
Can Coronaviruses Steal Genes from the Host as Evidenced in Western European Hedgehogs by EriCoV Genetic Characterization?

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
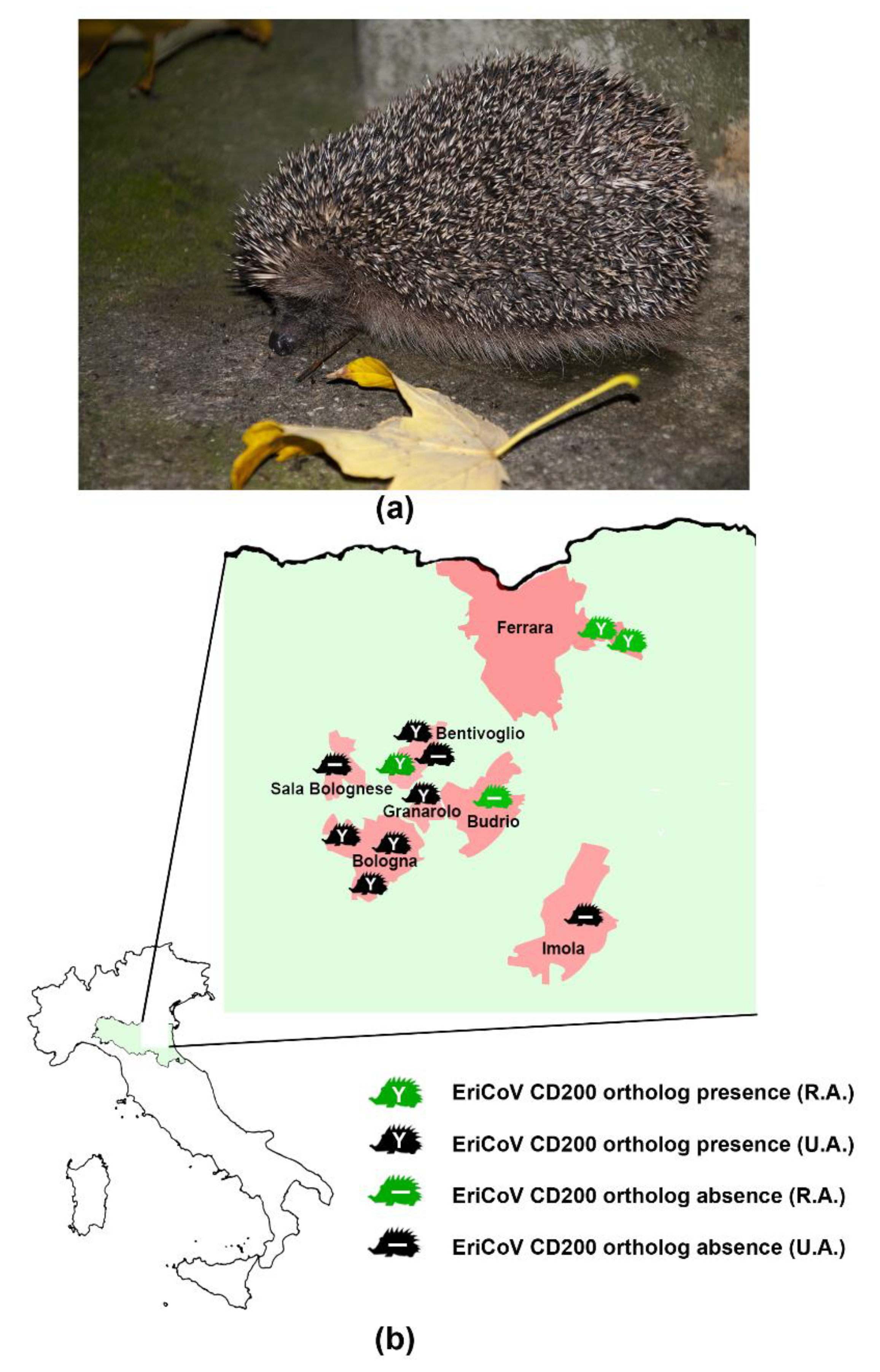
2.1. Samples
2.2. Whole-Genome Sequencing
2.3. Complete Genome Assembling
2.4. Recombination Analysis
2.5. CD200 Detection by Reverse Transcription-PCR
2.6. Phylogenetic Analysis
2.7. Structural Modelling
3. Results
3.1. Complete Genome Sequences and Genome Organization
3.2. CD200 Ortholog Detection by RT-PCR
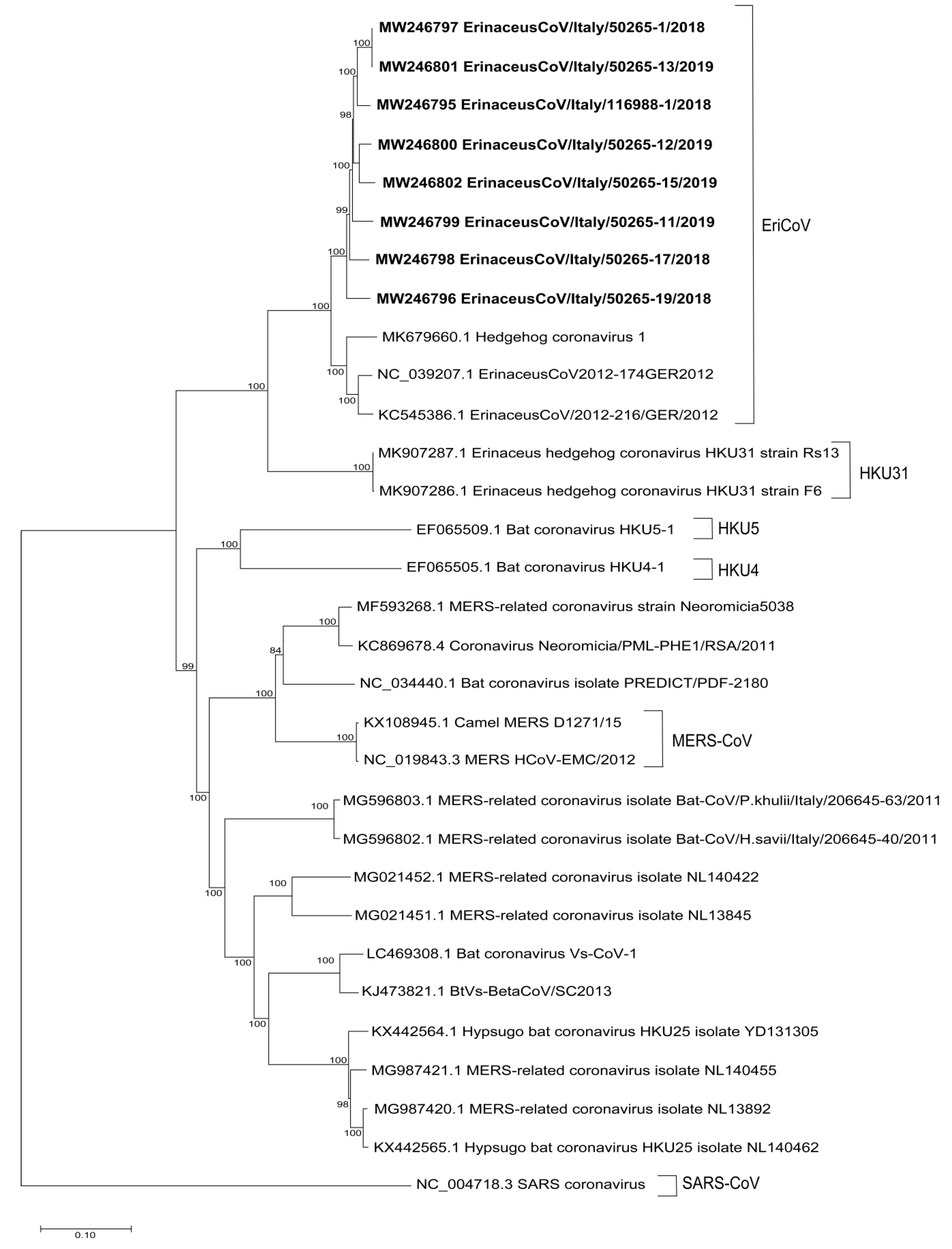
3.3. Phylogenetic Analysis of the Complete Genomes
3.4. Spike Protein Sequence Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, L.; Zhang, Y. Animal coronaviruses: A brief introduction. In Animal Coronaviruses; Springer Protocols Handbooks; Wang, L., Ed.; Humana Press: New York, NY, USA, 2016; pp. 3–11. [Google Scholar]
- Greenberg, S.B. Update on human rhinovirus and coronavirus infections. Semin. Respir. Crit. Care Med. 2016, 37, 555–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global epidemiology of bat coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Disease Outbreak News Update: Middle East Respiratory Syndrome Coronavirus (MERS-CoV). Available online: https://www.who.int/csr/don/02-jul-2020-mers-saudi-arabia/en/ (accessed on 16 November 2020).
