
Potentially Infectious Novel Hepatitis A Virus Strains Detected in Selected Treated Wastewater Discharge Sources, South Africa


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Processing
2.2. Initial HAV Screening by RT-qPCR
2.3. HAV Quantification by vPCR
2.4. Partial Genome Amplification
2.5. Cloning and Sequencing
2.6. Phylogenetic Analysis
3. Results
3.1. Screening
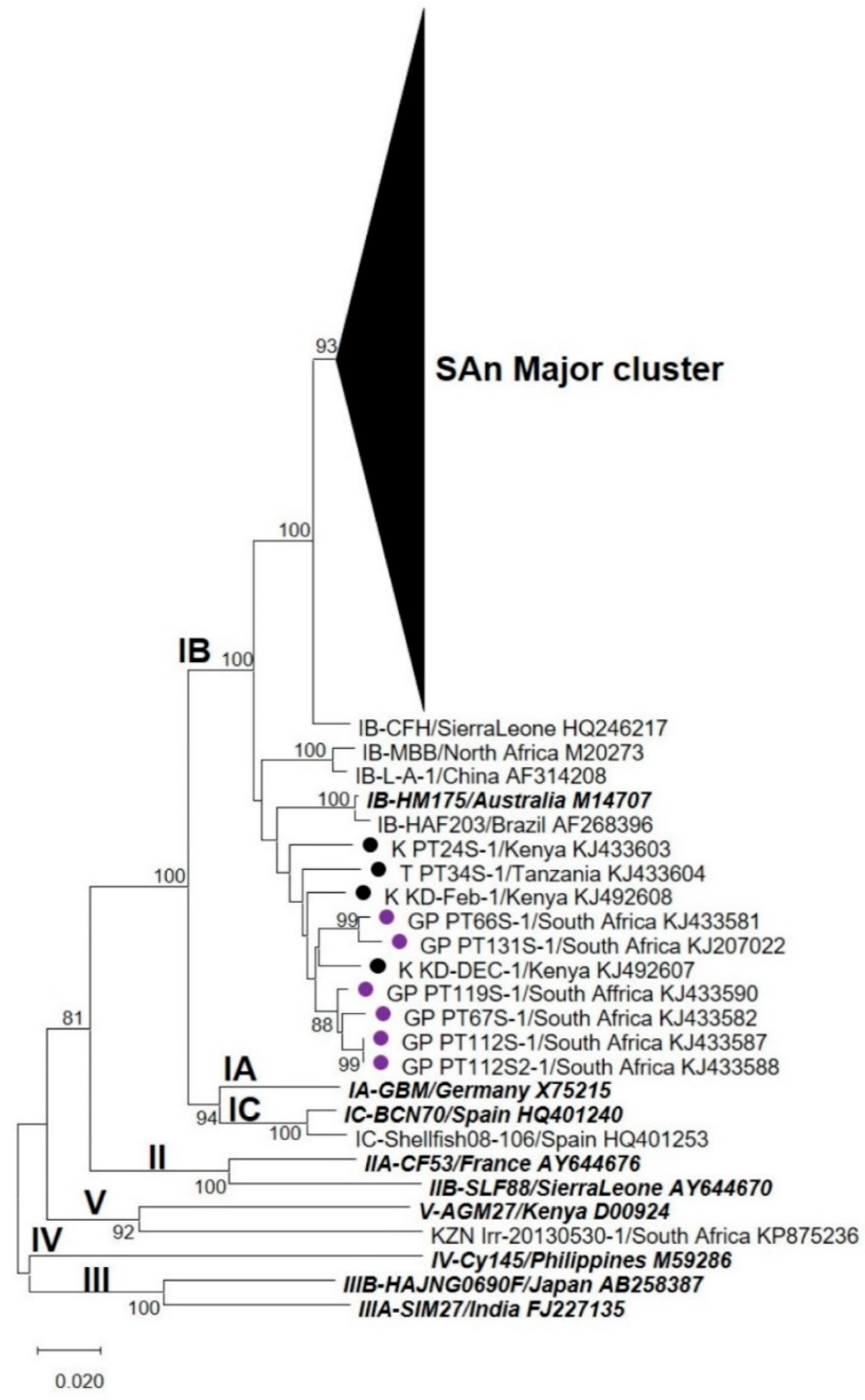
3.2. Nucleotide Sequence and Phylogenetic Analyses
3.3. Amino Acid Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Ethical Approval
References
- Lemon, S.M.; Ott, J.J.; Van Damme, P.; Shouval, D. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 2018, 68, 167–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, K.E. Viral pathogens in water: Occurrence, public health impact, and available control strategies. Curr. Opin. Virol. 2014, 4, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.; Santos, R. Enzymatic and viability RT-qPCR assays for evaluation of enterovirus, hepatitis A virus and norovirus inactivation: Implications for public health risk assessment. J. Appl. Microbiol. 2018, 124, 965–976. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Lázaro, D.; Cook, N.; Ruggeri, F.M.; Sellwood, J.; Nasser, A.; Nascimento, M.S.; D’Agostino, M.; Santos, R.; Saiz, J.C.; Rzeżutka, A.; et al. Virus hazards from food, water and other contaminated environments. FEMS Microbiol. Rev. 2012, 36, 