
Identification of an Intermediate Step in Foamy Virus Fusion

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression Constructs and Virus Preparation
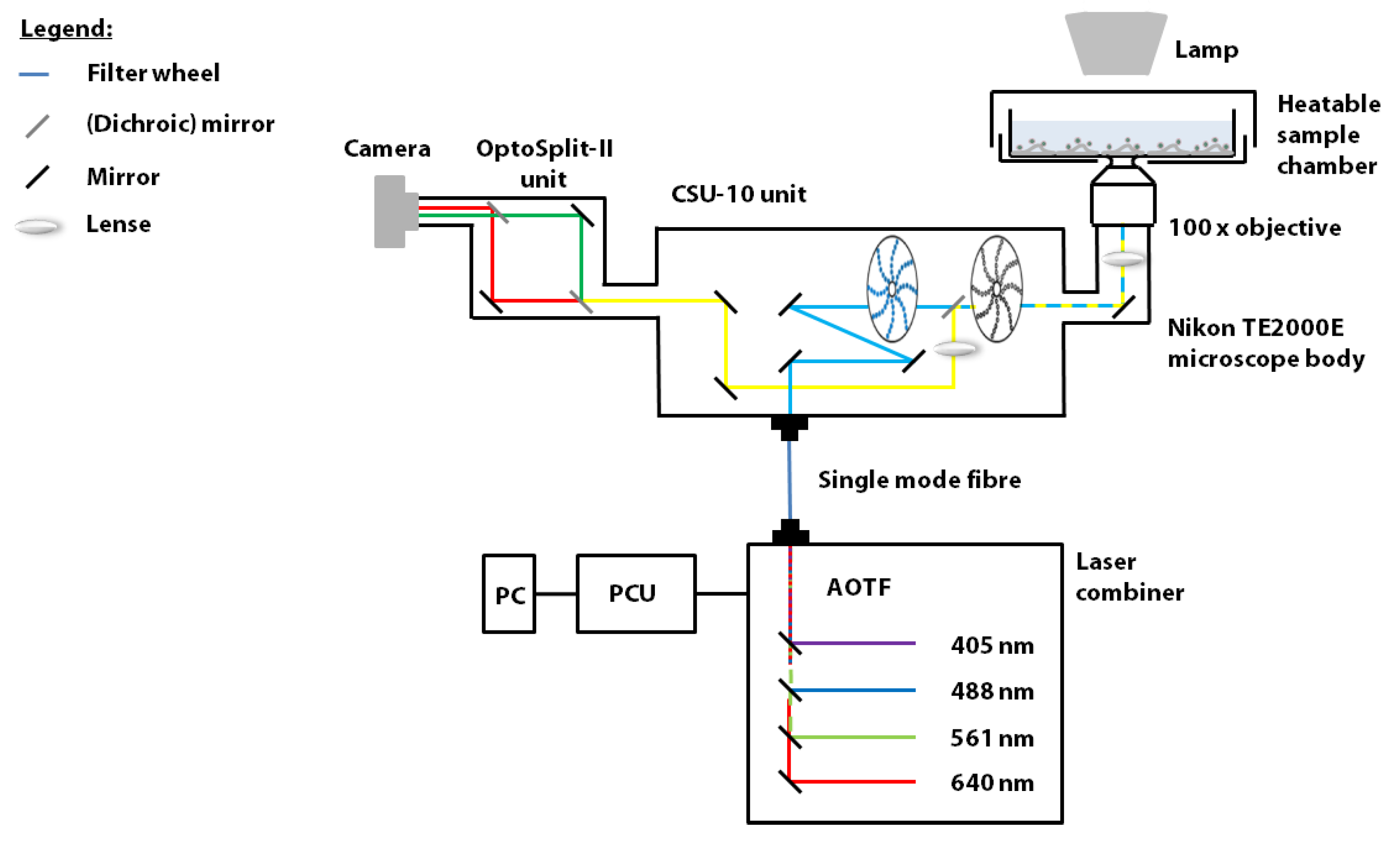
2.2. Spinning Disk Confocal Microscope
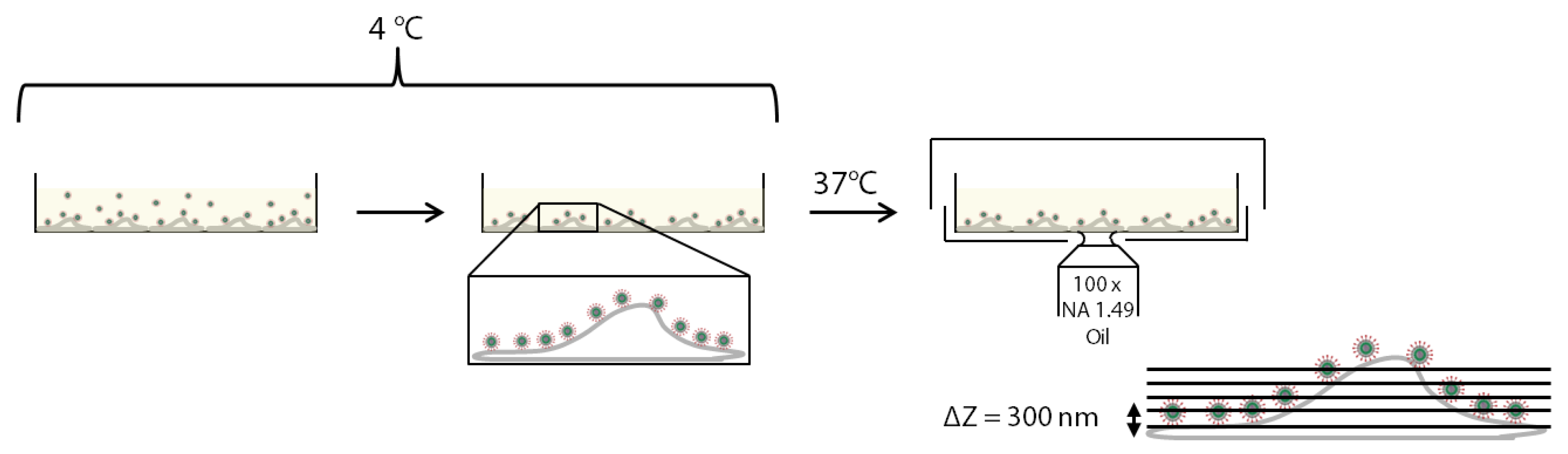
2.3. Live-Cell Imaging Experiments
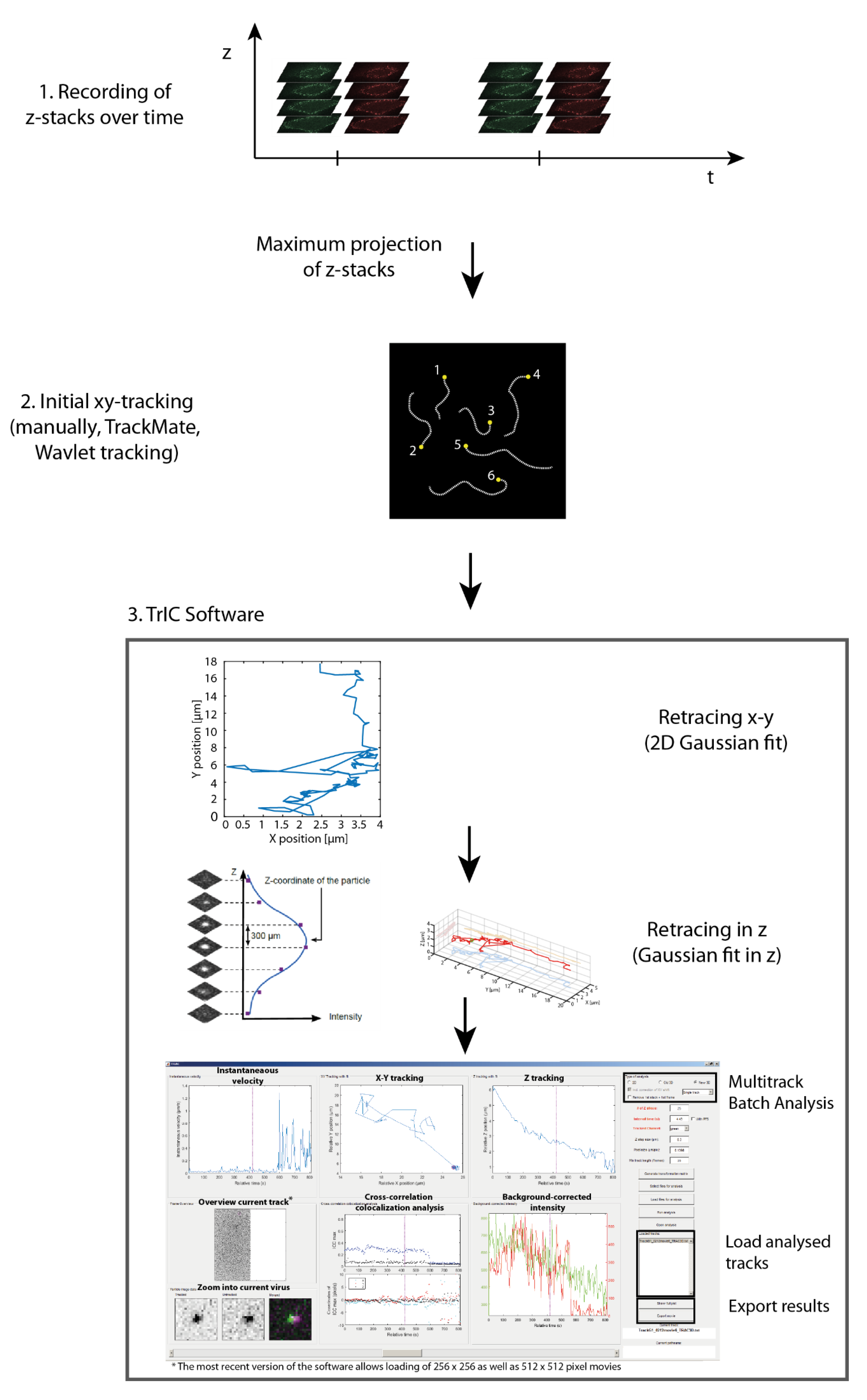
2.4. Data Analysis
2.5. Estimation of the Cell Surface
3. Results
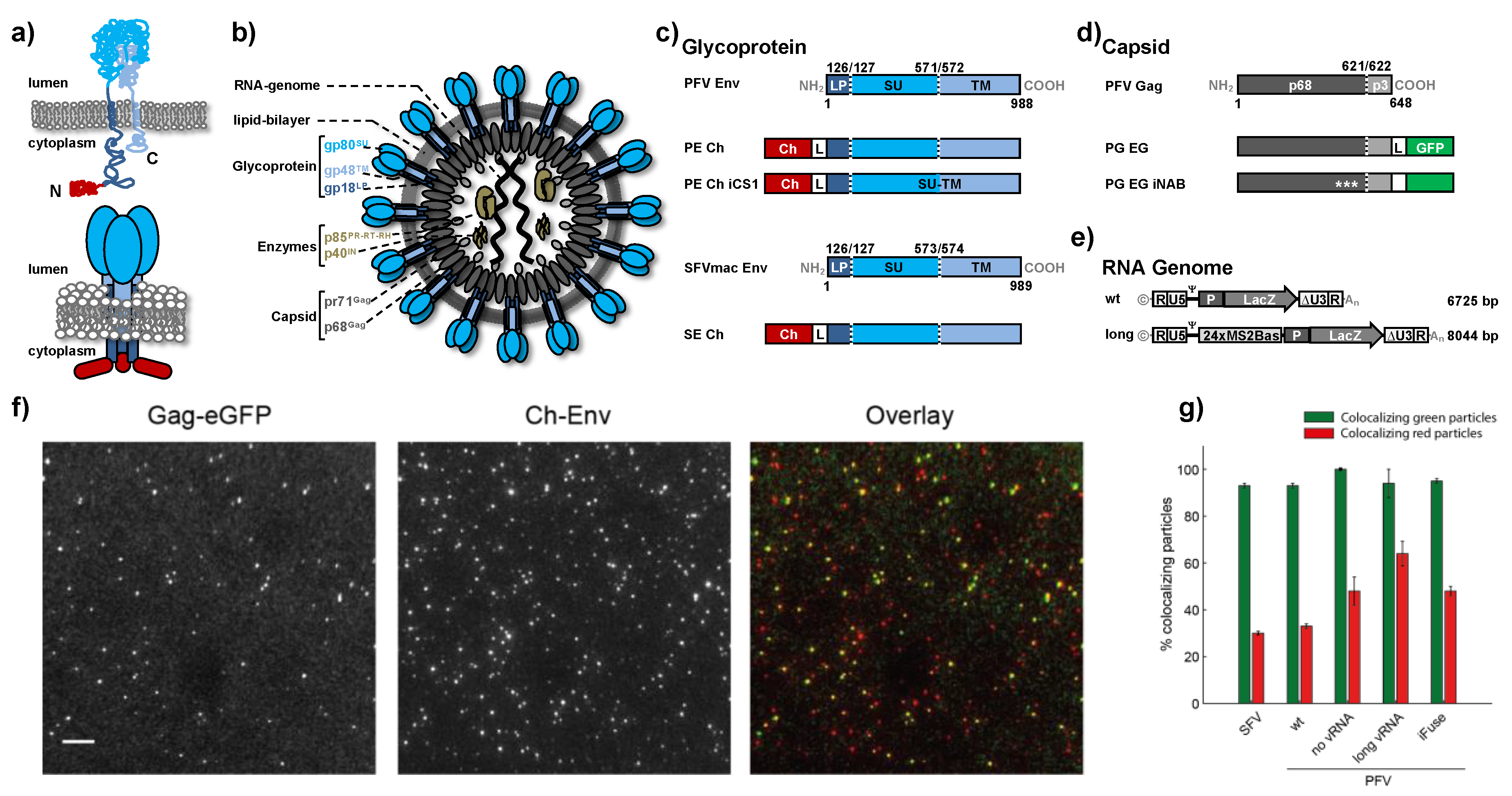
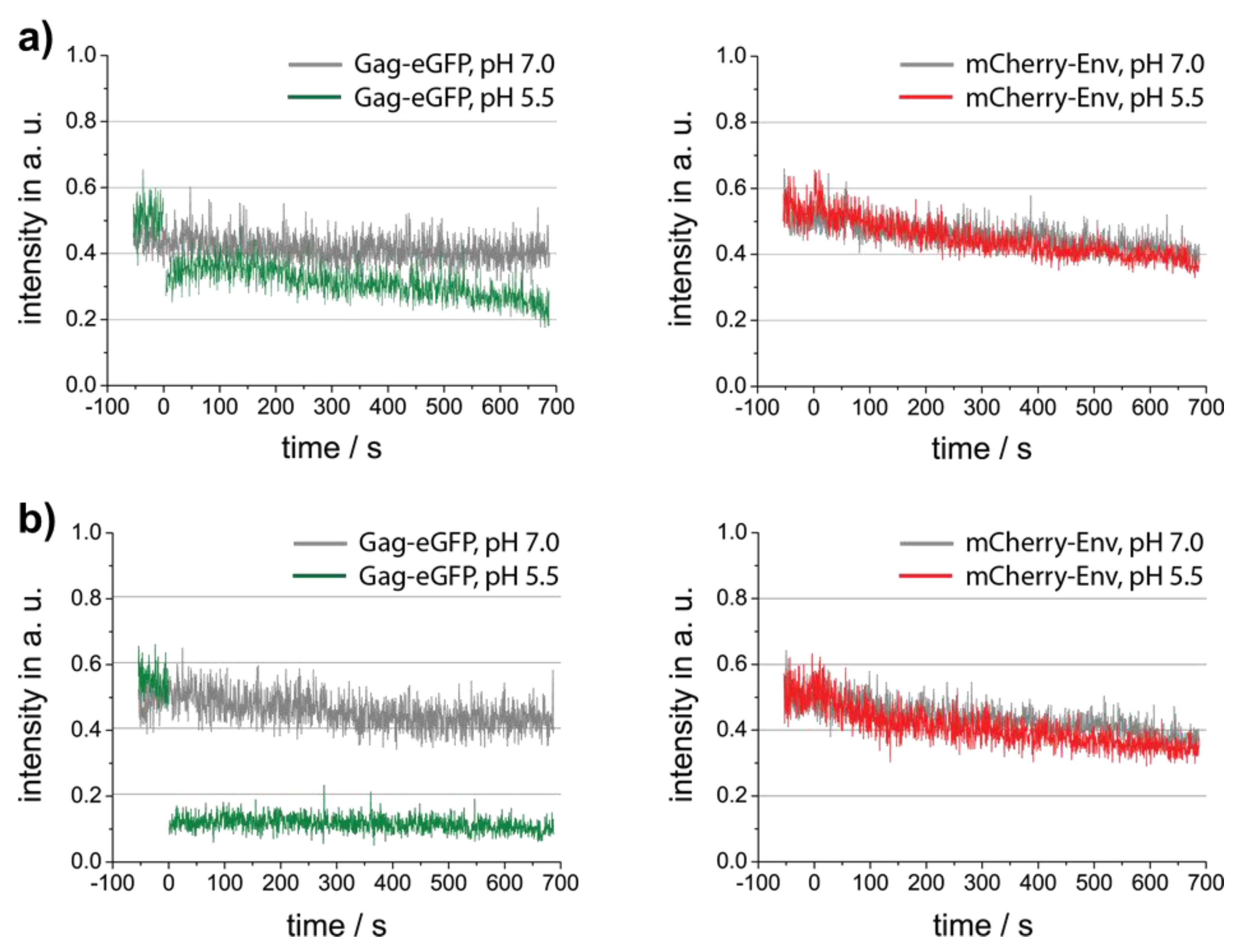
3.1. Characterization of Foamy Virus Particles
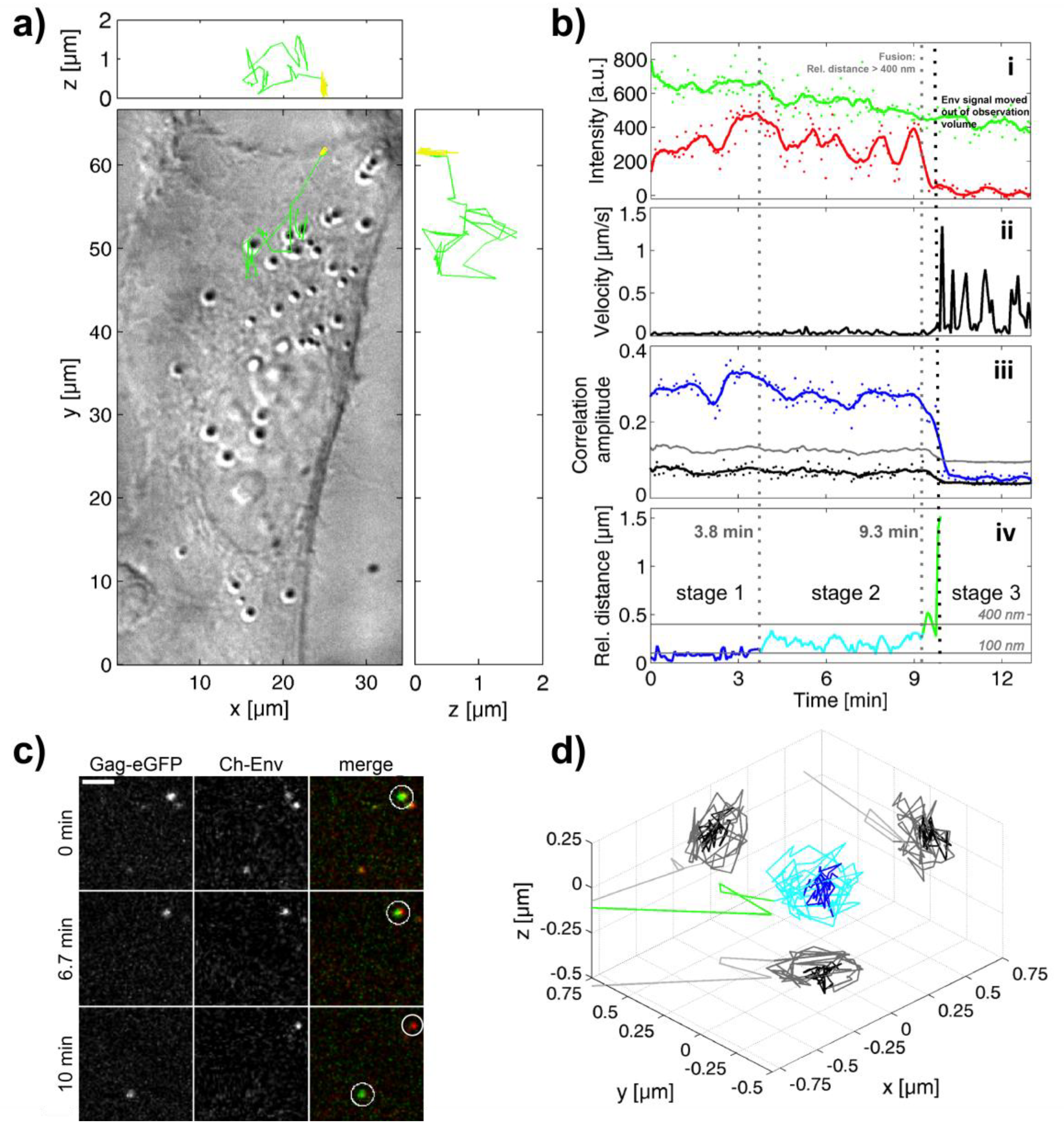
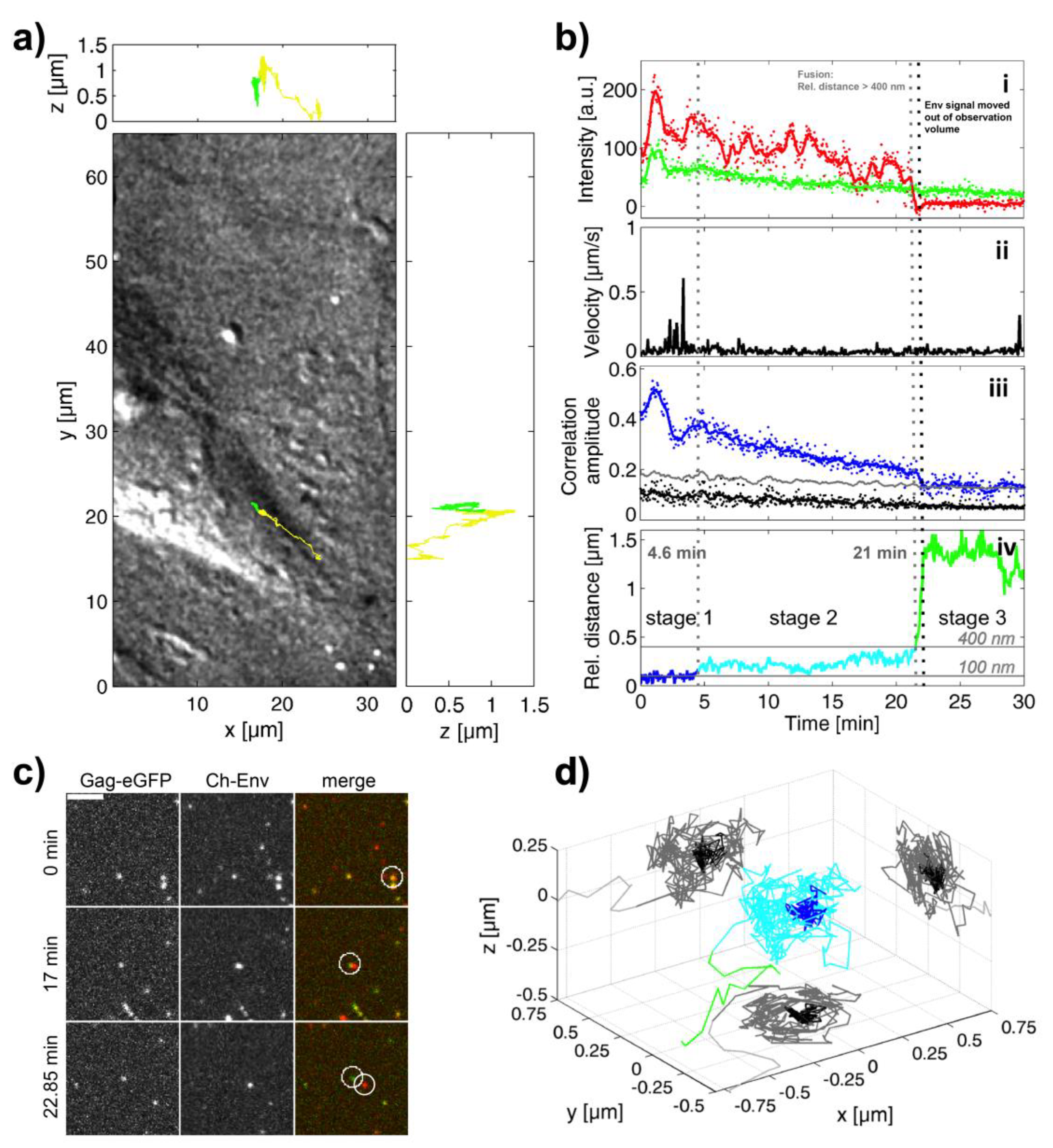
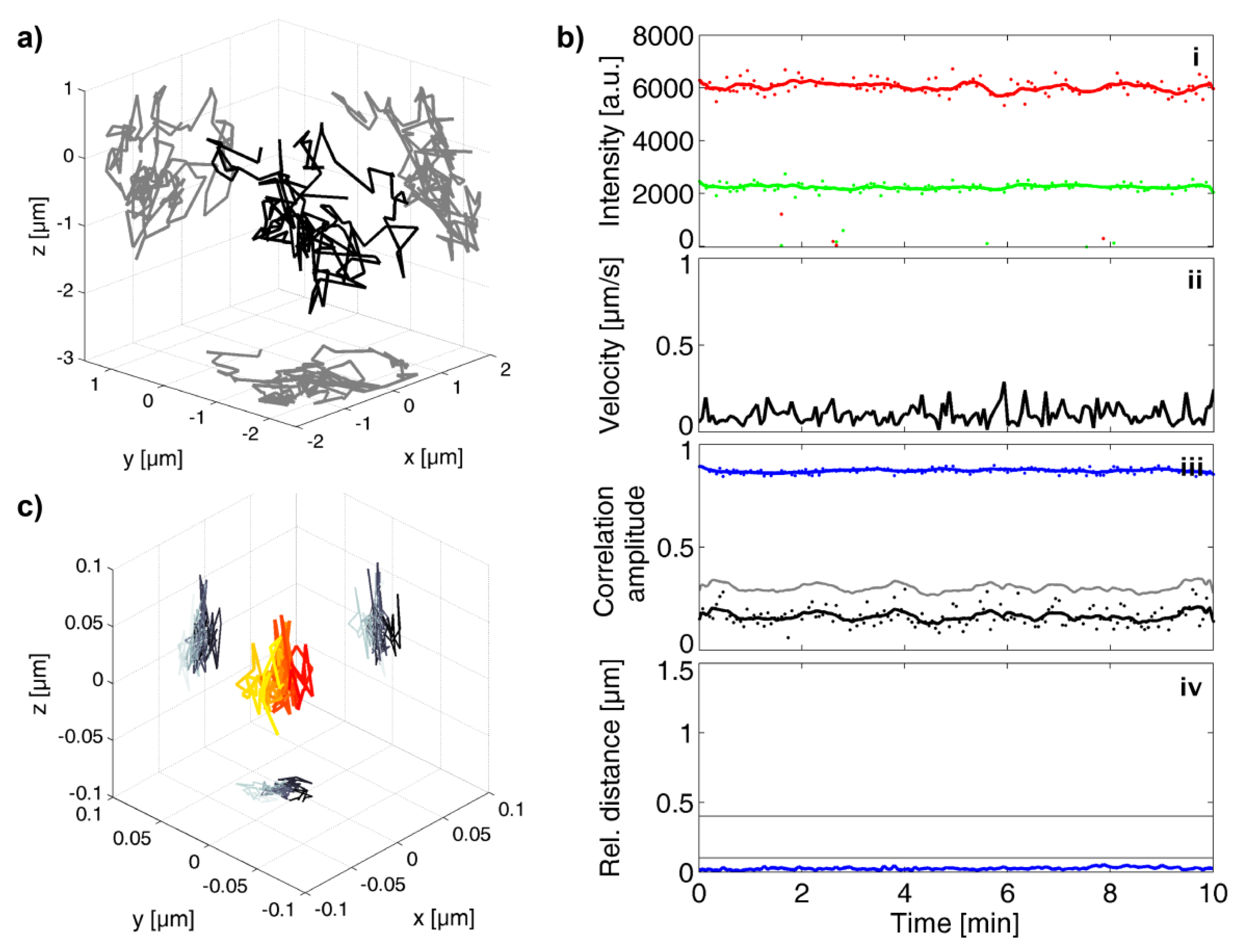
3.2. Investigation of Individual Prototype Foamy Virus Env-Mediated Fusion Events
3.3. Fusion Mediated by Simian Foamy Virus Env
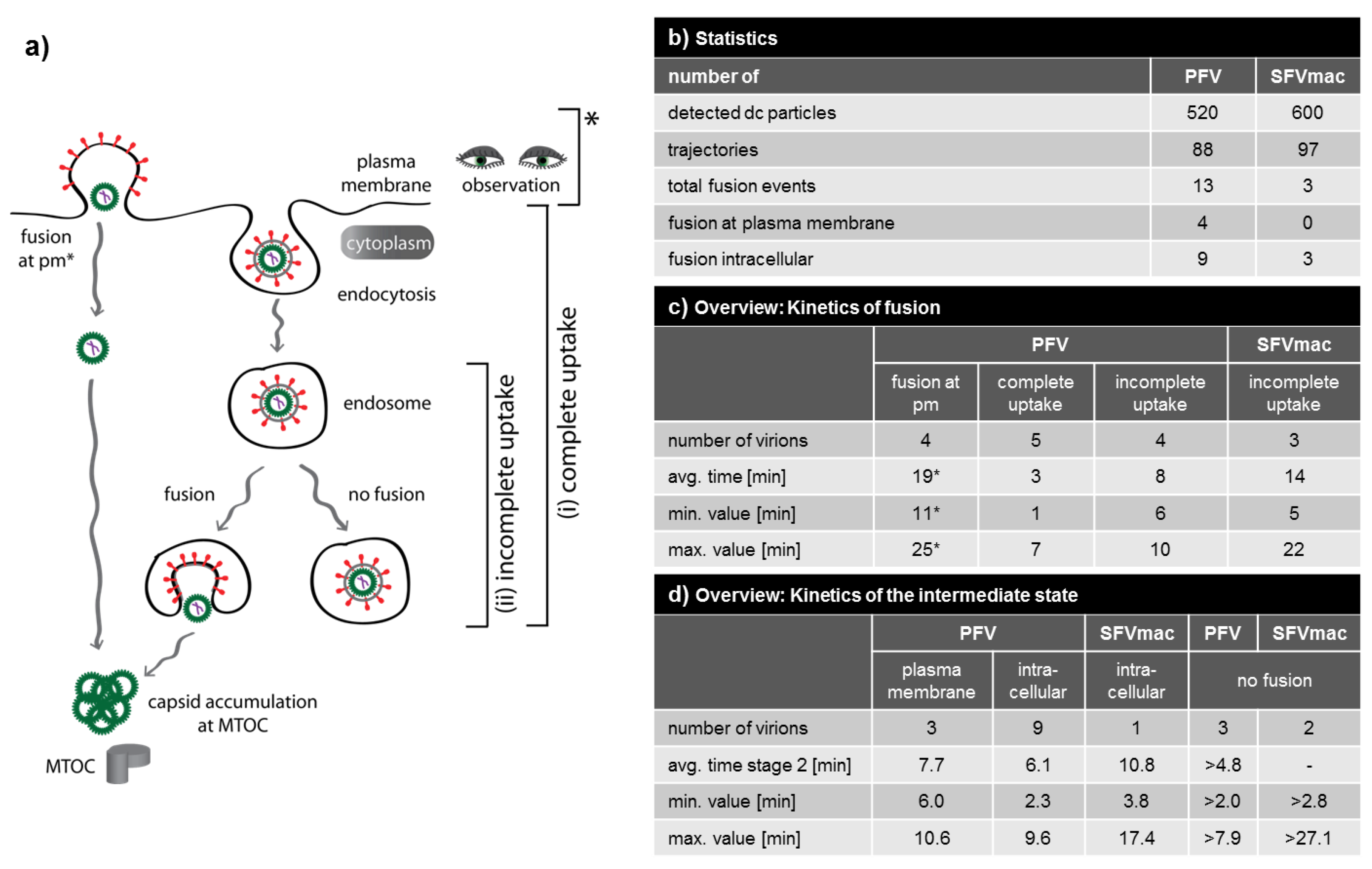
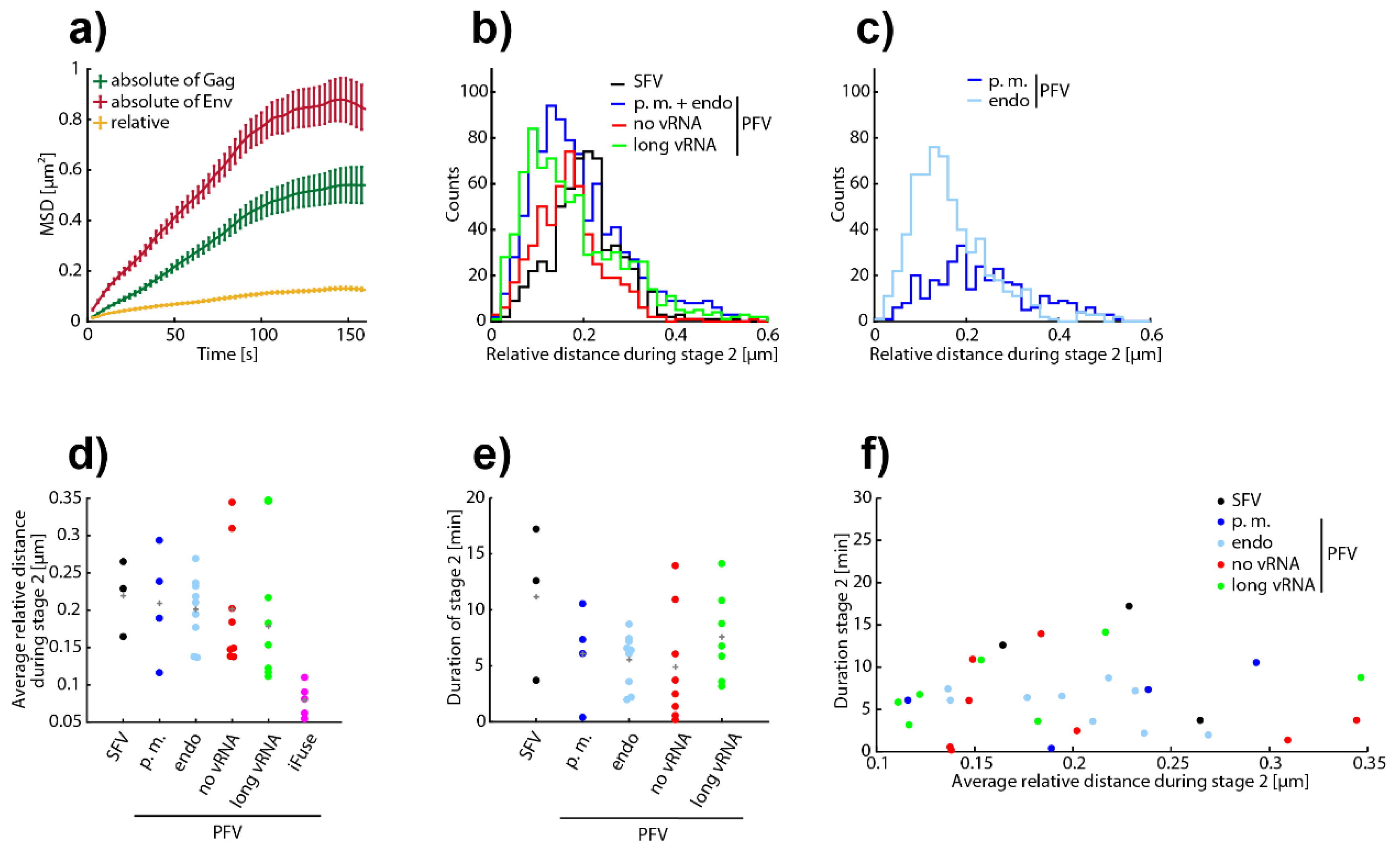
3.4. Analysis of All Observed Events
3.5. A Puzzling Delay in Fusion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DIC | differential interference contrast |
| DMEM | Dulbecco’s Modified Eagle Medium |
| eGFP | enhanced green fluorescent protein |
| Env | envelope |
| eMLV | ecotropic murine leukemia virus |
| FBS | fetal bovine serum |
| FV | foamy virus |
| Gag | group specific antigen |
| HIV-1 | human immunodeficiency virus 1 |
| INF | influenza A virus |
| LP | leader peptide |
| mRNA | messenger RNA |
| MSD | mean square displacement |
| PBS | phosphate-buffered saline |
| PFV | prototype foamy virus |
| Pol | polymerase |
| RNA | ribonucleic acid |
| SFVmac | macaque simian foamy virus |
| SFVmcy | simian foamy virus Macaca cyclopis |
| SFVpsc | simian foamy virus Pan troglodytes schweinfurthii |
| SU | surface |
| SVT | single virus tracing |
| TM | transmembrane |
| TrIC | tracking image correlation |
| vRNA | viral RNA |
