
Exposure Risk of Chronic Wasting Disease in Humans



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Risk of Exposure
3. PRNP Polymorphisms and Susceptibility
3.1. Humans
3.2. Elk
3.3. Mule Deer
3.4. White-Tailed Deer
3.5. Moose
3.6. Reindeer
4. The Role of Strains and Adaptation
4.1. Elk
4.2. Deer
4.3. Moose
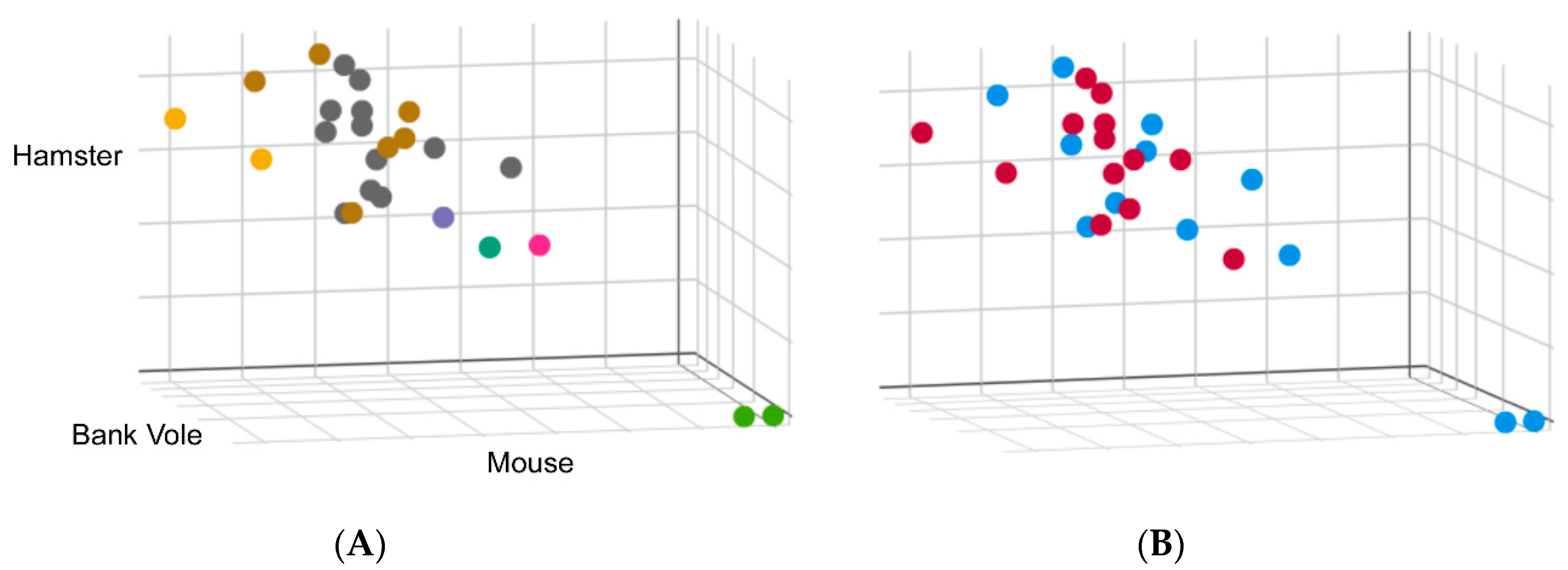
5. Crossing the Species Barrier
5.1. In Vitro Studies
5.2. In Vivo Experiments
5.3. Human Data
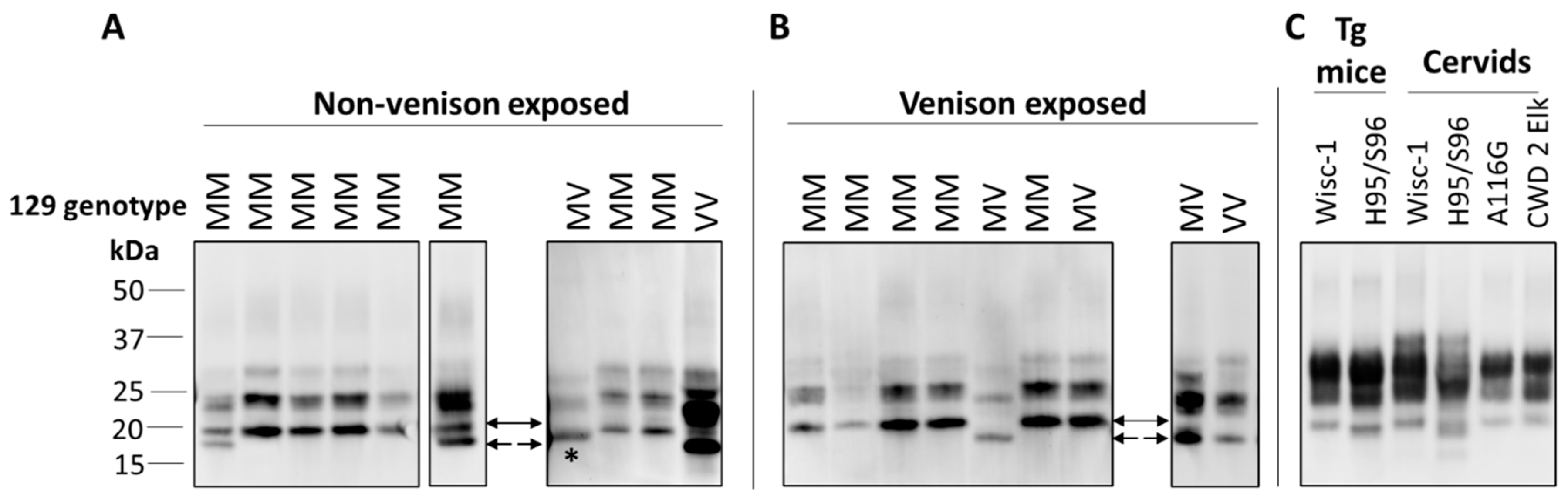
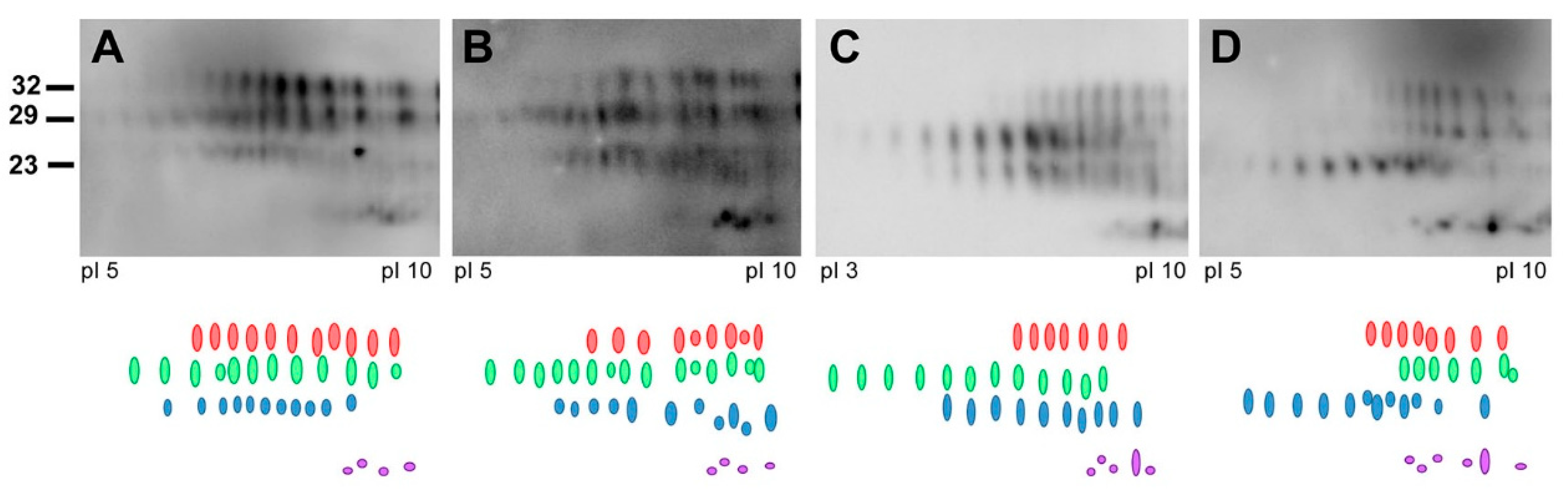
6. Detecting CWD in Humans; the Role of Surveillance and Strain Typing
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [Green Version]
- Gambetti, P.; Cali, I.; Notari, S.; Kong, Q.; Zou, W.Q.; Surewicz, W.K. Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol. 2011, 121, 79–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puoti, G.; Bizzi, A.; Forloni, G.; Safar, J.G.; Tagliavini, F.; Gambetti, P. Sporadic human prion diseases: Molecular insights and diagnosis. Lancet Neurol. 2012, 11, 618–628. [Google Scholar] [CrossRef]
- Schwartz, M. How the Cows Turned Mad by Maxime Schwartz—Paperback—University of California Press. Available online: https://www.ucpress.edu/book/9780520243378/how-the-cows-turned-mad (accessed on 5 September 2020).
- Anderson, R.M.; Donnelly, C.A.; Ferguson, N.M.; Woolhouse, M.E.J.; Watt, C.J.; Udy, H.J.; MaWhinney, S.; Dunstan, S.P.; Southwood, T.R.E.; Wilesmith, J.W.; et al. Transmission dynamics and epidemiology of BSE in British cattle. Nature 1996, 382, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Mathiason, C.K. Scrapie, CWD, and Transmissible Mink Encephalopathy. In Progress in Molecular Biology and Translational Science; Elsevier B.V.: Amsterdam, The Netherlands, 2017; Volume 150, pp. 267–292. [Google Scholar]
- Sigurdson, C.J.; Miller, M.W. Other animal prion diseases. Br. Med. Bull. 2003, 66, 199–212. [Google Scholar] [CrossRef]
- Babelhadj, B.; Di Bari, M.A.; Pirisinu, L.; Chiappini, B.; Gaouar, S.B.S.; Riccardi, G.; Marcon, S.; Agrimi, U.; Nonno, R.; Vaccari, G. Prion disease in dromedary camels, Algeria. Emerg. Infect. Dis. 2018, 24, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.S.; Young, S. Chronic wasting disease of captive mule deer: A spongiform encephalopathy. J. Wildl. Dis. 1980, 16, 89–98. [Google Scholar] [CrossRef] [Green Version]