786–814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollinger, F.B.; Emerson, S.U. Hepatitis A virus. In Fields Virology, 5th ed.; Knipe, D.M., Howley, P.M., Griffin, D.E., Lamb, R.A., Martin, M.A., Roizman, B., Straus, S.E., Eds.; Lippincott Williams and Wilkins, A Wolters Kluwer Business: Philadelphia, PA, USA, 2007; pp. 913–947. [Google Scholar]
- Vaughan, G.; Rossi, L.M.G.; Forbi, J.C.; de Paula, V.S.; Purdy, M.A.; Xia, G.; Khudyakov, Y.E. Hepatitis A virus: Host interactions, molecular epidemiology and evolution. Infect. Genet. Evol. 2014, 21, 227–243. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Sánchez, G.; Abbaszadegan, M.; Carducci, A.; Guix, S.; Le Guyader, F.S.; Netshikweta, R.; Pintó, R.M.; Van Der Poel, W.H.M.; Rutjes, S.; et al. Analytical methods for virus detection in water and food. Food Anal. Methods 2010, 4, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Hellmér, M.; Paxéus, N.; Magnius, L.; Enache, L.; Arnholm, B.; Johansson, A.; Bergström, T.; Norder, H. Detection of pathogenic viruses in sewage provided early warnings of hepatitis A virus and norovirus outbreaks. Appl. Environ. Microbiol. 2014, 80, 6771–6781. [Google Scholar] [CrossRef] [Green Version]
- International Committee on Taxonomy of Viruses (ICTV). Picornaviridae: The Family; 10th Report. 2019. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/positive-sense-rna-viruses/picornavirales/w/picornaviridae/709/genus-hepatovirus (accessed on 8 June 2020).