| VSV | vesicular stomatitis virus |
| YFP | yellow fluorescent protein |
Appendix A






References
- Khan, A.S.; Bodem, J.; Buseyne, F.; Gessain, A.; Johnson, W.; Kuhn, J.H.; Kuzmak, J.; Lindemann, D.; Linial, M.L.; Lochelt, M.; et al. Spumaretroviruses: Updated taxonomy and nomenclature. Virology 2018, 516, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Rethwilm, A.; Lindemann, D. Foamy Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins, a Wolters Kluwer Business: Philadelphia, PA, USA, 2013; Volume 2, pp. 1613–1632. [Google Scholar]
- Aiewsakun, P.; Katzourakis, A. Marine origin of retroviruses in the early Palaeozoic Era. Nat. Commun. 2017, 8, 13954. [Google Scholar] [CrossRef] [PubMed]
- Herchenröder, O.; Renne, R.; Loncar, D.; Cobb, E.K.; Murthy, K.K.; Schneider, J.; Mergia, A.; Luciw, P.A. Isolation, cloning, and sequencing of simian foamy viruses from chimpanzees (SFVcpz): High homology to human foamy virus (HFV). Virology 1994, 201, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Achong, B.G.; Mansell, P.W.; Epstein, M.A.; Clifford, P. An unusual virus in cultures from a human nasopharyngeal carcinoma. J. Natl. Cancer Inst. 1971, 46, 299–307. [Google Scholar] [PubMed]
- Duda, A.; Stange, A.; Lüftenegger, D.; Stanke, N.; Westphal, D.; Pietschmann, T.; Eastman, S.W.; Linial, M.L.; Rethwilm, A.; Lindemann, D. Prototype foamy virus envelope glycoprotein leader peptide processing is mediated by a furin-like cellular protease, but cleavage is not essential for viral infectivity. J. Virol. 2004, 78, 13865–13870. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindemann, D.; Pietschmann, T.; Picard-Maureau, M.; Berg, A.; Heinkelein, M.; Thurow, J.; Knaus, P.; Zentgraf, H.; Rethwilm, A. A particle-associated glycoprotein signal peptide essential for virus maturation and infectivity. J. Virol. 2001, 75, 5762–5771. [Google Scholar] [CrossRef] [Green Version]