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.Y.; Hu, B.; Shi, Z.L. Bat Coronaviruses. In Bats and Viruses: A New Frontier of Emerging Infectious Diseases; Wang, L.F., Cowled, C., Eds.; John Wiley Sons Inc.: Hoboken, NJ, USA, 2015. [Google Scholar]
- Beltz, L.A. Bats and Human Health: Ebola, SARS, Rabies and Beyond; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2018. [Google Scholar]
- Lau, S.K.P.; Zhang, L.; Luk, H.K.H.; Xiong, L.; Peng, X.; Li, K.S.M.; He, X.; Zhao, P.S.; Fan, R.Y.Y.; Wong, A.C.P.; et al. Receptor usage of a novel bat lineage c betacoronavirus reveals evolution of Middle East respiratory syndrome-related coronavirus spike proteins for human dipeptidyl peptidase 4 Binding. J. Infect. Dis. 2018, 218, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Onuma, M.; Cao, Y.; Hasegawa, M.; Kusakabe, S. A Close relationship of Chiroptera with Eulipotyphla (Core Insectivora) suggested by four mitochondrial genes. Zool. Sci. 2000, 17, 1327–1332. [Google Scholar] [CrossRef]
- Corman, V.M.; Kallies, R.; Philipps, H.; Göpner, G.; Müller, M.A.; Eckerle, I.; Brünink, S.; Drosten, C.; Drexler, J.F. Characterization of a novel betacoronavirus related to middle East respiratory syndrome coronavirus in European hedgehogs. J. Virol. 2014, 88, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Monchatre-Leroy, E.; Boué, F.; Boucher, J.M.; Renault, C.; Moutou, F.; Ar Gouilh, M.; Umhang, G. Identification of alpha and beta coronavirus in wildlife species in France: Bats, rodents, rabbits, and hedgehogs. Viruses 2017, 9, 364. [Google Scholar] [CrossRef] [Green Version]
- Saldanha, I.F.; Lawson, B.; Goharriz, H.; Fernandez, J.R.-R.; John, S.K.; Fooks, A.R.; Cunningham, A.A.; Johnson, N.; Horton, D.L. Extension of the known distribution of a novel clade C betacoronavirus in a wildlife host. Epidemiol. Infect. 2019, 147, e169. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.; Luk, H.; Wong, A.; Fan, R.; Lam, C.; Li, K.; Ahmed, S.S.; Chow, F.; Cai, J.P.; Zhu, X.; et al. Identification of a novel Betacoronavirus (Merbecovirus) in amur hedgehogs from China. Viruses 2019, 11, 980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delogu, M.; Cotti, C.; Lelli, D.; Sozzi, E.; Trogu, T.; Lavazza, A.; Garuti, G.; Castrucci, M.R.; Vaccari, G.; De Marco, M.A.; et al. Eco-virological preliminary study of potentially emerging pathogens in hedgehogs (Erinaceus europaeus) recovered at a wildlife treatment and rehabilitation center in northern Italy. Animals 2020, 10, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziebuhr, J.; Baric, R.S.; Baker, S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.; Haagmans, B.L.; Lauber, C.; Neuman, B.W.; Perlman, S.; et al. Create 12 New Species in the Family Coronaviridae. Taxonomic Proposal: 2015.003a-eS International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/ICTV/proposals/2015.003a-eS.A.v2.Coronaviridae_12sp.pdf (accessed on 18 November 2020).
