FV Vectors as Alternative Gene Vehicles for Gene Transfer in HSCs

Abstract
:1. Introduction
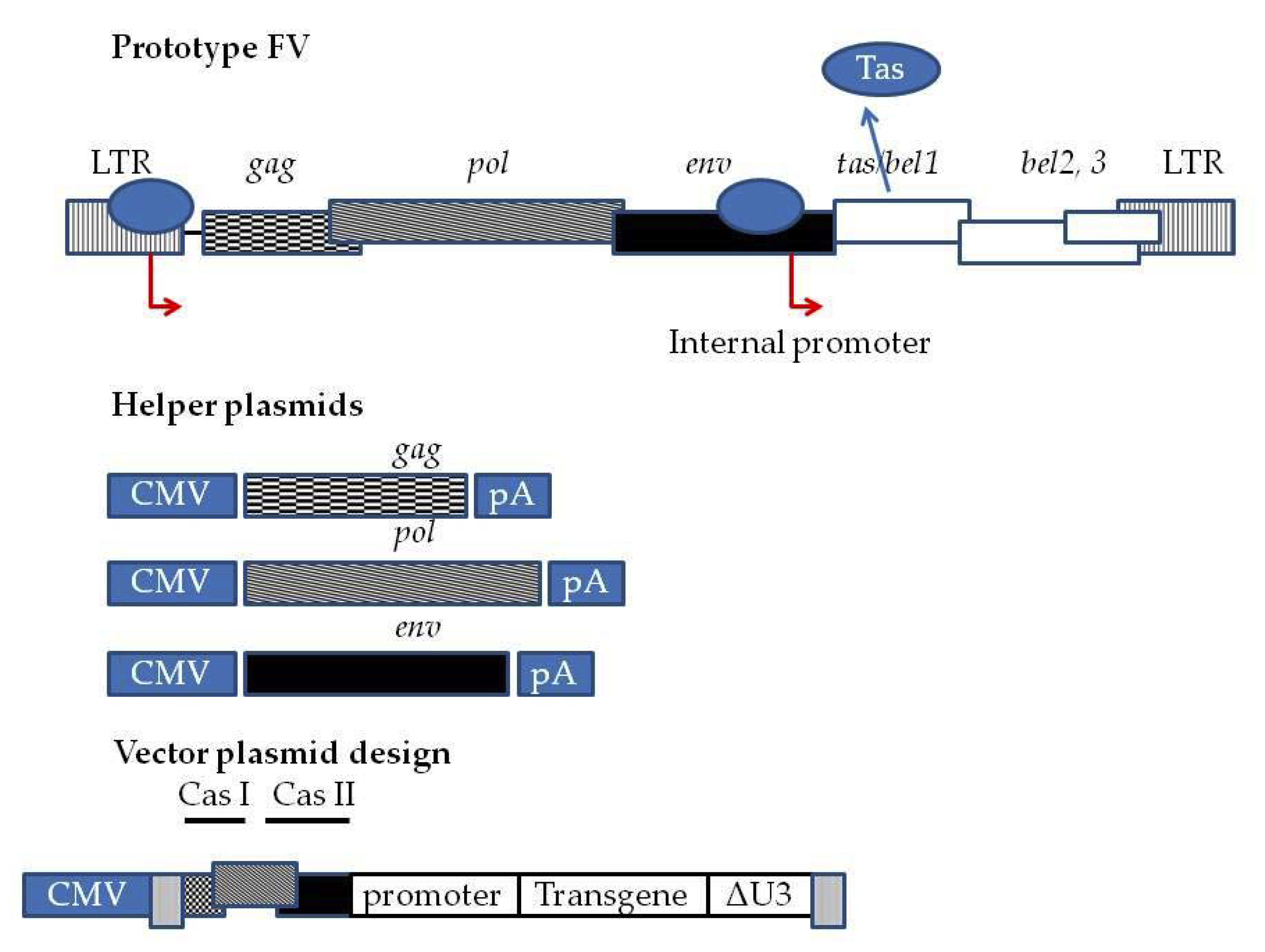
2. Features of FV Vectors for HSC Gene Delivery
3. Gene Marking Studies in Small Animals
4. Therapeutic Gene Transfer in Murine Preclinical Models
5. Large Animal Preclinical Models
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, D. The Bone Marrow Microenvironment for Hematopoietic Stem Cells. Adv. Exp. Med. Biol. 2017, 1041, 5–18. [Google Scholar] [PubMed]
- Heimfeld, S. Bone marrow transplantation: How important is CD34 cell dose in HLA-identical stem cell transplantation? Leukemia 2003, 17, 856–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucarelli, G.; Isgro, A.; Sodani, P.; Gaziev, J. Hematopoietic stem cell transplantation in thalassemia and sickle cell anemia. Cold Spring Harb. Perspect. Med. 2012, 2, a011825. [Google Scholar] [CrossRef] [PubMed]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef] [Green Version]
- Papapetrou, E.P.; Ziros, P.G.; Micheva, I.D.; Zoumbos, N.C.; Athanassiadou, A. Gene transfer into human hematopoietic progenitor cells with an episomal vector carrying an S/MAR element. Gene Ther. 2006, 13, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.S.; Bodem, J.; Buseyne, F.; Gessain, A.; Johnson, W.; Kuhn, J.H.; Kuzmak, J.; Lindemann, D.; Linial, M.L.; Lochelt, M.; et al. Corrigendum to “Spumaretroviruses: Updated taxonomy and nomenclature” [Virology 516 (2018) 158-164]. Virology 2019, 528. [Google Scholar] [CrossRef]
- Cavazzana-Calvo, M.; Hacein-Bey, S.; de Saint Basile, G.; Gross, F.; Yvon, E.; Nusbaum, P.; Selz, F.; Hue, C.; Certain, S.; Casanova, J.L.; et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. [Google Scholar] [CrossRef]
- Aiuti, A.; Cattaneo, F.; Galimberti, S.; Benninghoff, U.; Cassani, B.; Callegaro, L.; Scaramuzza, S.; Andolfi, G.; Mirolo, M.; Brigida, I.; et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009, 360, 447–458. [Google Scholar] [CrossRef] [Green Version]
- Hacein-Bey-Abina, S.; Garrigue, A.; Wang, G.P.; Soulier, J.; Lim, A.; Morillon, E.; Clappier, E.; Caccavelli, L.; Delabesse, E.; Beldjord, K.; et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Investig. 2008, 118, 3132–3142. [Google Scholar] [CrossRef]
- Lesbats, P.; Engelman, A.N.; Cherepanov, P. Retroviral DNA Integration. Chem. Rev. 2016, 116, 12730–12757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattoglio, C.; Facchini, G.; Sartori, D.; Antonelli, A.; Miccio, A.; Cassani, B.; Schmidt, M.; von Kalle, C.; Howe, S.; Thrasher, A.J.; et al. Hot spots of retroviral integration in human CD34+ hematopoietic cells. Blood 2007, 110, 1770–1778. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Miller, D.G.; Jacobs, M.A.; Allen, J.M.; Kiem, H.P.; Kaul, R.; Russell, D.W. Foamy virus vector integration sites in normal human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 1498–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowrouzi, A.; Dittrich, M.; Klanke, C.; Heinkelein, M.; Rammling, M.; Dandekar, T.; von Kalle, C.; Rethwilm, A. Genome-wide mapping of foamy virus vector integrations into a human cell line. J. Gen. Virol. 2006, 87 Pt 5, 1339–1347. [Google Scholar] [CrossRef]
- Lesbats, P.; Serrao, E.; Maskell, D.P.; Pye, V.E.; O’Reilly, N.; Lindemann, D.; Engelman, A.N.; Cherepanov, P. Structural basis for spumavirus GAG tethering to chromatin. Proc. Natl. Acad. Sci. USA 2017, 114, 5509–5514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Challita, P.M.; Kohn, D.B. Lack of expression from a retroviral vector after transduction of murine hematopoietic stem cells is associated with methylation in vivo. Proc. Natl. Acad. Sci. USA 1994, 91, 2567–2571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellis, J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum. Gene Ther. 2005, 16, 1241–1246. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Emery, D.W. The cHS4 chromatin insulator reduces gammaretroviral vector silencing by epigenetic modifications of integrated provirus. Gene Ther. 2008, 15, 49–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McInerney, J.M.; Nawrocki, J.R.; Lowrey, C.H. Long-term silencing of retroviral vectors is resistant to reversal by trichostatin A and 5-azacytidine. Gene Ther. 2000, 7, 653–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, H.; Smith, K.A.; Mosier, D.E.; Verma, I.M.; Torbett, B.E. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science 1999, 283, 682–686. [Google Scholar] [CrossRef] [Green Version]
- Vassilopoulos, G.; Trobridge, G.; Josephson, N.C.; Russell, D.W. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood 2001, 98, 604–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, D.; Rethwilm, A. Foamy virus biology and its application for vector development. Viruses 2011, 3, 561–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilopoulos, G.; Rethwilm, A. The usefulness of a perfect parasite. Gene Ther. 2008, 15, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lei, J.; Liu, Y.; Lukic, D.S.; Rathe, A.M.; Bao, Q.; Kehl, T.; Bleiholder, A.; Hechler, T.; Lochelt, M. Feline foamy virus-based vectors: Advantages of an authentic animal model. Viruses 2013, 5, 1702–1718. [Google Scholar] [CrossRef] [PubMed]