- Wilk, T.; Geiselhart, V.; Frech, M.; Fuller, S.D.; Flügel, R.M.; Löchelt, M. Specific interaction of a novel foamy virus env leader protein with the n-terminal gag domain. J. Virol. 2001, 75, 7995–8007. [Google Scholar] [CrossRef] [Green Version]
- Geiselhart, V.; Bastone, P.; Kempf, T.; Schnolzer, M.; Löchelt, M. Furin-mediated cleavage of the feline foamy virus Env leader protein. J. Virol. 2004, 78, 13573–13581. [Google Scholar] [CrossRef] [Green Version]
- Effantin, G.; Estrozi, L.F.; Aschman, N.; Renesto, P.; Stanke, N.; Lindemann, D.; Schoehn, G.; Weissenhorn, W. Cryo-electron Microscopy Structure of the Native Prototype Foamy Virus Glycoprotein and Virus Architecture. PLoS Pathog. 2016, 12, e1005721. [Google Scholar] [CrossRef] [Green Version]
- Wilk, T.; de Haas, F.; Wagner, A.; Rutten, T.; Fuller, S.; Flügel, R.M.; Löchelt, M. The intact retroviral Env glycoprotein of human foamy virus is a trimer. J. Virol. 2000, 74, 2885–2887. [Google Scholar] [CrossRef] [Green Version]
- Pietschmann, T.; Heinkelein, M.; Heldmann, M.; Zentgraf, H.; Rethwilm, A.; Lindemann, D. Foamy virus capsids require the cognate envelope protein for particle export. J. Virol. 1999, 73, 2613–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, N.; Heinkelein, M.; Lindemann, D.; Enssle, J.; Baum, C.; Werder, E.; Zentgraf, H.; Müller, J.G.; Rethwilm, A. Foamy virus particle formation. J. Virol. 1998, 72, 1610–1615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstone, D.C.; Flower, T.G.; Ball, N.J.; Sanz-Ramos, M.; Yap, M.W.; Ogrodowicz, R.W.; Stanke, N.; Reh, J.; Lindemann, D.; Stoye, J.P.; et al. A Unique Spumavirus Gag N-terminal Domain with Functional Properties of Orthoretroviral Matrix and Capsid. PLoS Pathog. 