- Wilesmith, J.W.; Ryan, J.B.; Hueston, W.D.; Hoinville, L.J. Bovine spongiform encephalopathy: Epidemiological features 1985 to 1990. Vet. Rec. 1992, 130, 90–94. [Google Scholar] [CrossRef]
- Houston, F.; Andréoletti, O. Animal prion diseases: The risks to human health. Brain Pathol. 2019, 29, 248–262. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.S.; Young, S. Spongiform encephalopathy of Rocky Mountain elk. J. Wildl. Dis. 1982, 18, 465–471. [Google Scholar] [CrossRef]
- Johnson, C.; Johnson, J.; Clayton, M.; McKenzie, D.; Aiken, J. Prion protein gene heterogeneity in free-ranging white-tailed deer within the chronic wasting disease affected region of Wisconsin. J. Wildl. Dis. 2003, 39, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Baeten, L.A.; Powers, B.E.; Jewell, J.E.; Spraker, T.R.; Miller, M.W. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). J. Wildl. Dis. 2007, 43, 309–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benestad, S.L.; Mitchell, G.; Simmons, M.; Ytrehus, B.; Vikøren, T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet. Res. 2016, 47. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L.; Beekes, M. Chronic wasting disease: Fingerprinting the culprit in risk assessments. Prion 2012, 6, 17–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakudo, A. Chronic wasting disease: Current assessment of transmissibility. Curr. Issues Mol. Biol. 2019, 36, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gough, K.C.; Maddison, B.C. Prion transmission: Prion excretion and occurrence in the environment. Prion 2010, 4, 275–282. [Google Scholar] [CrossRef] [Green Version]
- Osterholm, M.T.; Anderson, C.J.; Zabel, M.D.; Scheftel, J.M.; Moore, K.A.; Appleby, B.S. Chronic wasting disease in cervids: Implications for prion transmission to humans and other animal species. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Uehlinger, F.D.; Johnston, A.C.; Bollinger, T.K.; Waldner, C.L. Systematic review of management strategies to control chronic wasting disease in wild deer populations in North America. BMC Vet. Res. 2016, 12. [Google Scholar] [CrossRef]
- Gilch, S.; Chitoor, N.; Taguchi, Y.; Stuart, M.; Jewell, J.E.; Schätzl, H.M. Chronic wasting disease. Top. Curr. Chem. 2011, 305, 51–78. [Google Scholar] [CrossRef]
- Kurt, T.D.; Sigurdson, C.J. Cross-species transmission of CWD prions. Prion 2016, 10, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.A.; Vorberg, I.; Priola, S.A. Species barriers in prion diseases—Brief review. Arch. Virol. Suppl. 2005, 19, 187–202. [Google Scholar]