- Costa-Mattioli, M.; Cristina, J.; Romero, H.; Pérez-Bercof, R.; Casane, D.; Colina, R.; Garcia, L.; Vega, I.; Glikman, G.; Romanowsky, V.; et al. Molecular evolution of hepatitis A virus: A new classification based on the complete VP1 protein. J. Virol. 2002, 76, 9516–9525. [Google Scholar] [CrossRef] [Green Version]
- Nainan, O.V.; Brinton, M.A.; Margolis, S.H. Identification of amino acids located in the antibody binding sites of human hepatitis A virus. Virology 1992, 191, 984–987. [Google Scholar] [CrossRef]
- Ping, L.H.; Lemon, S.M. Antigenic structure of human hepatitis A virus defined by analysis of escape mutants selected against murine monoclonal antibodies. J. Virol. 1992, 66, 2208–2216. [Google Scholar] [CrossRef] [Green Version]
- Pintó, R.M.; D’Andrea, L.; Pérez-Rodriguez, F.J.; Costafreda, M.I.; Ribes, E.; Guix, S.; Bosch, A. Hepatitis A virus evolution and the potential emergence of new variants escaping the presently available vaccines. Future Microbiol. 2012, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhub, L.; Dang, M.; Hu, Z.; Gao, Q.; Yuan, S.; Sun, Y.; Zhang, B.; Ren, J.; Kotecha, A.; et al. Potent neutralization of hepatitis A virus reveals a receptor mimic mechanism and the receptor recognition site. Proc. Natl. Acad. Sci. USA 2017, 114, 770–775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aragonès, L.; Bosch, A.; Pintó, R.M. Hepatitis A virus mutant spectra under the selective pressure of monoclonal antibodies: Codon usage constraints limit capsid variability. J. Virol. 2008, 82, 1688–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.B. Molecular epidemiology of South African strains of hepatitis A virus:1982-1996. J. Med. Virol. 1997, 51, 273–279. [Google Scholar] [CrossRef]
- Taylor, M.B.; Cox, N.; Vrey, M.A.; Grabow, W.O.K. The occurrence of hepatitis A and astroviruses in selected river and dam waters in South Africa. Water Res. 2001, 35, 2653–2660. [Google Scholar] [CrossRef]
- Netshikweta, R. Optimisation and Assessment of Real-Time PCR Techniques for the Detection of Selected Food- and Waterborne Viruses. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2011. Available online: http://hdl.handle.net/2263/24883 (accessed on 24 March 2020).
- Chigor, V.N.; Okoh, A.I. Quantitative RT-PCR detection of hepatitis A virus, rotaviruses and enteroviruses in the Buffalo River and source water dams in the Eastern Cape province of South Africa. Int. J. Environ. Res. Public Health 2012, 9, 4017–4032. [Google Scholar] [CrossRef]
- Saïd, R.; Wolfaardt, M.; Taylor, M.B. Molecular characterisation of hepatitis A virus strains from water sources in South Africa. Water Sci. Technol. 2014, 69, 923–933. [Google Scholar] [CrossRef]
- Faber, M.S.; Stark, K.; Behnke, S.C.; Schreier, E.; Frank, C. Epidemiology of hepatitis A infection, Germany, 2007–2008. Emerg. Infect. Dis. 2009, 15, 1760–1768. [Google Scholar] [CrossRef] [Green Version]
- Saïd, R. Molecular Characterisation of Hepatitis A Virus Strains from Clinical and Environmental Sources in South Africa. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2014. [Google Scholar]
- Rachida, S.; Matsapola, P.N.; Wolfaardt, M.; Taylor, M.B. Genetic characterization of a novel hepatitis A virus strain in irrigation water in South Africa. J. Med. Virol. 2016, 88, 734–737. [Google Scholar] [CrossRef]