2013, 9, e1003376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reh, J.; Stange, A.; Götz, A.; Rönitz, M.; Grosse, A.; Lindemann, D. An N-terminal domain helical motif of Prototype Foamy Virus Gag with dual functions essential for particle egress and viral infectivity. Retrovirology 2013, 10, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, K.L.; Lindemann, D.; Mulligan, M.J.; Goepfert, P.A. Foamy virus envelope glycoprotein is sufficient for particle budding and release. J. Virol. 2003, 77, 2338–2348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, N.; Stange, A.; Lüftenegger, D.; Zentgraf, H.; Lindemann, D. Ubiquitination of the Prototype Foamy Virus Envelope Glycoprotein Leader Peptide Regulates Subviral Particle Release. J. Virol. 2005, 79, 15074–15083. [Google Scholar] [CrossRef] [Green Version]
- Stirnnagel, K.; Schupp, D.; Dupont, A.; Kudryavtsev, V.; Reh, J.; Müllers, E.; Lamb, D.C.; Lindemann, D. Differential pH-dependent cellular uptake pathways among foamy viruses elucidated using dual-colored fluorescent particles. Retrovirology 2012, 9, 71. [Google Scholar] [CrossRef] [Green Version]
- Picard-Maureau, M.; Jarmy, G.; Berg, A.; Rethwilm, A.; Lindemann, D. Foamy virus envelope glycoprotein-mediated entry involves a pH-dependent fusion process. J. Virol. 2003, 77, 4722–4730. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.L.; Wang, Z.G.; Xie, H.Y.; Liu, A.A.; Lamb, D.C.; Pang, D.W. Single-Virus Tracking: From Imaging Methodologies to Virological Applications. Chem. Rev. 2020, 120, 1936–1979. [Google Scholar] [CrossRef] [Green Version]
- Seisenberger, G.; Ried, M.U.; Endress, T.; Büning, H.; Hallek, M.; Bräuchle, C. Real-time single-molecule imaging of the infection pathway of an adeno-associated virus. Science 2001, 294, 1929–1932. [Google Scholar] [CrossRef]