- Collinge, J. Prion strain mutation and selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.W.; Williams, E.S. Chronic Wasting Disease of Cervids. Curr. Top. Microbiol. Immunol. 2004, 284, 193–214. [Google Scholar] [PubMed]
- Williams, E.S. Chronic wasting disease. Vet. Pathol. 2005, 42, 530–549. [Google Scholar] [CrossRef] [PubMed]
- Rivera, N.A.; Brandt, A.L.; Novakofski, J.E.; Mateus-Pinilla, N.E. Chronic Wasting Disease In Cervids: Prevalence, Impact And Management Strategies. Vet. Med. Res. Rep. 2019, 10, 123–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koutsoumanis, K.; Allende, A.; Alvarez-Ordoňez, A.; Bolton, D.; Bover-Cid, S.; Chemaly, M.; Davies, R.; De Cesare, A.; Herman, L.; Hilbert, F.; et al. Update on chronic wasting disease (CWD) III. EFSA J. 2019, 17. [Google Scholar] [CrossRef] [Green Version]
- Daus, M.L.; Breyer, J.; Wagenfuehr, K.; Wemheuer, W.M.; Thomzig, A.; Schulz-Schaeffer, W.J.; Beekes, M. Presence and seeding activity of pathological prion protein (PrPTSE) in skeletal muscles of white-tailed deer infected with chronic wasting disease. PLoS ONE 2011, 6, e18345. [Google Scholar] [CrossRef] [Green Version]
- Silva, C.J.; Erickson-Beltran, M.L.; Duque Velásquez, C.; Aiken, J.M.; Mckenzie, D. A General Mass Spectrometry-Based Method of Quantitating Prion Polymorphisms from Heterozygous Chronic Wasting Disease-Infected Cervids. Anal. Chem. 2020, 92, 1276–1284. [Google Scholar] [CrossRef]
- Haley, N.J.; Hoover, E.A. Chronic wasting disease of cervids: Current knowledge and future perspectives. Annu. Rev. Anim. Biosci. 2015, 3, 305–325. [Google Scholar] [CrossRef]
- Carlson, C.M.; Hopkins, M.C.; Nguyen, N.T.; Richards, B.J.; Walsh, D.P.; Walter, W.D. Chronic wasting disease—Status, science, and management support by the U.S. Geological Survey. Open-File Rep. 2018. [Google Scholar] [CrossRef] [Green Version]
- Benestad, S.L.; Telling, G.C. Chronic wasting disease: An evolving prion disease of cervids. In Handbook of Clinical Neurology; Elsevier B.V.: Amsterdam, The Netherlands, 2018; Volume 153, pp. 135–151. [Google Scholar]
- Robinson, S.J.; Samuel, M.D.; O’Rourke, K.I.; Johnson, C.J. The role of genetics in chronic wasting disease of North American cervids. Prion 2012, 6, 153–162. [Google Scholar] [CrossRef] [Green Version]
- Palmer, M.S.; Dryden, A.J.; Hughes, J.T.; Collinge, J. Homozygous prion protein genotype predisposes to sporadic Creutzfeldt-Jakob disease. Nature 1991, 352, 340–342. [Google Scholar] [CrossRef]