- Cangelosi, G.A.; Meschke, J.S. Dead or alive: Molecular assessment of microbial viability. Appl. Environ. Microbiol. 2014, 80, 5884–5891. [Google Scholar] [CrossRef] [Green Version]
- Hamza, I.A.; Bibby, K. Critical issues in application of molecular methods to environmental virology. J. Virol. Methods 2019, 266, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Moreno, L.; Aznar, R.; Sánchez, G. Application of viability PCR to discriminate the infectivity of hepatitis A virus in food samples. Int. J. Food Microbiol. 2015, 201, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Randazzo, W.; Piqueras, J.; Rodríguez-Díaz; Aznar, R.; Sánchez, G. Improving efficiency of viability-qPCR for selective detection of infectious HAV in food and water samples. J. Appl. Microbiol. 2018, 124, 958–964. [Google Scholar] [CrossRef] [PubMed]
- Enoch, A.; Hardie, D.R.; Hussey, G.D.; Kagina, B.M. Hepatitis A seroprevalence in Western Cape Province, South Africa: Are we in epidemiological transition? S. Afr. Med. J. 2019, 109, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haeri Mazanderani, A.; Motaze, N.V.; McCarthy, K.; Suchard, M.; du Plessis, N.M. Hepatitis A virus seroprevalence in South Africa—Estimates using routine laboratory data, 2005–2015. PLoS ONE 2019, 14, e0216033. [Google Scholar] [CrossRef]
- Kiulia, N.M.; Netshikweta, R.; Page, N.A.; van Zyl, W.B.; Kiraithe, M.M.; Nyachieo, A.; Mwenda, J.M.; Taylor, M.B. The detection of enteric viruses in selected urban and rural river water and sewage in Kenya, with special reference to rotaviruses. J. Appl. Microbiol. 2010, 109, 818–828. [Google Scholar] [CrossRef]
- Mans, J.; Netshikweta, R.; Magwalivha, M.; Van Zyl, W.B.; Taylor, M.B. Diverse norovirus genotypes identified in sewage-polluted river water in South Africa. Epidemiol. Infect. 2013, 141, 303–313. [Google Scholar] [CrossRef] [Green Version]
- ISO/TS 15216-2:2013(E). Microbiology of Food and Animal Feed—Horizontal Method for Determination of Hepatitis A Virus and Norovirus in Food Using Real-Time RT-PCR—Part 2: Methods for Qualitative Detection; ISO: Geneva, Switzerland, 2013. [Google Scholar]
- Mabasa, V.V. Characterisation and Histo-Blood Group Antigen Binding Profiles of South African Norovirus Genotype II Strains. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2017. [Google Scholar]
- Rachida, S. Genetic Assessment of Hepatitis A Virus Strains Detected in Selected Water Sources in Gauteng, South Africa. Ph.D. Thesis, University of Pretoria, Pretoria, South Africa, 2020. [Google Scholar]
- Nainan, O.; Xia, G.; Vaughan, G.; Margolis, H.S. Diagnosis of hepatitis A virus infection: A molecular approach. Clin. Microbiol. Rev. 2006, 19, 63–79. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Stats, S.A. Statistical Release: Mid-Year Population Estimates. 2019. Available online: http://www.statssa.gov.za/publications/P0302/P03022019.pdf (accessed on 3 May 2020).
- Sabrià, A.; Gregori, J.; Garcia-Cehic, D.; Guix, S.; Pumarola, T.; Manzanares-Laya, S.; Caylà, J.A.; Bosch, A.; Quer, J.; Pintó, R.M. Evidence for positive selection of hepatitis A virus antigenic variants in vaccinated men-having-sex-with men patients: Implications for immunization policies. EBioMedicine 2019, 39, 348–357. [Google Scholar] [CrossRef]
- World Health Organization. WHO position paper on hepatitis A vaccines—July 2012. WER 2012, 87, 261–276. Available online: https://www.who.int/wer/2012/wer8728_29.pdf?ua=1 (accessed on 30 March 2020).