- Floyd, D.L.; Ragains, J.R.; Skehel, J.J.; Harrison, S.C.; van Oijen, A.M. Single-particle kinetics of influenza virus membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 15382–15387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelkmans, L.; Burli, T.; Zerial, M.; Helenius, A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell 2004, 118, 767–780. [Google Scholar] [CrossRef] [Green Version]
- Pelkmans, L.; Puntener, D.; Helenius, A. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 2002, 296, 535–539. [Google Scholar] [CrossRef]
- Pelkmans, L.; Kartenbeck, J.; Helenius, A. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat. Cell Biol. 2001, 3, 473–483. [Google Scholar] [CrossRef]
- Elphick, G.F.; Querbes, W.; Jordan, J.A.; Gee, G.V.; Eash, S.; Manley, K.; Dugan, A.; Stanifer, M.; Bhatnagar, A.; Kroeze, W.K.; et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science 2004, 306, 1380–1383. [Google Scholar] [CrossRef] [Green Version]
- Pietiainen, V.; Marjomaki, V.; Upla, P.; Pelkmans, L.; Helenius, A.; Hyypia, T. Echovirus 1 endocytosis into caveosomes requires lipid rafts, dynamin II, and signaling events. Mol. Biol. Cell 2004, 15, 4911–4925. [Google Scholar] [CrossRef]
- Dupont, A.; Stirnnagel, K.; Lindemann, D.; Lamb, D.C. Tracking image correlation: Combining single-particle tracking and image correlation. Biophys. J. 2013, 104, 2373–2382. [Google Scholar] [CrossRef] [Green Version]
- Stirnnagel, K.; Lüftenegger, D.; Stange, A.; Swiersy, A.; Müllers, E.; Reh, J.; Stanke, N.; Grosse, A.; Chiantia, S.; Keller, H.; et al. Analysis of prototype foamy virus particle-host cell interaction with autofluorescent retroviral particles. Retrovirology 2010, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Müllers, E.; Uhlig, T.; Stirnnagel, K.; Fiebig, U.; Zentgraf, H.; Lindemann, D. Novel functions of prototype foamy virus Gag glycine-arginine-rich boxes in reverse transcription and particle morphogenesis. J. Virol. 