- Mead, S.; Poulter, M.; Uphill, J.; Beck, J.; Whitfield, J.; Webb, T.E.; Campbell, T.; Adamson, G.; Deriziotis, P.; Tabrizi, S.J.; et al. Genetic risk factors for variant Creutzfeldt-Jakob disease: A genome-wide association study. Lancet Neurol. 2009, 8, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.; Jaunmuktane, Z.; Joiner, S.; Campbell, T.; Morgan, C.; Wakerley, B.; Golestani, F.; Rudge, P.; Mead, S.; Jäger, H.R.; et al. Variant Creutzfeldt-Jakob disease in a patient with heterozygosity at PRNP codon 129. N. Engl. J. Med. 2017, 376, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Parchi, P.; Giese, A.; Capellari, S.; Brown, P.; Schulz-Schaeffer, W.; Windl, O.; Zerr, I.; Budka, H.; Kopp, N.; Piccardo, P.; et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann. Neurol. 1999, 46, 224–233. [Google Scholar] [CrossRef]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial CJD: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Rourke, K.I.; Besser, T.E.; Miller, M.W.; Cline, T.F.; Spraker, T.R.; Jenny, A.L.; Wild, M.A.; Zebarth, G.L.; Williams, E.S. PrP genotypes of captive and free-ranging Rocky Mountain elk (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 1999, 80, 2765–2769. [Google Scholar] [CrossRef]
- Spraker, T.R.; Balachandran, A.; Zhuang, D.; O’Rourke, K.I. Variable patterns of distribution of PrPCWD in the obex and cranial lymphoid tissues of Rocky Mountain elk (Cervus elaphus nelsoni) with subclinical chronic wasting disease. Vet. Rec. 2004, 155, 295–302. [Google Scholar] [CrossRef]
- Hamir, A.N.; Gidlewski, T.; Spraker, T.R.; Miller, J.M.; Creekmore, L.; Crocheck, M.; Cline, T.; O’Rourke, K.I. Preliminary observations of genetic susceptibility of elk (Cervus elaphus nelsoni) to chronic wasting disease by experimental oral inoculation. J. Vet. Diagnostic Investig. 2006, 18, 110–114. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, K.I.; Spraker, T.R.; Zhuang, D.; Greenlee, J.J.; Gidlewski, T.E.; Hamir, A.N. Elk with a long incubation prion disease phenotype have a unique PrPd profile. Neuroreport 2007, 18, 1935–1938. [Google Scholar] [CrossRef]
- Perucchini, M.; Griffin, K.; Miller, M.W.; Goldmann, W. PrP genotypes of free-ranging wapiti (Cervus elaphus nelsoni) with chronic wasting disease. J. Gen. Virol. 2008, 89, 1324–1328. [Google Scholar] [CrossRef]