- Pérez-Sautu, U.; Costafreda, M.I.; Caylà, J.; Tortajada, C.; Lite, J.; Bosch, A.; Pintó, R.M. Hepatitis A virus vaccine escape variants and potential new serotype emergence. Emerg. Infect. Dis. 2011, 17, 734–737. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genomic Region | PCR | Primer’s Name | Nucleotide Sequence (5′–3′) |
|---|---|---|---|
| VP1 | First round | HAV1 (Forward) | gTT TTg CTC CTC TTT ATC ATg CTA Tg |
| HAV2 (Reverse) | AgT CAC ACC TCT CCA ggA AAA CTT | ||
| Second round | 2172P (Forward) | gCT CCT CTT TAT CAT gCT ATg gAT | |
| 3125N (Reverse) | CCT gCA TTC TAT ATg ACT CT | ||
| VP1/P2B | First round | 2870P (Forward) | gAC AgA TTC TAC ATT Tgg ATT ggT |
| 3381N (Reverse) | CCA TTT CAA gAg TCC ACA CAC T | ||
| Second round | 2896P (Forward) | CTA TTC AgA TTg CAA ATA CAA T | |
| 3289N (Reverse) | AAC TTC ATT ATT TCA TgC TCC T |
| Genotype | Strain Name | Geographical Location | GenBank Accession Number | Genomic Region | Source of Characterisation |
|---|---|---|---|---|---|
| IA | GBM | Germany | X75215 | VP1, VP1/P2B | Clinical |
| IB | HM175 | Australia | M14707 | VP1, VP1/P2B | Clinical |
| IB | CFH-HAV | Sierra Leone | HQ246217 | VP1, VP1/P2B | Clinical |
| IB | MBB | North Africa | M20273 | VP1, VP1/P2B | Clinical |
| IB | HAF203 | Brazil | AF268396 | VP1, VP1/P2B | Unknown |
| IB | Banglane2000 | Thailand | LC128713 | VP1, VP1/P2B | Unknown |
| IB | HAV/Egy/BI-11/2015 | Egypt | KX228694 | VP1/P2B | Sewage |
| IB | L-A-1 | China | AF314208 | VP1, VP1/P2B | Unknown |
| IB | GP_66S-1, 67S-1, 106S-1, 109S-1, 110S-1, 111S-1, 112S-1, 112S2-1, 116S-1, 119S-1, 122S-1, 134S-1 | Gauteng, SA | KJ433581 to KJ433592 | VP1 | Clinical |
| IB | GP_131S-1 | Gauteng, SA | KJ207022 | VP1 | Clinical |
| IB | GP_PT66S, 67S, 106S, 109S, 110S, 111S, 112S, 112S2, 115S, 116S, 119S, 122S, 130S, 131S, 134S | Gauteng, SA | KJ492621 to KJ492635 | VP1/P2B | Clinical |
| IB | GP_RV2-20120618-col1 | Gauteng, SA | KJ492595 | VP1 | Surface water |
| IB | GP_11.1085-col7, 11.1085-col8, 11.1145-col1, 11.1145-col2 | Gauteng, SA | KJ492598 to KJ492601 | VP1 | Wastewater discharge |
| IB | GP_RV2-20121112, RV2-20130318, K19-20130225 | Gauteng, SA | KJ492654 to KJ492656 | VP1/P2B | Surface water |
| IB | GP_11.1051, 11.1145, 11.1147, 11.1085 | Gauteng, SA | KJ492657 to KJ492660 | VP1/P2B | Wastewater discharge |
| IB | SZ_PT126S | Swaziland | KJ492643 | VP1/P2B | Clinical |
| IB | SZ_PT29S-1, 126S-1 | Swaziland | KJ433605 to KJ433606 | VP1 | Clinical |
| IB | K_KD-FEB, KD-NOV, KD-DEC, MB-JUL, NR-AUG | Kenya | KJ492671 to KJ492675 | VP1/P2B | Surface water |
| IB | K_PT24S-1 | Kenya | KJ433603 | VP1 | Clinical |
| IB | K_KD-DEC-1, KD-Feb-1 | Kenya | KJ492607, KJ492608 | VP1 | Surface water |
| IB | T_PT34S-1 | Tanzania | KJ433604 | VP1 | Clinical |
| IC | BCN70 | Spain | HQ401240 | VP1 | Clinical |
| IC | Shellfish08-106 | Spain | HQ401253 | VP1 | Food |
| IIA | CF53/Berne | France | AY644676 | VP1, VP1/P2B | Clinical |