- Ledesma-Feliciano, C.; Hagen, S.; Troyer, R.; Zheng, X.; Musselman, E.; Slavkovic Lukic, D.; Franke, A.M.; Maeda, D.; Zielonka, J.; Munk, C.; et al. Replacement of feline foamy virus bet by feline immunodeficiency virus vif yields replicative virus with novel vaccine candidate potential. Retrovirology 2018, 15, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamsfus-Calle, A.; Daniel-Moreno, A.; Urena-Bailen, G.; Raju, J.; Antony, J.S.; Handgretinger, R.; Mezger, M. Hematopoietic stem cell gene therapy: The optimal use of lentivirus and gene editing approaches. Blood Rev. 2019, 100641. [Google Scholar] [CrossRef] [PubMed]
- Maetzig, T.; Galla, M.; Baum, C.; Schambach, A. Gammaretroviral vectors: Biology, technology and application. Viruses 2011, 3, 677–713. [Google Scholar] [CrossRef] [Green Version]
- Trobridge, G.D. Foamy virus vectors for gene transfer. Expert Opin. Biol. Ther. 2009, 9, 1427–1436. [Google Scholar] [CrossRef]
- Miyoshi, H.; Blomer, U.; Takahashi, M.; Gage, F.H.; Verma, I.M. Development of a self-inactivating lentivirus vector. J. Virol. 1998, 72, 8150–8157. [Google Scholar] [CrossRef] [Green Version]
- Thornhill, S.I.; Schambach, A.; Howe, S.J.; Ulaganathan, M.; Grassman, E.; Williams, D.; Schiedlmeier, B.; Sebire, N.J.; Gaspar, H.B.; Kinnon, C.; et al. Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency. Mol. Ther. 2008, 16, 590–598. [Google Scholar] [CrossRef]
- Goodman, M.A.; Arumugam, P.; Pillis, D.M.; Loberg, A.; Nasimuzzaman, M.; Lynn, D.; van der Loo, J.C.M.; Dexheimer, P.J.; Keddache, M.; Bauer, T.R., Jr.; et al. Foamy Virus Vector Carries a Strong Insulator in Its Long Terminal Repeat Which Reduces Its Genotoxic Potential. J. Virol. 2018, 92, e01639-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.L.; Malani, N.; Bushman, F.D. Gammaretroviral integration into nucleosomal target DNA in vivo. J. Virol. 2011, 85, 7393–7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, D.W.; Miller, A.D. Foamy virus vectors. J. Virol. 1996, 70, 217–222. [Google Scholar] [CrossRef] [Green Version]
- Hirata, R.K.; Miller, A.D.; Andrews, R.G.; Russell, D.W. Transduction of hematopoietic cells by foamy virus vectors. Blood 1996, 88, 3654–3661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plochmann, K.; Horn, A.; Gschmack, E.; Armbruster, N.; Krieg, J.; Wiktorowicz, T.; Weber, C.; Stirnnagel, K.; Lindemann, D.; Rethwilm, A.; et al. Heparan sulfate is an attachment factor for foamy virus entry. J. Virol. 2012, 86, 10028–10035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nasimuzzaman, M.; Persons, D.A. Cell Membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol. Ther. 2012, 20, 1158–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Josephson, N.C.; Vassilopoulos, G.; Trobridge, G.D.; Priestley, G.V.; Wood, B.L.; Papayannopoulou, T.; Russell, D.W. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc. Natl. Acad. Sci. USA 2002, 99, 8295–8300. [Google Scholar] [CrossRef] [Green Version]
