The Interactions Between HBV and the Innate Immunity of Hepatocytes


Abstract
:1. Introduction
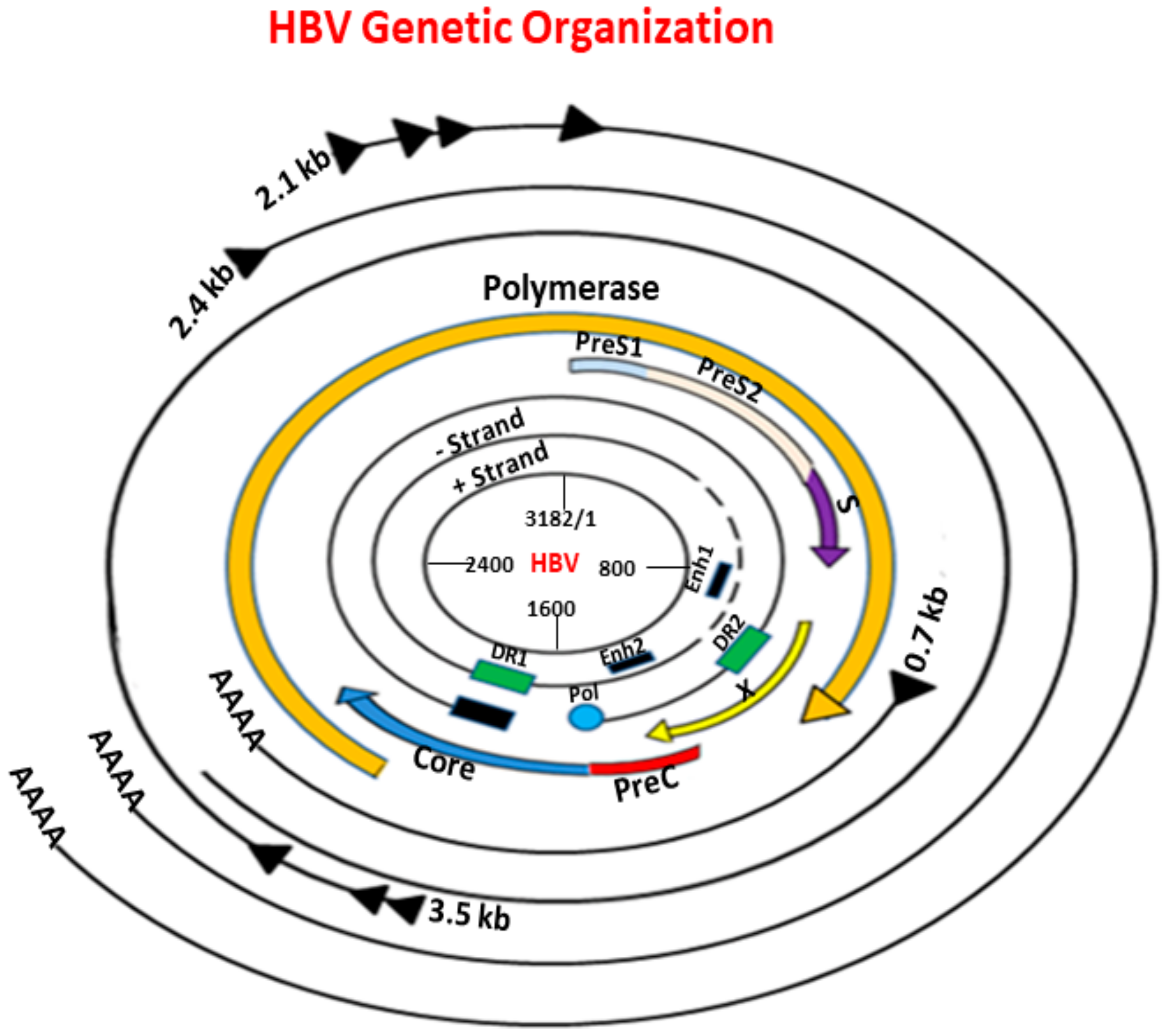
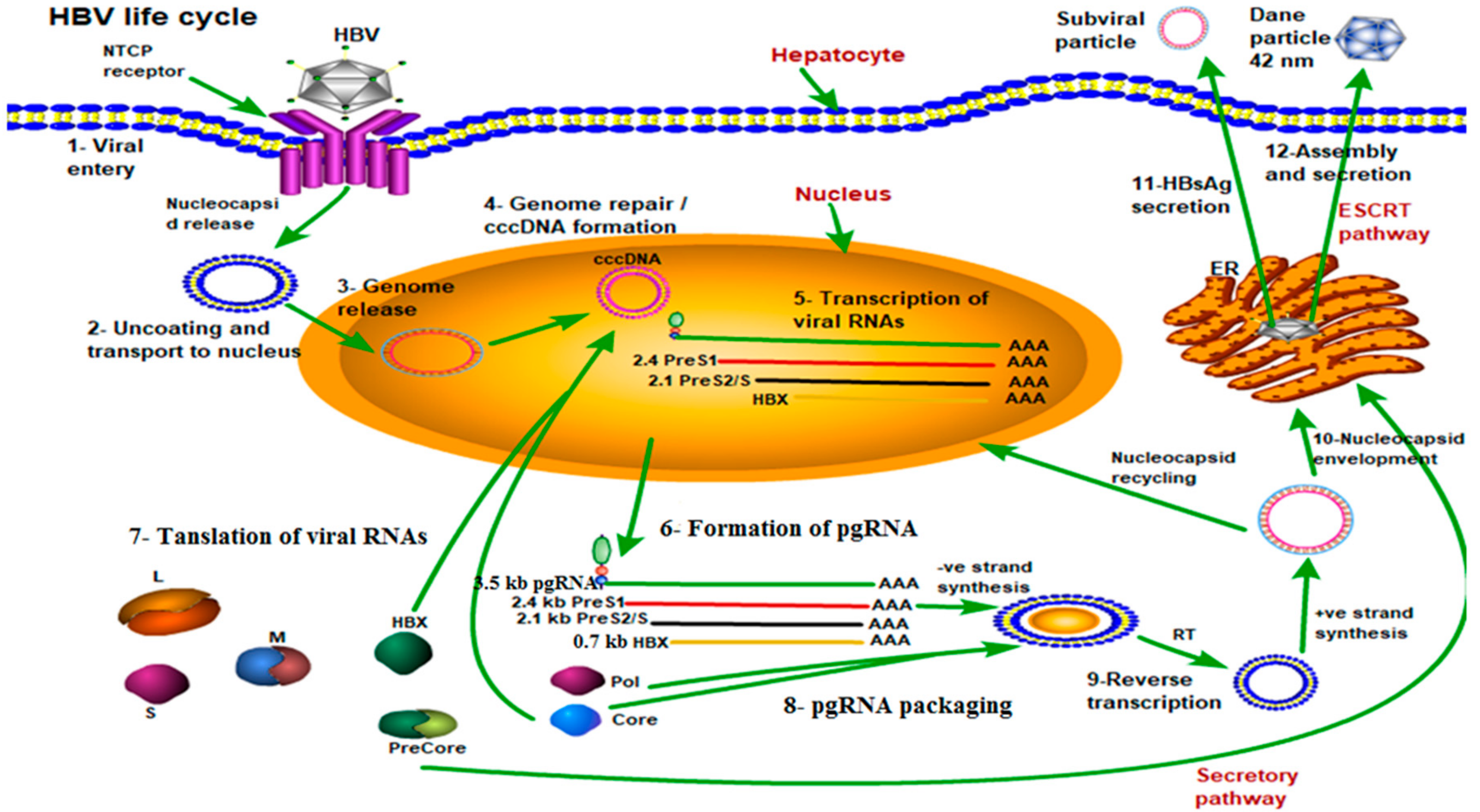
2. Genetic Organization, Life Cycle, and Global Epidemiology of HBV
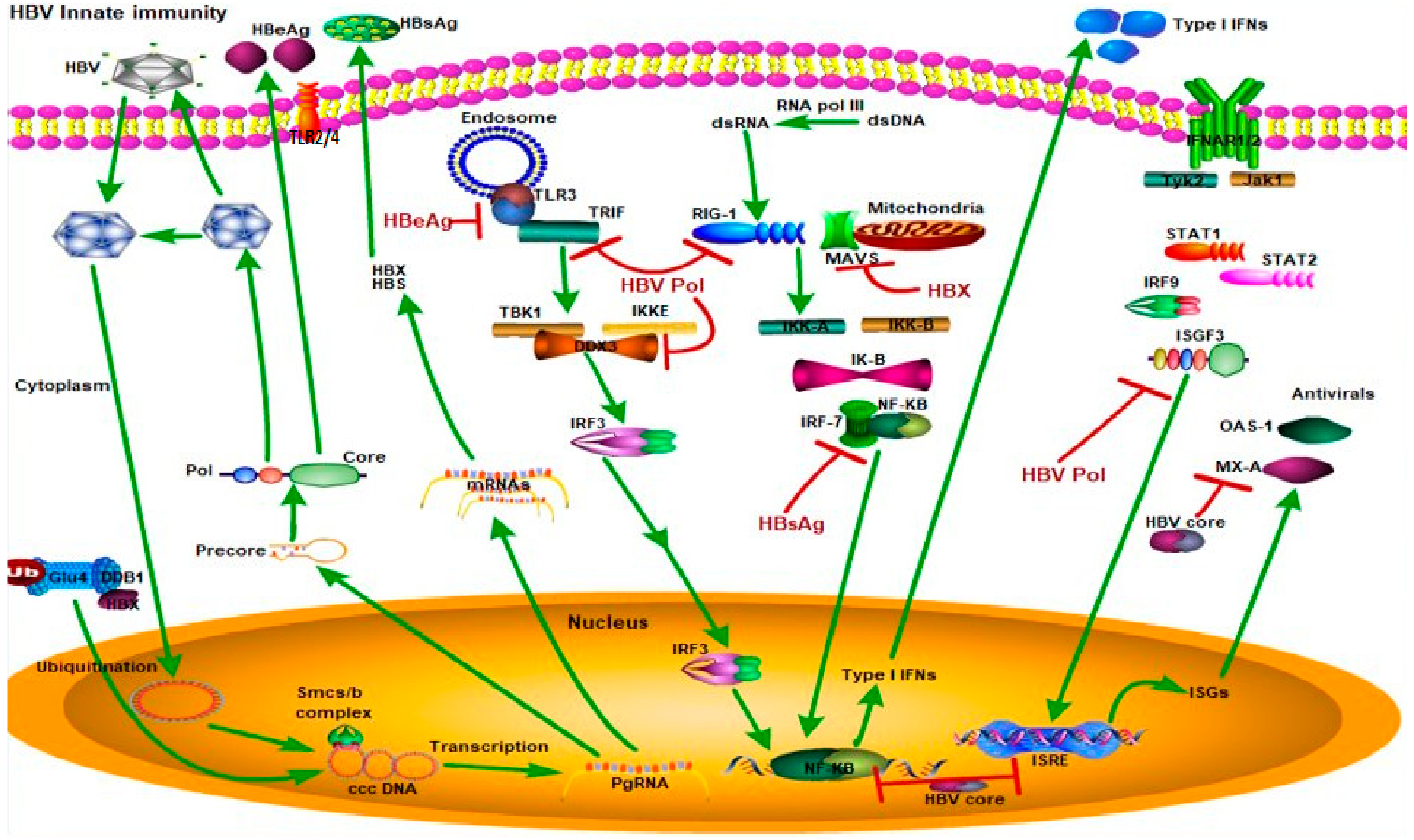
3. HBV Infection and Host Innate Immunity
3.1. Hepatitis B e Antigen (HBeAg)
3.2. HBV Core Protein (HBcAG) and Hepatitis B Splice Protein (HBSP)
3.3. Hepatitis B Virus X Protein (HBx)
3.4. Hepatitis B Virus Polymerase Inhibits Innate Immunity (HBV Pol)
4. Conflicts Regarding HBV and Innate Immunity
5. In Vitro Models to Study HBV Infection and Innate Immunity
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [CrossRef]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Innate immunity to virus infection. Immunol. Rev. 2009, 227, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Bertoletti, A.; Ferrari, C. Innate and adaptive immune responses in chronic hepatitis B virus infections: Towards restoration of immune control of viral infection. Gut 2012, 61, 1754–1764. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Block, T.M.; Guo, J.T. The innate immune response to hepatitis B virus infection: Implications for pathogenesis and therapy. Antivir. Res. 2012, 96, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Wieland, S.; Thimme, R.; Purcell, R.H.; Chisari, F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 6669–6674. [Google Scholar] [CrossRef] [Green Version]
- Thomsen, M.K.; Nandakumar, R.; Stadler, D.; Malo, A.; Valls, R.M.; Wang, F.; Reinert, L.S.; Dagnaes-Hansen, F.; Hollensen, A.K.; Mikkelsen, J.G.; et al. Lack of immunological DNA sensing in hepatocytes facilitates hepatitis B virus infection. Hepatology 2016, 64, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.C.; Tjwa, E.T.; Biesta, P.J.; Boonstra, A.; Xie, Q.; Janssen, H.L.; Woltman, A.M. Hepatitis B virus suppresses the functional interaction between natural killer cells and plasmacytoid dendritic cells. J. Viral Hepat. 2012, 19, e26–e33. [Google Scholar] [CrossRef]
- Yang, Y.; Han, Q.; Zhang, C.; Xiao, M.; Zhang, J. Hepatitis B virus antigens impair NK cell function. Int. Immunopharmacol. 2016, 38, 291–297. [Google Scholar] [CrossRef]
- Miller, R.H.; Kaneko, S.; Chung, C.T.; Girones, R.; Purcell, R.H. Compact organization of the hepatitis B virus genome. Hepatology 1989, 9, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Summers, J.; Mason, W.S. Replication of the genome of a hepatitis B—Like virus by reverse transcription of an RNA intermediate. Cell 1982, 29, 403–415. [Google Scholar] [CrossRef]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.C.; Kao, J.H. Persistence of hepatitis B virus covalently closed circular DNA in hepatocytes: Molecular mechanisms and clinical significance. Emerg. Microbes Infect. 2014, 3, e64. [Google Scholar] [CrossRef]