- Jewell, J.E.; Conner, M.M.; Wolfe, L.L.; Miller, M.W.; Williams, E.S. Low frequency of PrP genotype 225SF among free-ranging mule deer (Odocoileus hemionus) with chronic wasting disease. J. Gen. Virol. 2005, 86, 2127–2134. [Google Scholar] [CrossRef]
- Wilson, G.A.; Nakada, S.M.; Bollinger, T.K.; Pybus, M.J.; Merrill, E.H.; Coltman, D.W. Polymorphisms at the PRNP gene influence susceptibility to chronic wasting disease in two species of deer (Odocoileus Spp.) in Western Canada. J. Toxicol. Environ. Heal. Part A Curr. Issues 2009, 72, 1025–1029. [Google Scholar] [CrossRef] [PubMed]
- Duque Velásquez, C.; Kim, C.; Herbst, A.; Daude, N.; Garza, M.C.; Wille, H.; Aiken, J.; McKenzie, D. Deer Prion Proteins Modulate the Emergence and Adaptation of Chronic Wasting Disease Strains. J. Virol. 2015, 89, 12362–12373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velásquez, C.D.; Kim, C.; Haldiman, T.; Kim, C.; Herbst, A.; Aiken, J.; Safar, J.G.; McKenzie, D. Chronic wasting disease (CWD) prion strains evolve via adaptive diversification of conformers in hosts expressing prion protein polymorphisms. J. Biol. Chem. 2020, 295, 4985–5001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.J.; Herbst, A.; Duque-Velasquez, C.; Vanderloo, J.P.; Bochsler, P.; Chappell, R.; McKenzie, D. Prion protein polymorphisms affect chronic wasting disease progression. PLoS ONE 2011, 6, e17450. [Google Scholar] [CrossRef] [PubMed]
- Race, B.; Meade-White, K.; Miller, M.W.; Fox, K.A.; Chesebro, B. In Vivo Comparison of Chronic Wasting Disease Infectivity from Deer with Variation at Prion Protein Residue 96. J. Virol. 2011, 85, 9235–9238. [Google Scholar] [CrossRef] [Green Version]
- Haley, N.J.; Merrett, K.; Stein, A.B.; Simpson, D.; Carlson, A.; Mitchell, G.; Staskevicius, A.; Nichols, T.; Lehmkuhl, A.D.; Thomsen, B.V. Estimating relative CWD susceptibility and disease progression in farmed white-tailed deer with rare PRNP alleles. PLoS ONE 2019, 14, e0224342. [Google Scholar] [CrossRef] [Green Version]
- Wik, L.; Mikko, S.; Klingeborn, M.; Stéen, M.; Simonsson, M.; Linné, T. Polymorphisms and variants in the prion protein sequence of European moose (Alces alces), reindeer (Rangifer tarandus), roe deer (Capreolus capreolus) and fallow deer (Dama dama) in Scandinavia. Prion 2012, 6, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Kreeger, T.J.; Montgomery, D.L.; Jewell, J.E.; Schultz, W.; Williams, E.S. Oral transmission of chronic wasting disease in captive Shira’s moose. J. Wildl. Dis. 2006, 42, 640–645. [Google Scholar] [CrossRef] [Green Version]