- Trobridge, G.; Russell, D.W. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J. Virol. 2004, 78, 2327–2335. [Google Scholar] [CrossRef] [Green Version]
- Lehmann-Che, J.; Renault, N.; Giron, M.L.; Roingeard, P.; Clave, E.; Tobaly-Tapiero, J.; Bittoun, P.; Toubert, A.; de The, H.; Saib, A. Centrosomal latency of incoming foamy viruses in resting cells. PLoS Pathog. 2007, 3, e74. [Google Scholar] [CrossRef]
- Hendrie, P.C.; Huo, Y.; Stolitenko, R.B.; Russell, D.W. A rapid and quantitative assay for measuring neighboring gene activation by vector proviruses. Mol. Ther. 2008, 16, 534–540. [Google Scholar] [CrossRef]
- Ong, C.T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Heneine, W.; Switzer, W.M.; Sandstrom, P.; Brown, J.; Vedapuri, S.; Schable, C.A.; Khan, A.S.; Lerche, N.W.; Schweizer, M.; Neumann-Haefelin, D.; et al. Identification of a human population infected with simian foamy viruses. Nat. Med. 1998, 4, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Bhullar, V.; Shanmugam, V.; Cong, M.E.; Parekh, B.; Lerche, N.W.; Yee, J.L.; Ely, J.J.; Boneva, R.; Chapman, L.E.; et al. Frequent simian foamy virus infection in persons occupationally exposed to nonhuman primates. J. Virol. 2004, 78, 2780–2789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trobridge, G.; Josephson, N.; Vassilopoulos, G.; Mac, J.; Russell, D.W. Improved foamy virus vectors with minimal viral sequences. Mol. Ther. 2002, 6, 321–328. [Google Scholar] [CrossRef]
- Nasimuzzaman, M.; Kim, Y.S.; Wang, Y.D.; Persons, D.A. High-titer foamy virus vector transduction and integration sites of human CD34+ cell-derived SCID-repopulating cells. Mol. Ther. Methods Clin. Dev. 2014, 1, 14020. [Google Scholar] [CrossRef]
- Bock, M.; Heinkelein, M.; Lindemann, D.; Rethwilm, A. Cells expressing the human foamy virus (HFV) accessory Bet protein are resistant to productive HFV superinfection. Virology 1998, 250, 194–204. [Google Scholar] [CrossRef] [Green Version]
- Heinkelein, M.; Dressler, M.; Jarmy, G.; Rammling, M.; Imrich, H.; Thurow, J.; Lindemann, D.; Rethwilm, A. Improved primate foamy virus vectors and packaging constructs. J. Virol. 2002, 76, 3774–3783. [Google Scholar] [CrossRef] [Green Version]
- Lochelt, M.; Zentgraf, H.; Flugel, R.M. Construction of an infectious DNA clone of the full-length human spumaretrovirus genome and mutagenesis of the bel 1 gene. Virology 1991, 184, 43–54. [Google Scholar] [CrossRef]
- Kiem, H.P.; Wu, R.A.; Sun, G.; von Laer, D.; Rossi, J.J.; Trobridge, G.D. Foamy combinatorial anti-HIV vectors with MGMTP140K potently inhibit HIV-1 and SHIV replication and mediate selection in vivo. Gene Ther. 2010, 17, 37–49. [Google Scholar] [CrossRef] [Green Version]