- Hu, J.; Liu, K. Complete and Incomplete Hepatitis B Virus Particles: Formation, Function, and Application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Tsai, K.N.; Kuo, C.F.; Ou, J.J. Mechanisms of Hepatitis B Virus Persistence. Trends Microbiol. 2018, 26, 33–42. [Google Scholar] [CrossRef]
- Terrault, N.A.; Bzowej, N.H.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Murad, M.H. AASLD guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. [Google Scholar] [CrossRef]
- Collaborators, G.B. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990–2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 386, 743–800. [Google Scholar]
- Goldstein, S.T.; Zhou, F.; Hadler, S.C.; Bell, B.P.; Mast, E.E.; Margolis, H.S. A mathematical model to estimate global hepatitis B disease burden and vaccination impact. Int. J. Epidemiol. 2005, 34, 1329–1339. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.S.; Fan, J.G.; Zhang, Z.; Gao, B.; Wang, H.Y. The global burden of liver disease: The major impact of China. Hepatology 2014, 60, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Sun, L.; Liu, H.H.; Chen, X.; Seth, R.B.; Forman, J.; Chen, Z.J. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006, 24, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, A.; Flach, T.L.; Shi, Y.; Mydlarski, P.R. Toll-like receptors: Role in dermatological disease. Mediat. Inflamm. 2010, 2010, 437246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, D.; Zhu, Z.; Zhou, A.Y.; Yun, C.H.; Lee, K.E.; Toms, A.V.; Li, Y.; Dunn, G.P.; Chan, E.; Thai, T.; et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep. 2013, 3, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Pervolaraki, K.; Rastgou Talemi, S.; Albrecht, D.; Bormann, F.; Bamford, C.; Mendoza, J.L.; Garcia, K.C.; McLauchlan, J.; Hofer, T.; Stanifer, M.L.; et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018, 14, e1007420. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Olagnier, D.; Lin, R. Host and Viral Modulation of RIG-I-Mediated Antiviral Immunity. Front. Immunol. 2016, 7, 662. [Google Scholar] [CrossRef] [Green Version]
- Pourcelot, M.; Arnoult, D. Mitochondrial dynamics and the innate antiviral immune response. FEBS J. 2014, 281, 3791–3802. [Google Scholar] [CrossRef]
- McWhirter, S.M.; Barbalat, R.; Monroe, K.M.; Fontana, M.F.; Hyodo, M.; Joncker, N.T.; Ishii, K.J.; Akira, S.; Colonna, M.; Chen, Z.J.; et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med. 2009, 206, 1899–1911. [Google Scholar] [CrossRef]
