Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts


Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Isolates
2.2. Recombination and Phylogenetic Analysis
2.3. Nucleotide Composition Analysis
2.4. Relative Synonymous Codon Usage (RSCU) Analysis
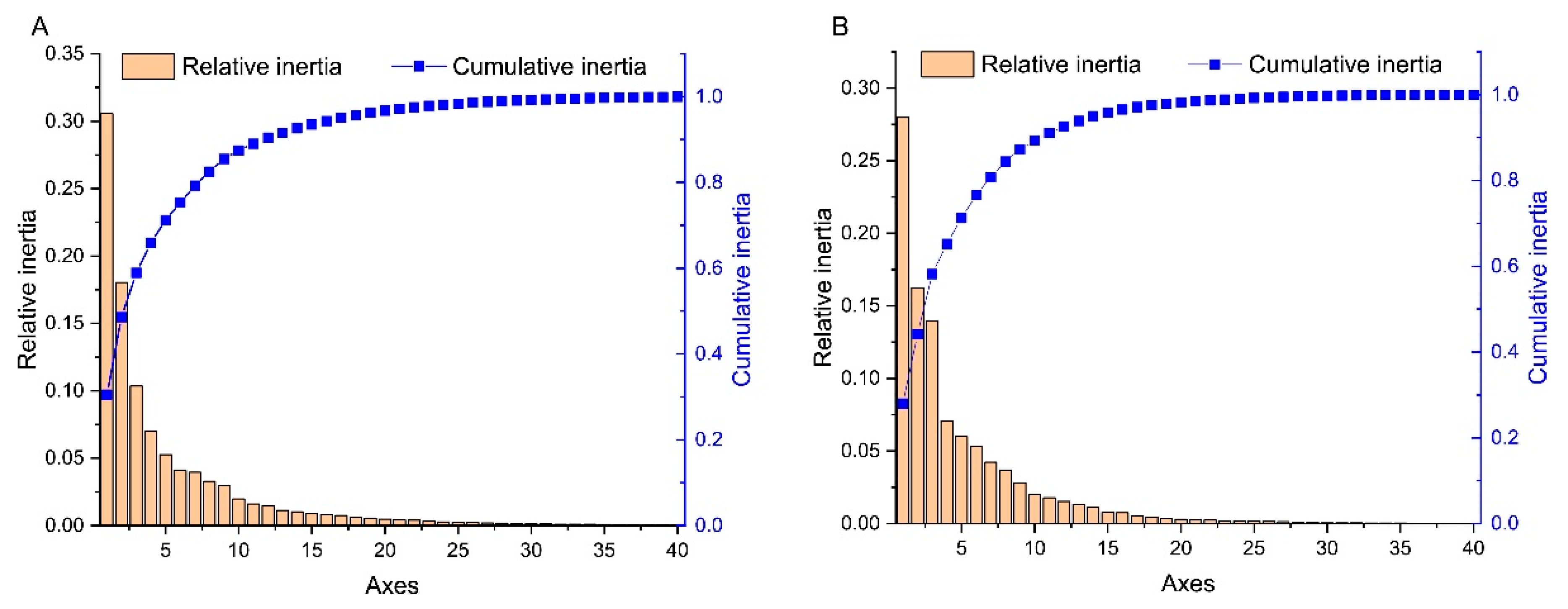
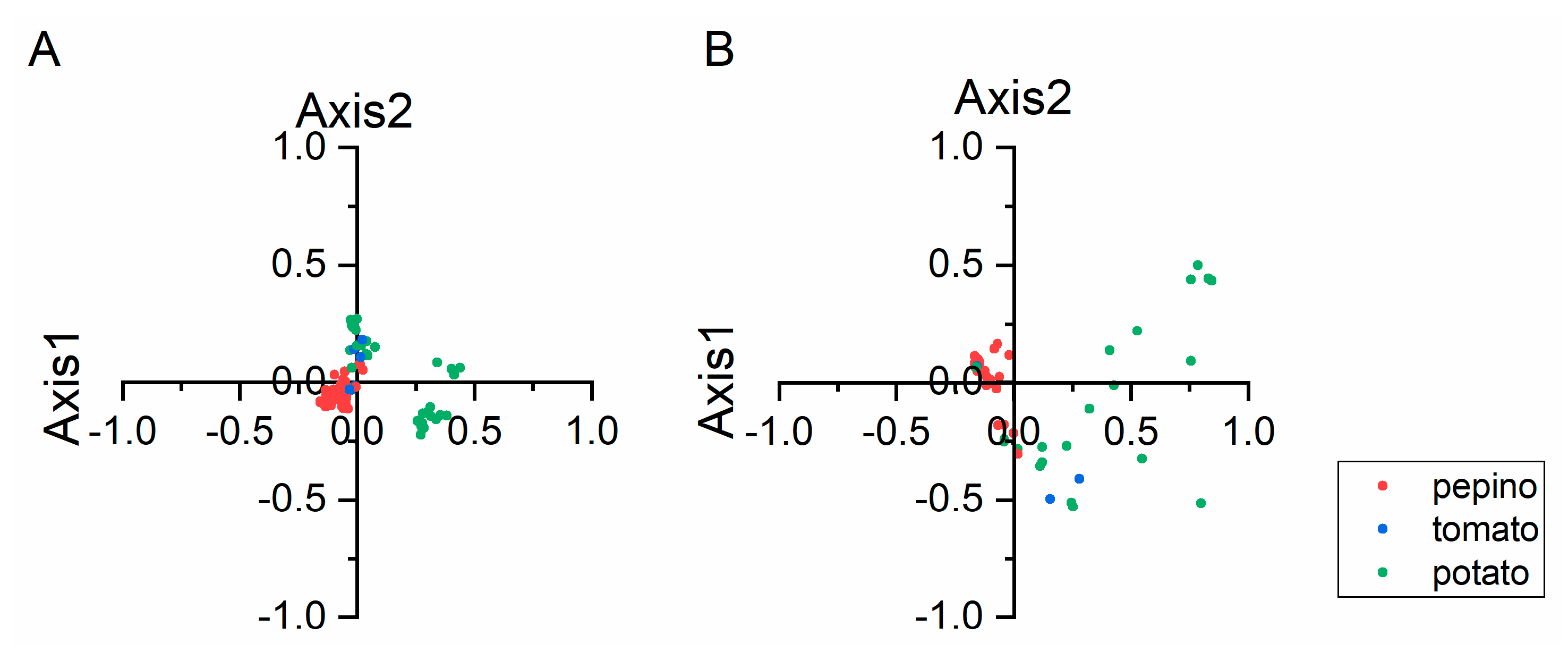
2.5. Principal Component Analysis (PCA)
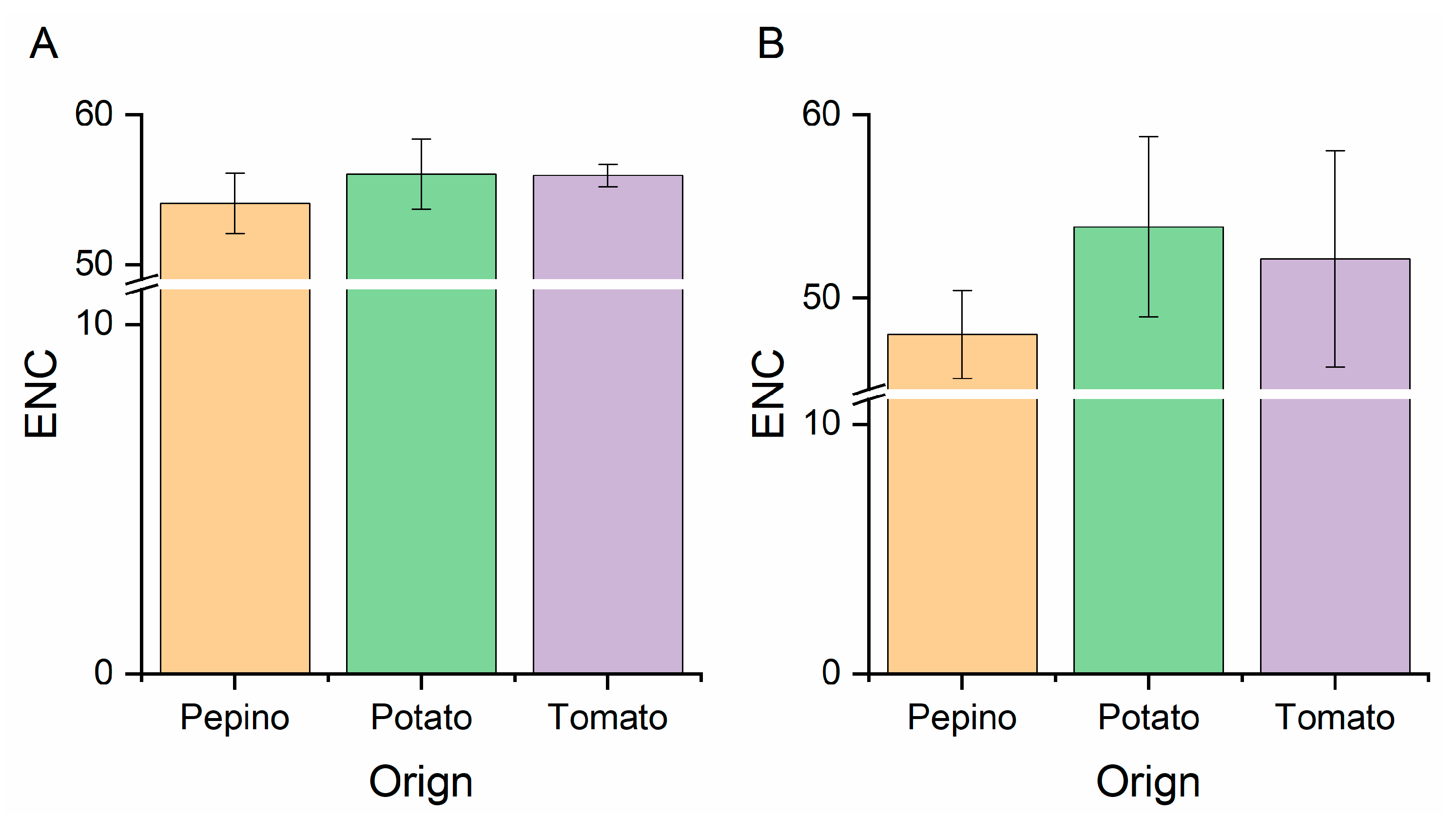
2.6. Effective Number of Codons (ENC) Analysis
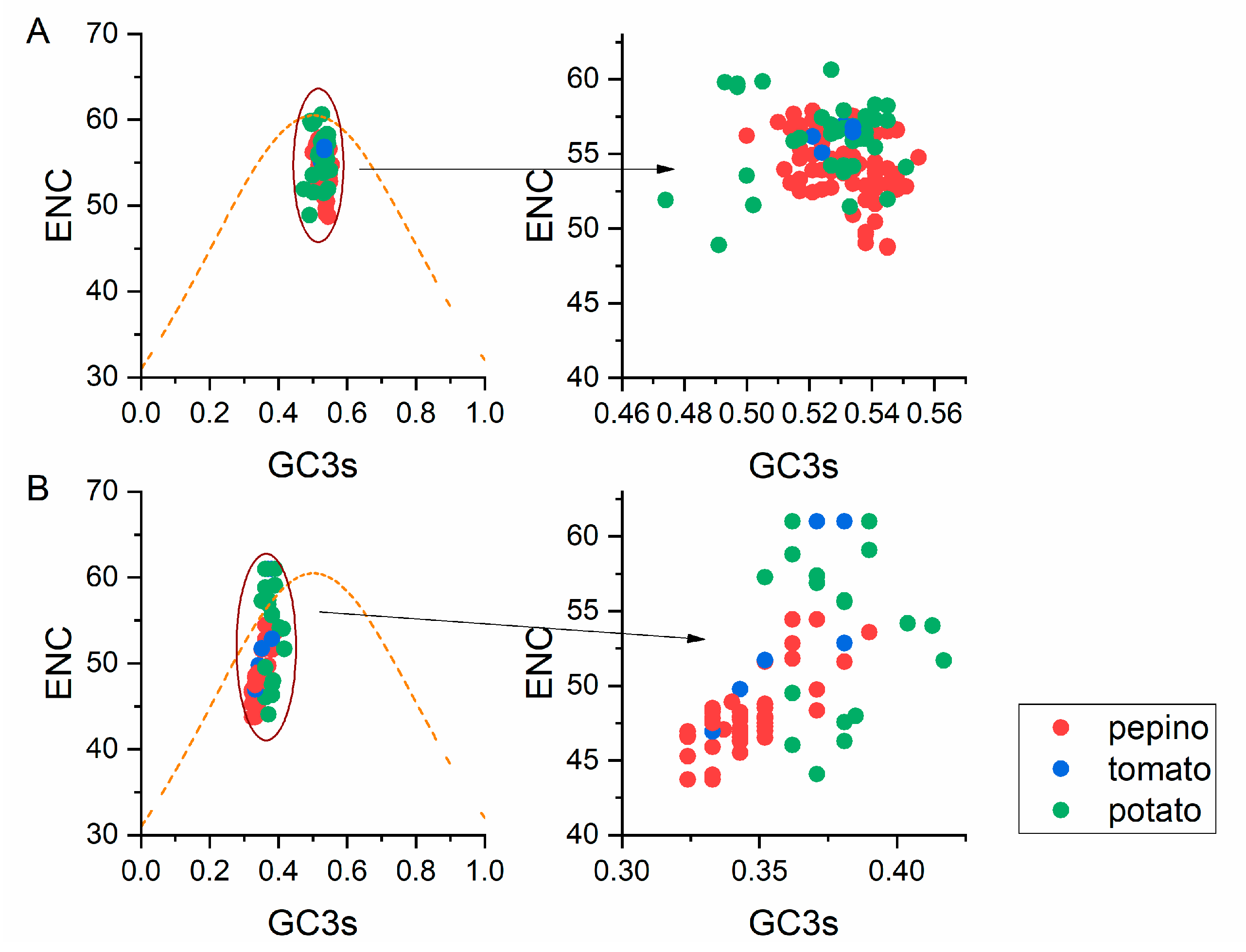
2.7. ENC-Plot Analysis
2.8. Parity Rule 2 (PR2) Analysis