- Pirisinu, L.; Tran, L.; Chiappini, B.; Vanni, I.; Di Bari, M.A.; Vaccari, G.; Vikøren, T.; Madslien, K.I.; Våge, J.; Spraker, T.; et al. Novel type of chronic wasting disease detected in moose (Alces alces), Norway. Emerg. Infect. Dis. 2018, 24, 2210–2218. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, G.B.; Sigurdson, C.J.; O’Rourke, K.I.; Algire, J.; Harrington, N.P.; Walther, I.; Spraker, T.R.; Balachandran, A. Experimental oral transmission of chronic wasting disease to reindeer (Rangifer tarandus tarandus). PLoS ONE 2012, 7, e39055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo Moore, S.; Kunkle, R.; West Greenlee, M.H.; Nicholson, E.; Richt, J.; Hamir, A.; Ray Waters, W.; Greenlee, J. Horizontal transmission of chronic wasting disease in Reindeer. Emerg. Infect. Dis. 2016, 22, 2142–2145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, M.T.; Will, R.G.; Manson, J.C. Defining sporadic Creutzfeldt-Jakob disease strains and their transmission properties. Proc. Natl. Acad. Sci. USA 2010, 107, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamgüney, G.; Miller, M.W.; Giles, K.; Lemus, A.; Glidden, D.V.; DeArmond, S.J.; Prusiner, S.B. Transmission of scrapie and sheep-passaged bovine spongiform encephalopathy prions to transgenic mice expressing elk prion protein. J. Gen. Virol. 2009, 90, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, J.; Tatum, T.; Hwang, S.; Vrentas, C.; West Greenlee, M.H.; Kong, Q.; Nicholson, E.; Greenlee, J. Novel Strain of the Chronic Wasting Disease Agent Isolated From Experimentally Inoculated Elk With LL132 Prion Protein. Sci. Rep. 2020, 10, 3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escobar, L.E.; Pritzkow, S.; Winter, S.N.; Grear, D.A.; Kirchgessner, M.S.; Dominguez-Villegas, E.; Machado, G.; Townsend Peterson, A.; Soto, C. The ecology of chronic wasting disease in wildlife. Biol. Rev. 2020, 95, 393–408. [Google Scholar] [CrossRef]
- Kurt, T.D.; Telling, G.C.; Zabel, M.D.; Hoover, E.A. Trans-species amplification of PrPCWD and correlation with rigid loop 170N. Virology 2009, 387, 235–243. [Google Scholar] [CrossRef] [Green Version]
- Barria, M.A.; Telling, G.C.; Gambetti, P.; Mastrianni, J.A.; Soto, C. Generation of a new form of human PrPScin vitro by interspecies transmission from cervid prions. J. Biol. Chem. 2011, 286, 7490–7495. [Google Scholar] [CrossRef] [Green Version]