- Cheng, X.; Xia, Y.; Serti, E.; Block, P.D.; Chung, M.; Chayama, K.; Rehermann, B.; Liang, T.J. Hepatitis B virus evades innate immunity of hepatocytes but activates cytokine production by macrophages. Hepatology 2017, 66, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.J.; et al. Temporal analysis of early immune responses in patients with acute hepatitis B virus infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutz, P.; Metz, P.; Lempp, F.A.; Bender, S.; Qu, B.; Schoneweis, K.; Seitz, S.; Tu, T.; Restuccia, A.; Frankish, J.; et al. HBV Bypasses the Innate Immune Response and Does Not Protect HCV From Antiviral Activity of Interferon. Gastroenterology 2018, 154, 1791–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Cao, Q.; Xiong, Y.; Zhang, E.; Lu, M. Interaction between Hepatitis B Virus and Toll-Like Receptors: Current Status and Potential Therapeutic Use for Chronic Hepatitis B. Vaccines 2018, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Dansako, H.; Ueda, Y.; Okumura, N.; Satoh, S.; Sugiyama, M.; Mizokami, M.; Ikeda, M.; Kato, N. The cyclic GMP-AMP synthetase-STING signaling pathway is required for both the innate immune response against HBV and the suppression of HBV assembly. FEBS J. 2016, 283, 144–156. [Google Scholar] [CrossRef]
- Liu, Y.; Li, J.; Chen, J.; Li, Y.; Wang, W.; Du, X.; Song, W.; Zhang, W.; Lin, L.; Yuan, Z. Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol. 2015, 89, 2287–2300. [Google Scholar] [CrossRef] [Green Version]
- Wu, M.; Xu, Y.; Lin, S.; Zhang, X.; Xiang, L.; Yuan, Z. Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 2007, 88, 3260–3269. [Google Scholar] [CrossRef]
- Wei, C.; Ni, C.; Song, T.; Liu, Y.; Yang, X.; Zheng, Z.; Jia, Y.; Yuan, Y.; Guan, K.; Xu, Y.; et al. The hepatitis B virus X protein disrupts innate immunity by downregulating mitochondrial antiviral signaling protein. J. Immunol. 2010, 185, 1158–1168. [Google Scholar] [CrossRef]
- Kumar, M.; Jung, S.Y.; Hodgson, A.J.; Madden, C.R.; Qin, J.; Slagle, B.L. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J. Virol. 2011, 85, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Zhang, C.; Zhang, J.; Tian, Z. The role of innate immunity in HBV infection. Semin. Immunopathol. 2013, 35, 23–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, A.; Cui, L.; Hao, R.; Wang, Y.; He, J.; Guo, D. Hepatitis B virus polymerase suppresses NF-kappaB signaling by inhibiting the activity of IKKs via interaction with Hsp90beta. PLoS ONE 2014, 9, e91658. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Kang, W.; Lei, X.; Li, Y.; Xiang, A.; Liu, Y.; Zhao, J.; Zhang, J.; Yan, Z. Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genom. 2012, 13, 563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ryu, W.S. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: Implications for immune evasion. PLoS Pathog. 2010, 6, e1000986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B virus polymerase inhibits RIG-I- and Toll-like receptor 3-mediated beta interferon induction in human hepatocytes through interference with interferon regulatory factor 3 activation and dampening of the interaction between TBK1/IKKepsilon and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar]