2011, 85, 1452–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamann, M.V.; Müllers, E.; Reh, J.; Stanke, N.; Effantin, G.; Weissenhorn, W.; Lindemann, D. The cooperative function of arginine residues in the Prototype Foamy Virus Gag C-terminus mediates viral and cellular RNA encapsidation. Retrovirology 2014, 11, 87. [Google Scholar] [CrossRef] [PubMed]
- Tinevez, J.Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Henss, A.K.; Sakong, S.; Messer, P.K.; Wiechers, J.; Schuster, R.; Lamb, D.C.; Gross, A.; Wintterlin, J. Density fluctuations as door-opener for diffusion on crowded surfaces. Science 2019, 363, 715–718. [Google Scholar] [CrossRef]
- Patterson, G.H.; Knobel, S.M.; Sharif, W.D.; Kain, S.R.; Piston, D.W. Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 1997, 73, 2782–2790. [Google Scholar] [CrossRef] [Green Version]
- Verdeny-Vilanova, I.; Wehnekamp, F.; Mohan, N.; Sandoval Alvarez, A.; Borbely, J.S.; Otterstrom, J.J.; Lamb, D.C.; Lakadamyali, M. 3D motion of vesicles along microtubules helps them to circumvent obstacles in cells. J. Cell Sci. 2017, 130, 1904–1916. [Google Scholar] [CrossRef] [Green Version]
- Lakadamyali, M.; Rust, M.J.; Zhuang, X. Ligands for clathrin-mediated endocytosis are differentially sorted into distinct populations of early endosomes. Cell 2006, 124, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Suomalainen, M.; Nakano, M.Y.; Keller, S.; Boucke, K.; Stidwill, R.P.; Greber, U.F. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J. Cell Biol. 1999, 144, 657–672. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.J.; White, P.J.; Pouton, C.W. Interaction of viruses with host cell molecular motors. Curr. Opin. Biotechnol. 2010, 21, 633–639. [Google Scholar] [CrossRef]
