCUUIAATT-3′ | [124] | |
| 6K3K | NMR | 5′-ATTCUCIAATT-3′ | [124] | ||
| 6BUX | 1.86 | 5′-AATCCCAAA-3′ | [112] | ||
| 6BWY | 2.9 | 5′-AGGTTAC-3′ | [125] | ||
| 4ROV | 1.8 | [126] | |||
| 4ROW | 1.7 | [126] | |||
| 3V4J | 2.04 | [127] | |||
| 3V4K | 1.38 | [127] | |||
| 3IR2 | 2.25 | [104] | |||
| 3IQS | 2.3 | [18] | |||
| 2KEM | NMR | [128] | |||
| 3E1U | 2.3 | [18] | |||
| 2JYW | NMR | [101] | |||
| 2KBO | NMR | [129] | |||
| A3GNTD | 2MZZ | NMR | [130] | ||
| rhA3GNTD 4 | 5K83 | 2.39 | 5′-TTTTTTTTTT-3′ | [113] | |
| 5K81 | 2.0 | [113] | |||
| 5K82 | 2.93 | [113] | |||
| rhA3G | 6P40 | 2.47 | [131] | ||
| full length | 6P3Z | 2.8 | [131] | ||
| chimera 5 | 6P3X | 2.4 | [131] | ||
| 6P3Y | 2.5 | [131] | |||
| A3H | 5W45 | 2.49 | [132] | ||
| 6B0B | 3.28 | 7bp duplex RNA with 1 nt overhang: 5′-UAAAAAAA-3′ + 5′-UUUUUUUUU-3′ | [108] | ||
| 6BBO | 3.43 | 7-bp duplex RNA with 1 nt overhang: 5′-UAAAAAAA-3′ + 5′-UUUUUUUU-3′ | [108] | ||
| pgtA3H 6 | 5W3V | 2.24 | 7-bp duplex RNA with 2 nt overhang: 5′-AACCCGGGGA-3′ + 5′-AACCCCGGGC-3′ | [107] | |
| cpzA3H 7 | 5Z98 | 2.2 | 9-bp duplex RNA with 1 nt overhang: 5′-CUGCCGGGUA-3′ + 5′-AUACCCGGCA-3′ | [133] | |
| Vif-CBFβ-Cul5-ElOB-ElOC | 4N9F | 3.3 | [134] | ||
| Vif SOCS-box and ElOBC | 2MA9 | NMR | [135] | ||
| Vif BC-box with ElOB | 3DCG | 2.4 | [136] | ||
| VifSIVRCM-CBFβ-ElOB-ElOC 8 | 6P59 | 2.9 | [137] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delviks-Frankenberry, K.A.; Desimmie, B.A.; Pathak, V.K. Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses 2020, 12, 587. https://doi.org/10.3390/v12060587
Delviks-Frankenberry KA, Desimmie BA, Pathak VK. Structural Insights into APOBEC3-Mediated Lentiviral Restriction. Viruses. 2020; 12(6):587. https://doi.org/10.3390/v12060587
Chicago/Turabian StyleDelviks-Frankenberry, Krista A., Belete A. Desimmie, and Vinay K. Pathak. 2020. "Structural Insights into APOBEC3-Mediated Lentiviral Restriction" Viruses 12, no. 6: 587. https://doi.org/10.3390/v12060587