- Chen, J.; Wu, M.; Zhang, X.; Zhang, W.; Zhang, Z.; Chen, L.; He, J.; Zheng, Y.; Chen, C.; Wang, F.; et al. Hepatitis B virus polymerase impairs interferon-alpha-induced STA T activation through inhibition of importin-alpha5 and protein kinase C-delta. Hepatology 2013, 57, 470–482. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Hepatitis B Virus X and Regulation of Viral Gene Expression. Cold Spring Harb. Perspect. Med. 2016, 6, a021402. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.H.; Park, E.S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.H.; Kim, C.W.; et al. Suppression of interferon-mediated anti-HBV response by single CpG methylation in the 5′-UTR of TRIM22. Gut 2018, 67, 166–178. [Google Scholar] [CrossRef]
- Fernandez, M.; Quiroga, J.A.; Carreno, V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J. Gen. Virol. 2003, 84, 2073–2082. [Google Scholar] [CrossRef]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2007, 45, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Lang, T.; Lo, C.; Skinner, N.; Locarnini, S.; Visvanathan, K.; Mansell, A. The hepatitis B e antigen (HBeAg) targets and suppresses activation of the toll-like receptor signaling pathway. J. Hepatol. 2011, 55, 762–769. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Kanda, T.; Imazeki, F.; Arai, M.; Yonemitsu, Y.; Nakamoto, S.; Fujiwara, K.; Fukai, K.; Nomura, F.; Yokosuka, O. Hepatitis B virus e antigen downregulates cytokine production in human hepatoma cell lines. Viral Immunol. 2010, 23, 467–476. [Google Scholar] [CrossRef]
- Twu, J.S.; Lee, C.H.; Lin, P.M.; Schloemer, R.H. Hepatitis B virus suppresses expression of human beta-interferon. Proc. Natl. Acad. Sci. USA 1988, 85, 252–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosmorduc, O.; Sirma, H.; Soussan, P.; Gordien, E.; Lebon, P.; Horisberger, M.; Brechot, C.; Kremsdorf, D. Inhibition of interferon-inducible MxA protein expression by hepatitis B virus capsid protein. J. Gen. Virol. 1999, 80((Pt. 5)), 1253–1262. [Google Scholar] [CrossRef]
- Soussan, P.; Garreau, F.; Zylberberg, H.; Ferray, C.; Brechot, C.; Kremsdorf, D. In vivo expression of a new hepatitis B virus protein encoded by a spliced RNA. J. Clin. Investig. 2000, 105, 55–60. [Google Scholar] [CrossRef] [Green Version]