- Barria, M.A.; Libori, A.; Mitchell, G.; Head, M.W. Susceptibility of human Prion protein to conversion by chronic wasting disease Prions. Emerg. Infect. Dis. 2018, 24, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Davenport, K.A.; Henderson, D.M.; Bian, J.; Telling, G.C.; Mathiason, C.K.; Hoover, E.A. Insights into Chronic Wasting Disease and Bovine Spongiform Encephalopathy Species Barriers by Use of Real-Time Conversion. J. Virol. 2015, 89, 9524–9531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandberg, M.K.; Al-Doujaily, H.; Sigurdson, C.J.; Glatzel, M.; O’Malley, C.; Powell, C.; Asante, E.A.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.F.; et al. Chronic wasting disease prions are not transmissible to transgenic mice overexpressing human prion protein. J. Gen. Virol. 2010, 91, 2651–2657. [Google Scholar] [CrossRef] [PubMed]
- Kong, Q.; Huang, S.; Zou, W.; Vanegas, D.; Wang, M.; Wu, D.; Yuan, J.; Zheng, M.; Bai, H.; Deng, H.; et al. Chronic wasting disease of elk: Transmissibility to humans examined by transgenic mouse models. J. Neurosci. 2005, 25, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Marsh, R.F.; Kincaid, A.E.; Bessen, R.A.; Bartz, J.C. Interspecies Transmission of Chronic Wasting Disease Prions to Squirrel Monkeys (Saimiri sciureus). J. Virol. 2005, 79, 13794–13796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Race, B.; Meade-White, K.D.; Miller, M.W.; Barbian, K.D.; Rubenstein, R.; LaFauci, G.; Cervenakova, L.; Favara, C.; Gardner, D.; Long, D.; et al. Susceptibilities of nonhuman primates to chronic wasting disease. Emerg. Infect. Dis. 2009, 15, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Hamir, A.N.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; O’Rourke, K.I.; Williams, E.S.; Miller, M.W.; Stack, M.J.; Chaplin, M.J.; Richt, J.A. Experimental transmission of chronic wasting disease agent from mule deer to cattle by the intracerebral route. J. Vet. Diagnostic Investig. 2005, 17, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Hamir, A.N.; Kunkle, R.A.; Cutlip, R.C.; Miller, J.M.; Williams, E.S.; Richt, J.A. Transmission of chronic wasting disease of mule deer to Suffolk sheep following intracerebral inoculation. J. Vet. Diagn. Investig. 2006, 18, 558–565. [Google Scholar] [CrossRef] [Green Version]