- Douady, C.J.; Douzery, E.J.P. Hedgehogs, shrews, moles, and solenodons (Eulipotyphla). In The Timetree of Life; Hedges, S.B., Kumar, S., Eds.; Oxford University Press: New York, NY, USA, 2009; pp. 495–498. [Google Scholar]
- Tsagkogeorga, G.; Parker, J.; Stupka, E.; Cotton, J.A.; Rossiter, S.J. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr. Biol. 2013, 23, 2262–2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondinini, C.; Capasso, S. Erinaceus europaeus Italian IUCN Committee, Ministry of the Environment and Protection of the Territory and the Sea, Federation of Parks [Comitato Italiano IUCN, Ministero dell’Ambiente e della Tutela del Territorio e del Mare, Federparchi] 2013. Available online: http://www.iucn.it/scheda.php?id=-944215748 (accessed on 16 November 2020).
- Rondinini, C.; Capasso, S. Erinaceus roumanicus Italian IUCN Committee, Ministry of the Environment and Protection of the Territory and the Sea, Federation of Parks [Comitato Italiano IUCN, Ministero dell’Ambiente e della Tutela del Territorio e del Mare, Federparchi] 2013. Available online: http://www.iucn.it/scheda.php?id=1308809394 (accessed on 16 November 2020).
- G.I.R.C. Gruppo Italiano Ricerca Chirotteri, Specie Associazione Teriologica Italiana, [G-I.R.C. Italian Chiroptera Research Group, Species, Italian Mammal Society]. Available online: https://www.mammiferi.org/girc/specie/ (accessed on 16 November 2020).
- Moreno, A.; Lelli, D.; de Sabato, L.; Zaccaria, G.; Boni, A.; Sozzi, E.; Prosperi, A.; Lavazza, A.; Cella, E.; Castrucci, M.R.; et al. Detection and full genome characterization of two beta CoV viruses related to Middle East respiratory syndrome from bats in Italy. Virol. J. 2018, 15, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djikeng, A.; Halpin, R.; Kuzmickas, R.; Depasse, J.; Feldblyum, J.; Sengamalay, N.; Afonso, C.; Zhang, X.; Anderson, N.G.; Ghedin, E.; et al. Viral genome sequencing by random priming methods. BMC Genom. 2008, 9, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the newENDscript server. Nucl. Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Makino, S.; Keck, J.G.; Stohlman, S.A.; Lai, M.M. High-frequency RNA recombination of murine coronaviruses. J. Virol. 1986, 57, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Seyran, M.; Pizzol, D.; Adadi, P.; El-Aziz, T.M.A.; Hassan, S.S.; Soares, A.; Kandimalla, R.; Lundstrom, K.; Tambuwala, M.; Aljabali, A.A.A.; et al. Questions concerning the proximal origin of SARS-CoV-2. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Vaine, C.A.; Soberman, R.J. The CD200-CD200R1 inhibitory signaling pathway: Immune regulation and host-pathogen interactions. Adv. Immunol. 2014, 121, 191–211. [Google Scholar]
- Mousavinezhad-Moghaddam, M.; Amin, A.A.; Rafatpanah, H.; Rezaee, S.A. A new insight into viral proteins as Immunomodulatory therapeutic agents: KSHV vOX2 a homolog of human CD200 as a potent anti-inflammatory protein. Iran. J. Basic Med. Sci. 2016, 19, 2–13. [Google Scholar] [PubMed]