- Nasimuzzaman, M.; Lynn, D.; Ernst, R.; Beuerlein, M.; Smith, R.H.; Shrestha, A.; Cross, S.; Link, K.; Lutzko, C.; Nordling, D.; et al. Production and purification of high-titer foamy virus vector for the treatment of leukocyte adhesion deficiency. Mol. Ther. Methods Clin. Dev. 2016, 3, 16004. [Google Scholar] [CrossRef] [Green Version]
- Spannaus, R.; Miller, C.; Lindemann, D.; Bodem, J. Purification of foamy viral particles. Virology 2017, 506, 28–33. [Google Scholar] [CrossRef]
- Leurs, C.; Jansen, M.; Pollok, K.E.; Heinkelein, M.; Schmidt, M.; Wissler, M.; Lindemann, D.; Von Kalle, C.; Rethwilm, A.; Williams, D.A.; et al. Comparison of three retroviral vector systems for transduction of nonobese diabetic/severe combined immunodeficiency mice repopulating human CD34+ cord blood cells. Hum. Gene Ther. 2003, 14, 509–519. [Google Scholar] [CrossRef]
- Morianos, I.; Siapati, E.K.; Pongas, G.; Vassilopoulos, G. Comparative analysis of FV vectors with human alpha- or beta-globin gene regulatory elements for the correction of beta-thalassemia. Gene Ther. 2012, 19, 303–311. [Google Scholar] [CrossRef]
- Kang, E.M.; Malech, H.L. Gene therapy for chronic granulomatous disease. Methods Enzymol. 2012, 507, 125–154. [Google Scholar]
- Chatziandreou, I.; Siapati, E.K.; Vassilopoulos, G. Genetic correction of X-linked chronic granulomatous disease with novel foamy virus vectors. Exp. Hematol. 2011, 39, 643–652. [Google Scholar] [CrossRef]
- Marciano, B.E.; Zerbe, C.S.; Falcone, E.L.; Ding, L.; DeRavin, S.S.; Daub, J.; Kreuzburg, S.; Yockey, L.; Hunsberger, S.; Foruraghi, L.; et al. X-linked carriers of chronic granulomatous disease: Illness, lyonization, and stability. J. Allergy Clin. Immunol. 2018, 141, 365–371. [Google Scholar] [CrossRef] [Green Version]
- Chiriaco, M.; Farinelli, G.; Capo, V.; Zonari, E.; Scaramuzza, S.; Di Matteo, G.; Sergi, L.S.; Migliavacca, M.; Hernandez, R.J.; Bombelli, F.; et al. Dual-regulated lentiviral vector for gene therapy of X-linked chronic granulomatosis. Mol. Ther. 2014, 22, 1472–1483. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, T.; Adriani, M.; Jagadeesh, G.J.; Paine, A.; Candotti, F. Foamy virus vector-mediated gene correction of a mouse model of Wiskott-Aldrich syndrome. Mol. Ther. 2012, 20, 1270–1279. [Google Scholar] [CrossRef] [Green Version]
- Humbert, O.; Chan, F.; Rajawat, Y.S.; Torgerson, T.R.; Burtner, C.R.; Hubbard, N.W.; Humphrys, D.; Norgaard, Z.K.; O’Donnell, P.; Adair, J.E.; et al. Rapid immune reconstitution of SCID-X1 canines after G-CSF/AMD3100 mobilization and in vivo gene therapy. Blood Adv. 2018, 2, 987–999. [Google Scholar] [CrossRef] [Green Version]