- Castanier, C.; Zemirli, N.; Portier, A.; Garcin, D.; Bidere, N.; Vazquez, A.; Arnoult, D. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012, 10, 44. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ma, Z.; Liu, H.; Liu, J.; Meng, Z.; Broering, R.; Yang, D.; Schlaak, J.F.; Roggendorf, M.; Lu, M. Role of Toll-like receptor 2 in the immune response against hepadnaviral infection. J. Hepatol. 2012, 57, 522–528. [Google Scholar] [CrossRef]
- Gane, E.J.; Lim, Y.S.; Gordon, S.C.; Visvanathan, K.; Sicard, E.; Fedorak, R.N.; Roberts, S.; Massetto, B.; Ye, Z.; Pflanz, S.; et al. The oral toll-like receptor-7 agonist GS-9620 in patients with chronic hepatitis B virus infection. J. Hepatol. 2015, 63, 320–328. [Google Scholar] [CrossRef]
- Christen, V.; Duong, F.; Bernsmeier, C.; Sun, D.; Nassal, M.; Heim, M.H. Inhibition of alpha interferon signaling by hepatitis B virus. J. Virol. 2007, 81, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Suslov, A.; Boldanova, T.; Wang, X.; Wieland, S.; Heim, M.H. Hepatitis B Virus Does Not Interfere With Innate Immune Responses in the Human Liver. Gastroenterology 2018, 154, 1778–1790. [Google Scholar] [CrossRef] [Green Version]
- Sells, M.A.; Chen, M.L.; Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proc. Natl. Acad. Sci. USA 1987, 84, 1005–1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fellig, Y.; Almogy, G.; Galun, E.; Ketzinel-Gilad, M. A hepatocellular carcinoma cell line producing mature hepatitis B viral particles. Biochem. Biophys. Res. Commun. 2004, 321, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Lechon, M.J.; Donato, M.T.; Castell, J.V.; Jover, R. Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Curr. Drug Metab. 2004, 5, 443–462. [Google Scholar] [CrossRef] [PubMed]
- Piryaei, A.; Valojerdi, M.R.; Shahsavani, M.; Baharvand, H. Differentiation of bone marrow-derived mesenchymal stem cells into hepatocyte-like cells on nanofibers and their transplantation into a carbon tetrachloride-induced liver fibrosis model. Stem Cell Rev. 2011, 7, 103–118. [Google Scholar] [CrossRef]
- Dolle, L.; Best, J.; Mei, J.; Al Battah, F.; Reynaert, H.; van Grunsven, L.A.; Geerts, A. The quest for liver progenitor cells: A practical point of view. J. Hepatol. 2010, 52, 117–129. [Google Scholar] [CrossRef]
- Sullivan, G.J.; Hay, D.C.; Park, I.H.; Fletcher, J.; Hannoun, Z.; Payne, C.M.; Dalgetty, D.; Black, J.R.; Ross, J.A.; Samuel, K.; et al. Generation of functional human hepatic endoderm from human induced pluripotent stem cells. Hepatology 2010, 51, 329–335. [Google Scholar] [CrossRef] [Green Version]
- Hay, D.C.; Fletcher, J.; Payne, C.; Terrace, J.D.; Gallagher, R.C.; Snoeys, J.; Black, J.R.; Wojtacha, D.; Samuel, K.; Hannoun, Z.; et al. Highly efficient differentiation of hESCs to functional hepatic endoderm requires ActivinA and Wnt3a signaling. Proc. Natl. Acad. Sci. USA 2008, 105, 12301–12306. [Google Scholar] [CrossRef] [Green Version]