- Schelhaas, M.; Ewers, H.; Rajamaki, M.L.; Day, P.M.; Schiller, J.T.; Helenius, A. Human papillomavirus type 16 entry: Retrograde cell surface transport along actin-rich protrusions. PLoS Pathog. 2008, 4, e1000148. [Google Scholar] [CrossRef] [Green Version]
- Bausinger, R.; von Gersdorff, K.; Braeckmans, K.; Ogris, M.; Wagner, E.; Brauchle, C.; Zumbusch, A. The transport of nanosized gene carriers unraveled by live-cell imaging. Angew. Chem. Int. Ed. Engl. 2006, 45, 1568–1572. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.J.; Sherer, N.M.; Marks, C.B.; Pypaert, M.; Mothes, W. Actin- and myosin-driven movement of viruses along filopodia precedes their entry into cells. J. Cell Biol. 2005, 170, 317–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbard, A.L. Endocytosis. Curr. Opin. Cell Biol. 1989, 1, 675–683. [Google Scholar] [CrossRef]
- Mellman, I.; Fuchs, R.; Helenius, A. Acidification of the endocytic and exocytic pathways. Annu. Rev. Biochem. 1986, 55, 663–700. [Google Scholar] [CrossRef]
- Daecke, J.; Fackler, O.T.; Dittmar, M.T.; Kräusslich, H.G. Involvement of clathrin-mediated endocytosis in human immunodeficiency virus type 1 entry. J. Virol. 2005, 79, 1581–1594. [Google Scholar] [CrossRef] [Green Version]
- Johannsdottir, H.K.; Mancini, R.; Kartenbeck, J.; Amato, L.; Helenius, A. Host cell factors and functions involved in vesicular stomatitis virus entry. J. Virol. 2009, 83, 440–453. [Google Scholar] [CrossRef] [Green Version]
- Ruigrok, R.W.; Hewat, E.A.; Wade, R.H. Low pH deforms the influenza virus envelope. J. Gen. Virol. 1992, 73, 995–998. [Google Scholar] [CrossRef]
- Koch, P.; Lampe, M.; Godinez, W.J.; Müller, B.; Rohr, K.; Kräusslich, H.G.; Lehmann, M.J. Visualizing fusion of pseudotyped HIV-1 particles in real time by live cell microscopy. Retrovirology 2009, 6, 84. [Google Scholar] [CrossRef] [Green Version]