- Horino, S.; Uchiyama, T.; So, T.; Nagashima, H.; Sun, S.L.; Sato, M.; Asao, A.; Haji, Y.; Sasahara, Y.; Candotti, F.; et al. Gene therapy model of X-linked severe combined immunodeficiency using a modified foamy virus vector. PLoS ONE 2013, 8, e71594. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Ernstberger, A.; Wang, H.; Bailey, B.J.; Hartwell, J.R.; Sinn, A.L.; Eckermann, O.; Linka, Y.; Goebel, W.S.; Hanenberg, H.; et al. In vivo selection of hematopoietic stem cells transduced at a low multiplicity-of-infection with a foamy viral MGMT (P140K) vector. Exp. Hematol. 2008, 36, 283–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszko, M.E.; Trobridge, G.D. Foamy virus vectors for HIV gene therapy. Viruses 2013, 5, 2585–2600. [Google Scholar] [CrossRef] [PubMed]
- Papadaki, M.; Siapati, E.K.; Vassilopoulos, G. A foamy virus vector system for stable and efficient RNAi expression in mammalian cells. Hum. Gene Ther. 2011, 22, 1293–1303. [Google Scholar] [CrossRef]
- Leukocyte Adhesion Deficiency Type 1. Available online: https://ghr.nlm.nih.gov/condition/leukocyte-adhesion-deficiency-type-1 (accessed on 3 March 2020).
- Kijas, J.M.; Bauer, T.R., Jr.; Gafvert, S.; Marklund, S.; Trowald-Wigh, G.; Johannisson, A.; Hedhammar, A.; Binns, M.; Juneja, R.K.; Hickstein, D.D.; et al. A missense mutation in the beta-2 integrin gene (ITGB2) causes canine leukocyte adhesion deficiency. Genomics 1999, 61, 101–107. [Google Scholar] [CrossRef]
- Bauer, T.R., Jr.; Allen, J.M.; Hai, M.; Tuschong, L.M.; Khan, I.F.; Olson, E.M.; Adler, R.L.; Burkholder, T.H.; Gu, Y.C.; Russell, D.W.; et al. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008, 14, 93–97. [Google Scholar] [CrossRef]
- Takegawa, S.; Fujii, H.; Miwa, S. Change of pyruvate kinase isozymes from M2- to L-type during development of the red cell. Br. J. Haematol. 1983, 54, 467–474. [Google Scholar] [CrossRef]
- Trobridge, G.D.; Beard, B.C.; Wu, R.A.; Ironside, C.; Malik, P.; Kiem, H.P. Stem cell selection in vivo using foamy vectors cures canine pyruvate kinase deficiency. PLoS ONE 2012, 7, e45173. [Google Scholar] [CrossRef] [Green Version]
- Burtner, C.R.; Beard, B.C.; Kennedy, D.R.; Wohlfahrt, M.E.; Adair, J.E.; Trobridge, G.D.; Scharenberg, A.M.; Torgerson, T.R.; Rawlings, D.J.; Felsburg, P.J.; et al. Intravenous injection of a foamy virus vector to correct canine SCID-X1. Blood 2014, 123, 3578–3584. [Google Scholar] [CrossRef] [Green Version]
- Macaulay, R. How CAR-T Cell and Gene Therapies Are Redefining the Traditional Pharmaceutical Pricing and Reimbursement Model. Available online: https://www.parexel.com/news-events-resources/blog/how-car-t-cell-and-gene-therapies-are-redefining-traditional-pharmaceutical-pricing-and-reimbursement-model (accessed on 16 March 2020).