- Foster-Cuevas, M.; Wright, G.J.; Puklavec, M.J.; Brown, M.H.; Barclay, A.N. Human herpesvirus 8 K14 protein mimics CD200 in down- regulating macrophage activation through CD200 receptor. J. Virol. 2004, 78, 7667–7676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblum, M.D.; Olasz, E.B.; Yancey, K.B.; Woodliff, J.E.; Lazarova, Z.; Gerber, K.A.; Truitt, R.L. Expression of CD200 on epithelial cells of the murine hair follicle: A role in tissue-specific immune tolerance? J. Investig. Dermatol. 2004, 123, 880–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Stanford, M.; Liu, J.; Barrett, C.; Jiang, L.; Barclay, A.N.; McFadden, G. Inhibition of macrophage activation by the myxoma virus M141 protein (vCD200). J. Virol. 2009, 83, 9602–9607. [Google Scholar] [CrossRef] [Green Version]
- Foster-Cuevas, M.; Westerholt, T.; Ahmed, M.; Brown, M.H.; Barclay, A.N.; Voigt, S. Cytomegalovirus e127 protein interacts with the inhibitory CD200 receptor. J. Virol. 2011, 85, 6055–6059. [Google Scholar] [CrossRef] [Green Version]
- Rezaee, S.A.; Gracie, J.A.; McInnes, I.B.; Blackbourn, D.J. Inhibition of neutrophil function by the Kaposi’s sarcoma-associated herpesvirus vOX2 protein. AIDS 2005, 19, 1907–1910. [Google Scholar] [CrossRef]
- Shiratori, I.; Yamaguchi, M.; Suzukawa, M.; Yamamoto, K.; Lanier, L.L.; Saito, T.; Arase, H. Down-regulation of basophil function by human CD200 and human herpesvirus-8 CD200. J. Immunol. 2005, 175, 4441–4449. [Google Scholar] [CrossRef]
- Misstear, K.; Chanas, S.A.; Rezaee, S.A.; Colman, R.; Quinn, L.L.; Long, H.M.; Goodyear, O.; Lord, J.M.; Hislop, A.D.; Blackbourn, D.J. Suppression of antigen-specific T cell responses by the Kaposi’s sarcoma-associated herpesvirus viral OX2 protein and its cellular orthologue, CD200. J. Virol. 2012, 86, 6246–6257. [Google Scholar] [CrossRef] [Green Version]
- Karnam, G.; Rygiel, T.P.; Raaben, M.; Grinwis, G.C.M.; Coenjaerts, F.E.; Ressing, M.E.; Rottier, P.J.M.; de Haan, C.A.M.; Meyaard, M. CD200 receptor controls sex-specific TLR7 responses to viral infection. PLoS Pathog. 2012, 8, e1002710. [Google Scholar] [CrossRef]
- Ceribelli, A.; Motta, F.; De Santis, M.; Ansari, A.A.; Ridgway, W.M.; Gershwin, M.E.; Selmi, C. Recommendations for coronavirus infection in rheumatic diseases treated with biologic therapy. J. Autoimmun. 2020, 109, 102442. [Google Scholar] [CrossRef]
- Cameron, C.M.; Barrett, J.W.; Liu, L.; Lucas, A.R.; McFadden, G. Myxoma virus M141R expresses a viral CD200 (vOX-2) that is responsible for down-regulation of macrophage and T-cell activation in vivo. J. Virol. 2005, 79, 6052–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akkaya, M.; Kwong, L.S.; Akkaya, E.; Hatherley, D.; Barclay, A.N. Rabbit CD200R binds host CD200 but not CD200-like proteins from poxviruses. Virology 2016, 488, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Qi, J.; Yuan, Y.; Xuan, Y.; Han, P.; Wan, Y.; Ji, W.; Li, Y.; Wu, Y.; Wang, J.; et al. Bat origins of MERS-CoV supported by bat coronavirus HKU4 usage of human receptor CD26. Cell Host Microbe 2014, 16, 328–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Riley, P.Y.; Chomel, B.B. Hedgehog zoonoses. Emerg. Infect. Dis. 2005, 11, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Morris, P.A. A study of home range and movements in the hedgehog (Erinaceus europaeus). J. Zool. Lond. 1988, 214, 433–499. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Sample ID | Municipality (Province) 1 | Recovery Site 2 | Total NGS Reads | N° CoVs Reads | % of Genome Built | CD200 Ortholog 3 |
|---|---|---|---|---|---|---|
| 116988/1 | Budrio (BO) | R.A. | 1,521,953 | 13,567 | 100% | - |