2.9. Neutrality Analysis
2.10. Codon Adaptation Index (CAI) Analysis
2.11. Relative Codon Deoptimization Index (RCDI) Analysis
2.12. Similarity Index (SiD) Analysis
2.13. Gravy and Aroma Statistics
2.14. Statistical Analysis
3. Results
3.1. Recombination and Phylogenetic Analysis
3.2. Nucleotide Composition Analysis
3.3. U- and G-Ending Codons Are Preferred in PVM CP and NABP Coding Sequences
3.4. Codon Usage Bias of the PVM CP and NABP Coding Sequences
3.5. Trends in Codon Usage Variations
3.6. ENC-Plot Analysis
3.7. Neutrality Plot
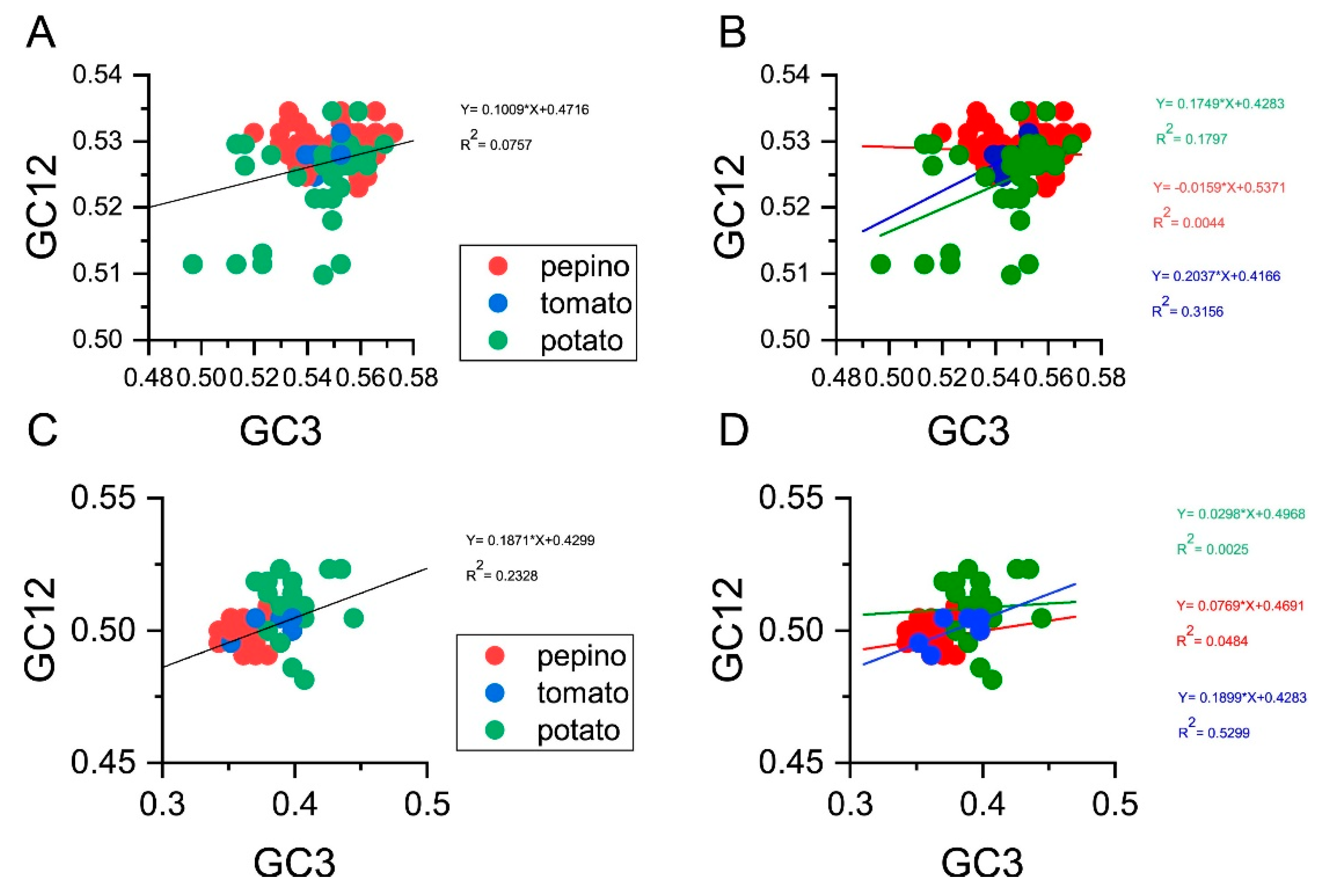
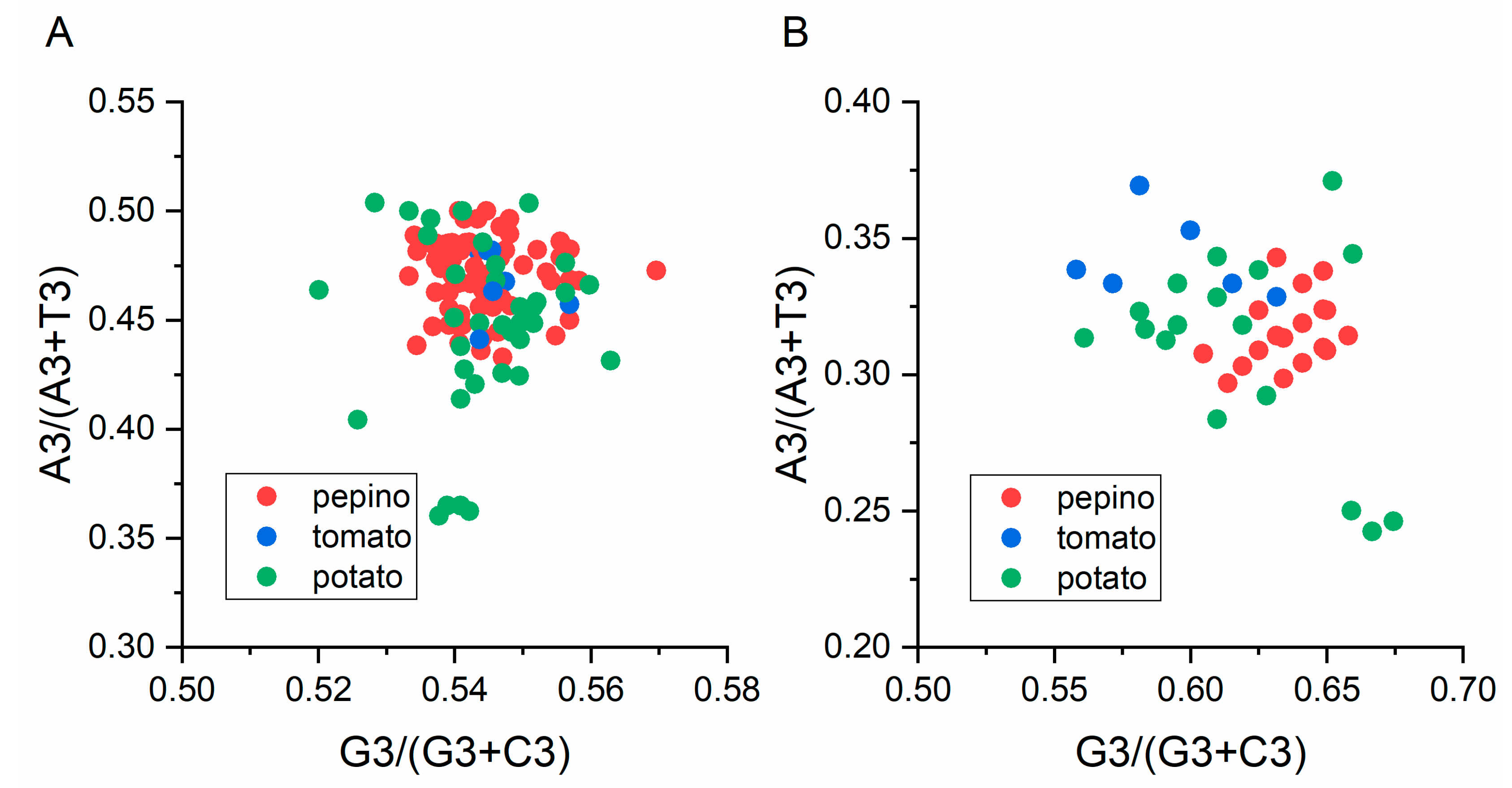
3.8. Parity Analysis
3.9. Natural Selection is a Major Player in Shaping PVM Codon Usage Patterns
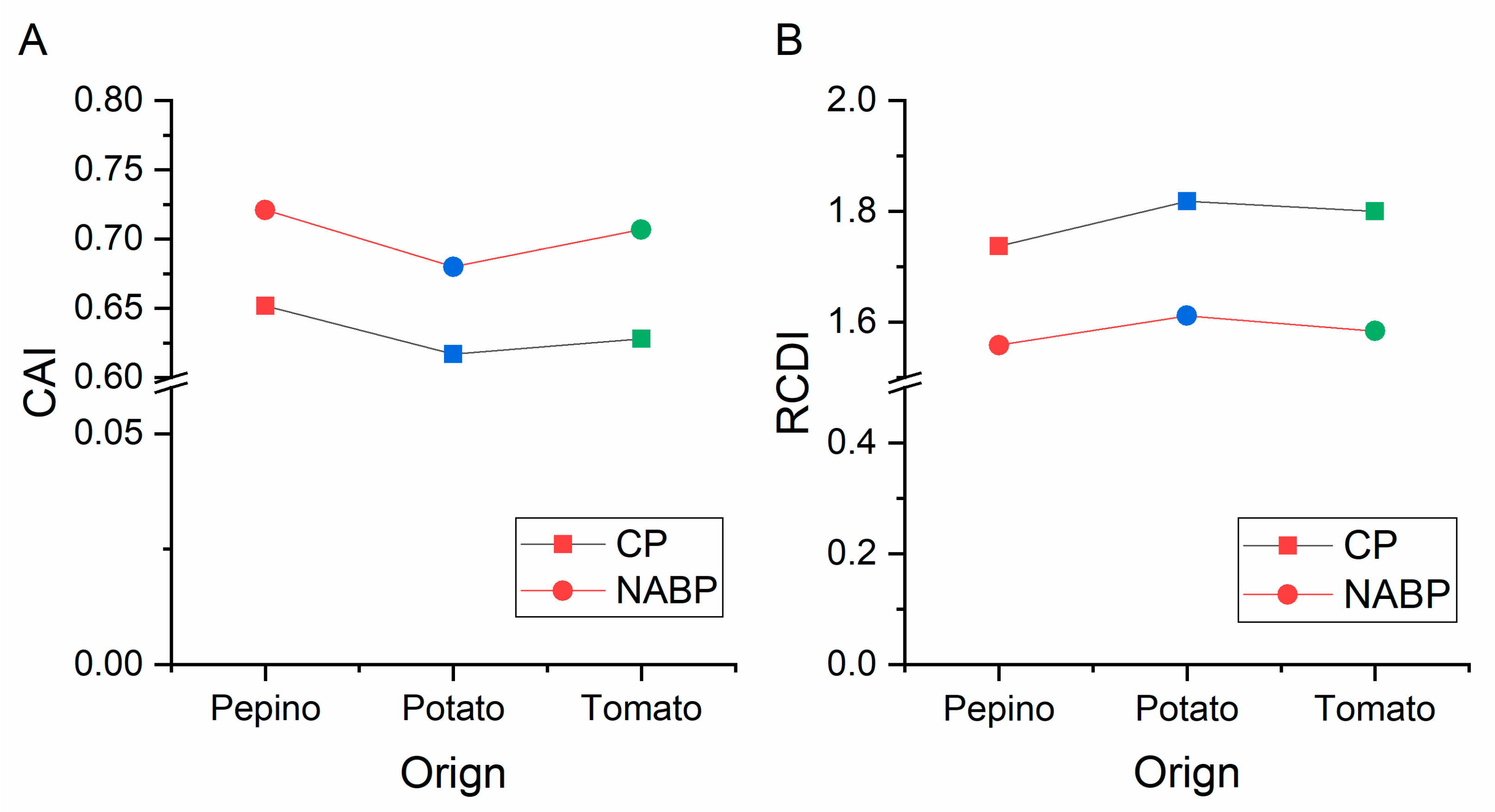
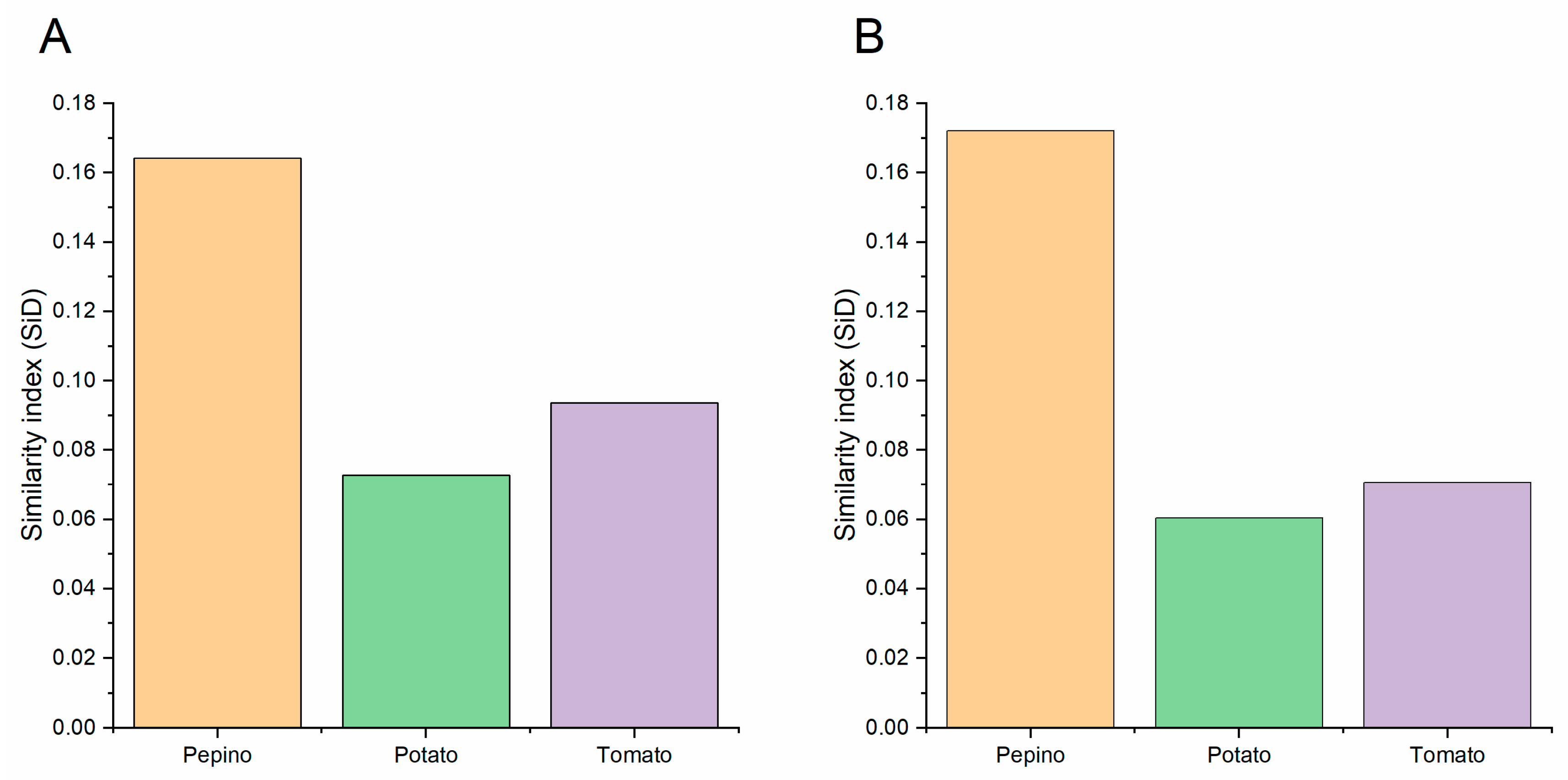
3.10. Codon Usage Adaptation in PVM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Schultz, E.; Folsom, D. Transmission, variation, and control of certain degeneration diseases of Irish potatoes. J. Agric. Res. 1923, 25, 43–147. [Google Scholar]