- Barrett, C.T.; Dutch, R.E. Viral Membrane Fusion and the Transmembrane Domain. Viruses 2020, 12, 693. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479–480, 498–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Label of Virus Prep | vgRNA | Viral Gag | Viral Env | Gag Ratio Tagged: Untagged | ∑ Green Particles | % Coloc. Green ± SEM | ∑ Red Particles | % Coloc. Red ± SEM |
|---|---|---|---|---|---|---|---|---|
| PFV wt 1,2 | wt | PG EG | PE Ch | 1:0 | 6666 | 97 ± 0.8 | 12,649 | 55 ± 0.5 |
| PFV wt 2 | wt | PG EG | PE Ch | 1:3 | 2365 | 98 ± 0.2 | 7683 | 33 ± 0.7 |
| PFV wt 1,2 | wt | PG EG | PE Ch | 1:3 | 1476 | 93 ± 1 | 4022 | 33 ± 1 |
| PFV iFuse 1 | wt | PG EG | PE Ch iCS1 | 1:3 | 542 | 95 ± 1 | 1159 | 48 ± 2 |
| PFV no vRNA | - | PG EG | PE Ch | 1:3 | 168 | 100 ± 0 | 351 | 48 ± 6 |
| PFV long vRNA | long | PG EG | PE Ch | 1:3 | 152 | 94 ± 6 | 235 | 64 ± 5 |
| SFV wt 1 | wt | PG EG | SE Ch | 1:3 | 944 | 93 ± 0.8 | 3290 | 30 ± 1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dupont, A.; Glück, I.M.; Ponti, D.; Stirnnagel, K.; Hütter, S.; Perrotton, F.; Stanke, N.; Richter, S.; Lindemann, D.; Lamb, D.C. Identification of an Intermediate Step in Foamy Virus Fusion. Viruses 2020, 12, 1472. https://doi.org/10.3390/v12121472
Dupont A, Glück IM, Ponti D, Stirnnagel K, Hütter S, Perrotton F, Stanke N, Richter S, Lindemann D, Lamb DC. Identification of an Intermediate Step in Foamy Virus Fusion. Viruses. 2020; 12(12):1472. https://doi.org/10.3390/v12121472
Chicago/Turabian StyleDupont, Aurélie, Ivo M. Glück, Dorothee Ponti, Kristin Stirnnagel, Sylvia Hütter, Florian Perrotton, Nicole Stanke, Stefanie Richter, Dirk Lindemann, and Don C. Lamb. 2020. "Identification of an Intermediate Step in Foamy Virus Fusion" Viruses 12, no. 12: 1472. https://doi.org/10.3390/v12121472