| 50265/1 | Bentivoglio (BO) | R.A. | 1,246,304 | 13,094 | 100% | Y |
| 50265/3 | Sala Bolognese (BO) | U.A. | 1,531,553 | 45 | - | - |
| 50265/4 | Bologna (BO) | U.A. | 1,679,321 | 320 | - | Y |
| 50265/10 | Ferrara (FE) | R.A. | 628,517 | 135 | - | Y |
| 50265/11 | Ferrara (FE) | R.A. | 1,506,517 | 174,733 | 100% | Y |
| 50265/12 | Bentivoglio (BO) | U.A. | 979,563 | 78,706 | 100% | Y |
| 50265/13 | Granarolo (BO) | U.A. | 1,635,986 | 16,679 | 100% | Y |
| 50265/15 | Imola (BO) | U.A. | 1,046,044 | 85,354 | 100% | - |
| 50265/16 | Bentivoglio (BO) | U.A. | 1,406,884 | 89 | - | - |
| 50265/17 | Bologna (BO) | U.A. | 1,677,168 | 136,279 | 100% | Y |
| 50265/19 | Bologna (BO) | U.A. | 1,627,896 | 753,529 | 100% | Y |
| CD200 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | T33 | Q35 | N44 | T47 | N94 | S41 | P42 | Q57 | T95 | G97 | S98 | Q99 | K100 | F96 |
| 50265/1 | V52 | Q54 | N63 | T66 | N113 | G60 | P61 | Q76 | T114 | G116 | S117 | G118 | K119 | F115 |
| 50265/4 | T52 | Q54 | N63 | T66 | N112 | I60 | P61 | Q76 | T113 | - | S115 | I116 | K120 | Y114 |
| 50265/10 | T52 | Q54 | N63 | T66 | N113 | S60 | T61 | Q76 | T114 | G116 | S117 | G118 | S119 | F115 |
| 50265/11 | T52 | Q54 | N63 | T66 | N113 | G60 | P61 | Q76 | I114 | G116 | P117 | V118 | M119 | F115 |
| 50265/12 | T52 | T54 | N63 | T66 | - 1 | I60 | P61 | L76 | - | - | - | - | - | - |
| 50265/14 | V52 | Q54 | N63 | T66 | N113 | G60 | P61 | Q76 | T114 | G116 | S117 | G118 | K119 | F115 |
| 50265/17 | T52 | Q54 | N63 | T66 | N113 | V60 | P61 | Q76 | T114 | G116 | S117 | G118 | R119 | F115 |
| 50265/19 | T52 | Q54 | N63 | T66 | N113 | S60 | P61 | Q76 | T114 | - | S116 | R117 | K119 | F115 |
| Hedgehog | T60 | Q62 | N71 | T74 | N121 | S68 | P69 | Q84 | T122 | G124 | S125 | G126 | K127 | F123 |
| MERS-CoV 1 | Y 499 | N 501 | K 502 | L 506 | D 510 | R 511 | E 513 | P 515 | W 535 | E 536 | D 537 | D 539 | Y 540 | R 542 | W 553 | V 555 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Italian EriCoVs | Y 491 | S 493 | R 494 | - | G 500 | K 501 | - | P 502 | K 523 | K 524 | D 525 | - | - | G 530 | Y 538 | V 540 |
| HKU31 2 | Y 491 | S 493 | R 494 | G 497 | K 501 | P 502 | - | L 504 | N 524 | D 525 | V 526 | - | - | D 527 | F 538 | I 540 |
| HKU4 3 | Y 503 | S 505 | K 506 | L 510 | N 514 | Q 515 | E 518 | P 520 | S 540 | E 541 | D 542 | Q 544 | V 545 | K 547 | L 558 | I 560 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Sabato, L.; Di Bartolo, I.; De Marco, M.A.; Moreno, A.; Lelli, D.; Cotti, C.; Delogu, M.; Vaccari, G. Can Coronaviruses Steal Genes from the Host as Evidenced in Western European Hedgehogs by EriCoV Genetic Characterization? Viruses 2020, 12, 1471. https://doi.org/10.3390/v12121471
De Sabato L, Di Bartolo I, De Marco MA, Moreno A, Lelli D, Cotti C, Delogu M, Vaccari G. Can Coronaviruses Steal Genes from the Host as Evidenced in Western European Hedgehogs by EriCoV Genetic Characterization? Viruses. 2020; 12(12):1471. https://doi.org/10.3390/v12121471
Chicago/Turabian StyleDe Sabato, Luca, Ilaria Di Bartolo, Maria Alessandra De Marco, Ana Moreno, Davide Lelli, Claudia Cotti, Mauro Delogu, and Gabriele Vaccari. 2020. "Can Coronaviruses Steal Genes from the Host as Evidenced in Western European Hedgehogs by EriCoV Genetic Characterization?" Viruses 12, no. 12: 1471. https://doi.org/10.3390/v12121471