{kind=link}
| Vector System | Lenti- | Gammaretro- | Foamy- |
|---|---|---|---|
| Transgene Capacity (kb) | 9 [26] | 10 [27] | At least 9 [28] |
| Self-Inactivating (SIN) design | + [29] | + [30] | + [31] |
| Generation | 3rd | 3rd | 3rd |
| Presence of insulators in design | + | + | + |
| Pseudotyping | + | + | - |
| Cell cycle requirement | |||
| cGMP complience | + | + | possible |
| Preferred integration sites in host genome | Active transcriptional units [11] | Transcriptional start sites and CpG islands [11,32] | Constitutively lamina associated regions (cLAD) and less often CpGs [15] |
| Disease | Animal Model | FV Vector Systems | Promoter | Transgene | Target Cells | Method of Application | Outcome | Reference |
|---|---|---|---|---|---|---|---|---|
| β-thalassemia | β-Thal3 mice | 3rd | Hu-α-globin HS40-short hu-β-globin | hu-β-globin | Lin- BM HSCs | Ex vivo | Conversion to thalassemia carrier phenotype | [53] |
| 3rd | Hu-β-globin HS2-HS3 LCR-short hu-β-globin | |||||||
| Chronic Granulomatus Disease (CGD) | B6.129S-Cybbtm1Din/J mice | 3rd | PGK | c-o hu-gp91phox | Lin-BM HSCs | Ex vivo | Complete phenotypic restoration | [55] |
| 3rd | MSCV-LTR | c-o hu-gp91phox IRES.EGFP | ||||||
| Wiskott-Aldrich Syndrome (WAS) | WAS KO mice | 3rd | Native promoter | hu-was | Lin- BM HSCs | Ex vivo | Complete phenotypic restoration | [58] |
| 3rd | UCO631 | |||||||
| SCID-X1 | NOD/SCID γc KO mice | 3rd | UCO631 | Human γc gene (IL2RG) | Lin- BM HSCs | Ex vivo | Reconstitution of T and B cells. No NK correction | [60] |
| SCID-X1 | SCID-X1 dogs | 3rd | EF1a (intronless) | GFP.T2A.hIL2RG | i.v. infusion | In vivo | Partial lymphocyte reconstitution | [69] |
| i.v. infusion in HSC mobilized animals | Lymphocyte reconstitution. Successful treatment | [59] | ||||||
| SCID-X1 | SCID-X1 dogs | 3rd | PGK | GFP.T2A.hIL2RG | i.v. infusion in mobilized HSC | In vivo | Lymphocyte reconstitution. Phenotypic correction | |
| Leukocyte adhesion deficiency | CLAD dogs | 3rd | MSCV | hu-CD18 | BM derived CD34+ cells | Ex vivo | Phenotypic correction | [66] |
| Pyruvate Kinase deficiency | Basenji Dog PKD | 3rd | PGK | SFFVMGMT.T2AEGFP.PGK.cPK | Mobilized CD34+ HSCs | Ex vivo | Phenotypic correction | [67] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simantirakis, E.; Tsironis, I.; Vassilopoulos, G. FV Vectors as Alternative Gene Vehicles for Gene Transfer in HSCs. Viruses 2020, 12, 332. https://doi.org/10.3390/v12030332
Simantirakis E, Tsironis I, Vassilopoulos G. FV Vectors as Alternative Gene Vehicles for Gene Transfer in HSCs. Viruses. 2020; 12(3):332. https://doi.org/10.3390/v12030332
Chicago/Turabian StyleSimantirakis, Emmanouil, Ioannis Tsironis, and George Vassilopoulos. 2020. "FV Vectors as Alternative Gene Vehicles for Gene Transfer in HSCs" Viruses 12, no. 3: 332. https://doi.org/10.3390/v12030332