- Cayo, M.A.; Mallanna, S.K.; Di Furio, F.; Jing, R.; Tolliver, L.B.; Bures, M.; Urick, A.; Noto, F.K.; Pashos, E.E.; Greseth, M.D.; et al. A Drug Screen using Human iPSC-Derived Hepatocyte-like Cells Reveals Cardiac Glycosides as a Potential Treatment for Hypercholesterolemia. Cell Stem Cell 2017, 20, 478–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Sun, P.; Lucendo-Villarin, B.; Angus, A.G.; Szkolnicka, D.; Cameron, K.; Farnworth, S.L.; Patel, A.H.; Hay, D.C. Modulating innate immunity improves hepatitis C virus infection and replication in stem cell-derived hepatocytes. Stem Cell Rep. 2014, 3, 204–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; Ploss, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Dao Thi, V.L.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-Genotype Hepatitis E Virus Replication in Stem Cell-Derived Hepatocellular Systems. Gastroenterology 2018, 154, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [Green Version]
- Schulze-Bergkamen, H.; Untergasser, A.; Dax, A.; Vogel, H.; Buchler, P.; Klar, E.; Lehnert, T.; Friess, H.; Buchler, M.W.; Kirschfink, M.; et al. Primary human hepatocytes—A valuable tool for investigation of apoptosis and hepatitis B virus infection. J. Hepatol. 2003, 38, 736–744. [Google Scholar] [CrossRef]
- Yoneda, M.; Hyun, J.; Jakubski, S.; Saito, S.; Nakajima, A.; Schiff, E.R.; Thomas, E. Hepatitis B Virus and DNA Stimulation Trigger a Rapid Innate Immune Response through NF-kappaB. J. Immunol. 2016, 197, 630–643. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Tang, L.; Shu, S.; Sehgal, M.; Sheraz, M.; Liu, B.; Zhao, Q.; Cheng, J.; Zhao, X.; Zhou, T.; et al. Activation of Stimulator of Interferon Genes in Hepatocytes Suppresses the Replication of Hepatitis B Virus. Antimicrob. Agents Chemother. 2017, 61, e00771-17. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Liu, H.; Ikeda, Y.; Amiot, B.P.; Rinaldo, P.; Duncan, S.A.; Nyberg, S.L. Hepatocyte-like cells differentiated from human induced pluripotent stem cells: Relevance to cellular therapies. Stem Cell Res. 2012, 9, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, D.M.; Foldes, G.; Gatheral, T.; Paschalaki, K.E.; Lendvai, Z.; Bagyura, Z.; Nemeth, T.; Skopal, J.; Merkely, B.; Telcian, A.G.; et al. Pathogen sensing pathways in human embryonic stem cell derived-endothelial cells: Role of NOD1 receptors. PLoS ONE 2014, 9, e91119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulze, A.; Mills, K.; Weiss, T.S.; Urban, S. Hepatocyte polarization is essential for the productive entry of the hepatitis B virus. Hepatology 2012, 55, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, M.; Watashi, K.; Tsukuda, S.; Aly, H.H.; Fukasawa, M.; Fujimoto, A.; Suzuki, R.; Aizaki, H.; Ito, T.; Koiwai, O.; et al. Evaluation and identification of hepatitis B virus entry inhibitors using HepG2 cells overexpressing a membrane transporter NTCP. Biochem. Biophys. Res. Commun. 