- Race, B.; Williams, K.; Orrú, C.D.; Hughson, A.G.; Lubke, L.; Chesebro, B. Lack of Transmission of Chronic Wasting Disease to Cynomolgus Macaques. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Olszowy, K.M.; Lavelle, J.; Rachfal, K.; Hempstead, S.; Drouin, K.; Darcy, J.M.; Reiber, C.; Garruto, R.M. Six-year follow-up of a point-source exposure to CWD contaminated venison in an Upstate New York community: Risk behaviours and health outcomes 2005–2011. Public Health 2014, 128, 860–868. [Google Scholar] [CrossRef]
- Belay, E.D.; Gambetti, P.; Schonberger, L.B.; Parchi, P.; Lyon, D.R.; Capellari, S.; McQuiston, J.H.; Bradley, K.; Dowdle, G.; Crutcher, J.M.; et al. Creutzfeldt-Jakob disease in unusually young patients who consumed venison. Arch. Neurol. 2001, 58, 1673–1678. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.A.; Bosque, P.; Filley, C.M.; Arciniegas, D.B.; Kleinschmidt-DeMasters, B.K.; Pape, W.J.; Tyler, K.L. Colorado surveillance program for chronic wasting disease transmission to humans: Lessons from 2 highly suspicious but negative cases. Arch. Neurol. 2007, 64, 439–441. [Google Scholar] [CrossRef] [PubMed]
- MaWhinney, S.; Pape, W.J.; Forster, J.E.; Anderson, C.A.; Bosque, P.; Miller, M.W. Human prion disease and relative risk associated with chronic wasting disease. Emerg. Infect. Dis. 2006, 12, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Barria, M.A.; Ironside, J.W.; Head, M.W. Exploring the zoonotic potential of animal prion diseases in vivo and in vitro approaches. Prion 2014, 8, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Notari, S.; Capellari, S.; Giese, A.; Westner, I.; Baruzzi, A.; Ghetti, B.; Gambetti, P.; Kretzschmar, H.A.; Parchi, P. Effects of different experimental conditions on the PrPSc core generated by protease digestion: Implications for strain typing and molecular classification of CJD. J. Biol. Chem. 2004, 279, 16797–16804. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemani, S.K.; Myskiw, J.L.; Lamoureux, L.; Booth, S.A.; Sim, V.L. Exposure Risk of Chronic Wasting Disease in Humans. Viruses 2020, 12, 1454. https://doi.org/10.3390/v12121454
Nemani SK, Myskiw JL, Lamoureux L, Booth SA, Sim VL. Exposure Risk of Chronic Wasting Disease in Humans. Viruses. 2020; 12(12):1454. https://doi.org/10.3390/v12121454
Chicago/Turabian StyleNemani, Satish K., Jennifer L. Myskiw, Lise Lamoureux, Stephanie A. Booth, and Valerie L. Sim. 2020. "Exposure Risk of Chronic Wasting Disease in Humans" Viruses 12, no. 12: 1454. https://doi.org/10.3390/v12121454