| IIB | SLF88 | Sierra Leone | AY644670 | VP1, VP1/P2B | Clinical |
| IIIA | SIM27 | India | FJ227135 | VP1, VP1/P2B | Clinical |
| IIIB | HA-JNG06-90F | Japan | AB258387 | VP1, VP1/P2B | Clinical |
| IV | Cy145 | Philippines | M59286 | VP1, VP1/P2B | Simian |
| V | AGM-27 | Kenya | D00924 | VP1, VP1/P2B | Simian |
| V | KZN_Irr-20130530-1 | KwaZulu-Natal, SA | KP875236 | VP1 | Surface water |
| V | KZN_Irr-20130530-2B | KwaZulu-Natal, SA | KP875241 | VP1/P2B | Surface water |
| Initial Screening | After Viability Treatment | ||
|---|---|---|---|
| Sewage | WWTP 1 | 10/11 | 8/9 |
| WWTP 2 | 11/12 | 8/8 | |
| WWTP 3 | 5/12 | 1/5 | |
| WWTP 4 | 7/7 | 6/7 | |
| WWTP 5 | 10/12 | 7/8 | |
| Total | 43/54 (80%) | 30/37 (81%) | |
| Treated wastewater discharge | WWTP 1 | 10/11 | 9/10 |
| WWTP 2 | 12/12 | 11/11 | |
| WWTP 3 | 3/11 | 0/3 | |
| WWTP 4 | 7/7 | 7/7 | |
| WWTP 5 | 11/11 | 11/11 | |
| Total | 43/52 (83%) | 38/42 (90%) | |
| Dam water | 0/12 (0%) | − | |
| Name of Strain | a,b 102 | a,b 171 | a,b 176 | c 217 | b 221 | ||
|---|---|---|---|---|---|---|---|
| WWTP 1 | d R | DR4-1-2 MT721459 | - | - | - | G217C | - |
| R | DR5-1-F MT721466 | - | - | - | - | K221E | |
| WWTP 2 | R | OR4-1-E MT721551 | - | V171E | - | - | - |
| e E | OE7-1-302 MT721608 | - | - | - | - | K221R | |
| WWTP 4 | R | FR6-1-B MT380571 | - | - | A176T | - | - |
| R | FR1-1-3 MT380587 | S102P | - | - | - | - | |
| E | FE8-1-A MT380605 | - | - | - | - | K221E | |
| WWTP 5 | R | VLR4-1-A MT721649 | - | - | - | - | K221E |
| R | VLR8-1-F MT721671 | - | - | - | - | K221R | |
| E | VLE10-1-C MT721704 | - | - | - | - | K221E | |
| E | VLE11-1-D MT721712 | - | - | - | G217A | - | |
| In-Frame Deletion | Amino Acid Changes | ||||
|---|---|---|---|---|---|
| Before | After | Before | After | ||
| VP1 | FR1 | Two a aa deletion: position 241 to 242; FR1-1-2 (MT380586) and FR1-1-3 (MT380587) (Figure 6D) | Two a aa deletion: position 241 to 242; FR1-1v-a3 (MT380634) (Figure 6D) | S102P, R298K | S102P, G217D, R298K |
| FE8 | 105 aa deletion: position 145 to 249; FE8-1-F (MT380609) on Figure 6B–D | − | V251T, V251I | V251I | |
| VP1/P2B | FR1 | − | − | R63K, R71S | R63K, C70S, R71S, M104I |
| FE8 | − | − | R63K, C70S, R71S, M104I | C70S, M104I | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachida, S.; Taylor, M.B. Potentially Infectious Novel Hepatitis A Virus Strains Detected in Selected Treated Wastewater Discharge Sources, South Africa. Viruses 2020, 12, 1468. https://doi.org/10.3390/v12121468
Rachida S, Taylor MB. Potentially Infectious Novel Hepatitis A Virus Strains Detected in Selected Treated Wastewater Discharge Sources, South Africa. Viruses. 2020; 12(12):1468. https://doi.org/10.3390/v12121468
Chicago/Turabian StyleRachida, Saïd, and Maureen Beatrice Taylor. 2020. "Potentially Infectious Novel Hepatitis A Virus Strains Detected in Selected Treated Wastewater Discharge Sources, South Africa" Viruses 12, no. 12: 1468. https://doi.org/10.3390/v12121468