2014, 443, 808–813. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef] [Green Version]
- Sato, S.; Li, K.; Kameyama, T.; Hayashi, T.; Ishida, Y.; Murakami, S.; Watanabe, T.; Iijima, S.; Sakurai, Y.; Watashi, K.; et al. The RNA Sensor RIG-I Dually Functions as an Innate Sensor and Direct Antiviral Factor for Hepatitis B Virus. Immunity 2015, 42, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Israelow, B.; Narbus, C.M.; Sourisseau, M.; Evans, M.J. HepG2 cells mount an effective antiviral interferon-lambda based innate immune response to hepatitis C virus infection. Hepatology 2014, 60, 1170–1179. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Chen, Z.; Kato, N.; Gale, M., Jr.; Lemon, S.M. Distinct poly(I-C) and virus-activated signaling pathways leading to interferon-beta production in hepatocytes. J. Biol. Chem. 2005, 280, 16739–16747. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Jiang, D.; Ma, D.; Chang, J.; Dougherty, A.M.; Cuconati, A.; Block, T.M.; Guo, J.T. Activation of pattern recognition receptor-mediated innate immunity inhibits the replication of hepatitis B virus in human hepatocyte-derived cells. J. Virol. 2009, 83, 847–858. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Xu, Y.Y.; Zhou, J.M.; Wu, Y.Y.; Qun, E.; Zhu, Y.Y. TLR3 dsRNA agonist inhibits growth and invasion of HepG2.2.15 HCC cells. Oncol. Rep. 2012, 28, 200–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Zhang, C.; Zhang, J.; Tian, Z. Involvement of activation of PKR in HBx-siRNA-mediated innate immune effects on HBV inhibition. PLoS ONE 2011, 6, e27931. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| HBV Proteins | Cellular Innate Immunity Targets | References |
|---|---|---|
| Polymerase | RIG-I, TLR3/TBK1, IKKɛ, DDX3/IFN-β, IFN/JAK-STAT | [45,46,47] |
| Hepatitis B virus X protein | RIG-I, melanoma differentiation-associated gene 5 (MDA5)/MAVS/IFN-β/Trim22 | [40,41,48,49] |
| Core/precore | IFN/myxovirus resistance A (MxA) | [50] |
| Hepatitis B e antigen | TLR2/ MyD88-Adapter-Like (MAL) | [51,52] |
| Cell Models | HBV Infection Efficiency | Innate Immunity Pathways Present | Advantages Limitations | References |
|---|---|---|---|---|
| Primary human hepatocytes (PHHs) | Average 50% in the presence of 5% polyethylene glycol (PEG) | Low TLR expression | Gold standard for investigation of HBV infection; limited availability and unpredictable variability | [77,78,79] |
| Low stimulator of interferon genes(STING) expression | ||||
| RIG-I/MDA5 | ||||
| NF-κB pathway | ||||
| IRF pathway | ||||
| IFN pathway | ||||
| Human embryonic stem cell (hESC)/human induced-pluripotent stem cell (hiPSC)-derived hepatocytes | 25–90% | Low TLR expression | Close to PHHs depending on differentiation status; can support long-term infection, can be generated from donors with different genetic backgrounds; immature status needs to be improved | [72,76,80,81,82] |
| Low STING expression | ||||
| NF-κB pathway | ||||
| IRF pathway | ||||
| IFN pathway | ||||
| HepaRG | ~10% in the presence of PEG, maximum rate is 20% | Low TLR expression | Close to PHHs; | [78,79,83,84] |
| Low STING expression | suitable for drug metabolism and HBV infection; can be differentiated into both biliary cells and hepatocytes; a long period for differentiation is needed (at least two weeks) | |||
| RIG-I/MDA5 | ||||
| NF-κB pathway | ||||
| IRF pathway | ||||
| IFN pathway | ||||
| HepG2-NTCP | ~70% infection efficiency at 4% PEG and 2.5% dimethyl sulfoxide (DMSO) | Poorly characterized | Can be used to screen novel drugs and elucidate host–virus interaction; high concentrations of HBV genome equivalents are needed for a high infection rate | [37,64,65,79,85,86,87,88,89,90] |
| TLR expression | ||||
| No cyclic GMP-AMP synthase (cGAS) | ||||
| expression and low STING expression | ||||
| RIG-I/MDA5 | ||||
| NF-κB pathway | ||||
| IRF pathway | ||||
| IFN pathway | ||||
| Huh7-NTCP | ~5% infection efficiency at 4% PEG and 2.5% DMSO | Poorly characterized | Low HBV infection rate; defects in some innate immunity pathways | [37,64,65,79,88,89,90] |
| TLR expression | ||||
| No cGAS and STING expression | ||||
| Both RIG-I/MDA-5 and IFN pathway are present but weaker than HepG2 | ||||
| HepG2.2.15 | HBV genome integrated the into HepG2 genome | No cGAS expression and low | Can be used to screen anti-HBV drugs Unsuitable for studying HBV entry and uncoating | [37,62,86,87,91,92] |
| STING expression | ||||
| RIG-I/MDA5 | ||||
| NF-κB pathway | ||||
| IRF pathway | ||||
| IFN pathway |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Megahed, F.A.K.; Zhou, X.; Sun, P. The Interactions Between HBV and the Innate Immunity of Hepatocytes. Viruses 2020, 12, 285. https://doi.org/10.3390/v12030285
Megahed FAK, Zhou X, Sun P. The Interactions Between HBV and the Innate Immunity of Hepatocytes. Viruses. 2020; 12(3):285. https://doi.org/10.3390/v12030285
Chicago/Turabian StyleMegahed, Fayed Attia Koutb, Xiaoling Zhou, and Pingnan Sun. 2020. "The Interactions Between HBV and the Innate Immunity of Hepatocytes" Viruses 12, no. 3: 285. https://doi.org/10.3390/v12030285