- Ge, B.B.; He, Z.; Jiang, D.M.; Zhang, Z.X.; Liu, G.J.; Wang, H.Q. Characterization and complete nucleotide sequence of Potato virus M isolated from tomato in China. Acta Virol. 2012, 56, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; He, Z.; Zhang, Z.; Wang, H.; Li, S. Genetic variation in Potato virus M isolates infecting pepino (Solanum muricatum) in China. Arch. Virol. 2014, 159, 3197–3210. [Google Scholar] [CrossRef] [PubMed]
- Tabasinejad, F.; Jafarpour, B.; Zakiaghl, M.; Siampour, M.; Rouhani, H.; Mehrvar, M. Genetic structure and molecular variability of Potato virus M populations. Arch. Virol. 2014, 159, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Tabasinejad, F.; Jafarpour, B.; Zakiaghl, M.; Siampour, M.; Rowhani, H.; Mehrvar, M. Molecular variability in the cysteine rich protein of Potato virus M. Virus Dis. 2015, 26, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Wu, K.; Zhang, L.Z.; Rahman, M.S.; Zheng, K.; Li, T.; Zhang, Z.; Dong, J. Complete genome sequence of a new isolate of potato virus M. in Yunnan, China. Arch. Virol. 2017, 162, 2485–2488. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; D’Aubin, J.; Nie, J. Genomic variability in potato virus M and the development of RT-PCR and RFLP procedures for the detection of this virus in seed potatoes. Virol. J. 2010, 7, 25. [Google Scholar] [CrossRef]
- Zavriev, S.K.; Kanyuka, K.V.; Levay, K.E. The genome organization of Potato virus M RNA. J. Gen. Virol. 1991, 72, 9–14. [Google Scholar] [CrossRef]
- Flatken, S.; Ungewickell, V.; Menzel, W.; Maiss, E. Construction of an infectious full-length cDNA clone of potato virus M. Arch. Virol. 2008, 153, 1385–1389. [Google Scholar] [CrossRef]
- Rozanov, M.N.; Koonin, E.V.; Gorbalenya, A.E. Conservation of the putative methyltransferase domain: A hallmark of the “Sindbis-like” supergroup of positive-strand RNA viruses. J. Gen. Virol. 1992, 73, 2129–2134. [Google Scholar] [CrossRef]
- Koonin, E.V.; Dolja, V.V.; Morris, T.J. Evolution and taxonomy of positive-strand RNA viruses: Implications of comparative analysis of amino acid sequences. Crit. Rev. Biochem. Mol. Biol. 1993, 28, 375–430. [Google Scholar] [CrossRef] [PubMed]
- Morozov, S.Y.; Solovyev, A.G.; Kalinina, N.O.; Fedorkin, O.N.; Samuilova, O.V.; Schiemann, J.; Atabekov, J.G. Evidence for Two Nonoverlapping Functional Domains in the Potato Virus X 25K Movement Protein. Virology 1999, 260, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ju, H.J.; Samuels, T.D.; Wang, Y.S.; Blancaflor, E.; Payton, M.; Mitra, R.; Krishnamurthy, K.; Nelson, R.S.; Verchot-Lubicz, J. The potato virus X TGBp2 movement protein associates with endoplasmic reticulum-derived vesicles during virus infection. Plant Physiol. 2005, 138, 1877–1895. [Google Scholar] [CrossRef] [PubMed]
- Senshu, H.; Yamaji, Y.; Minato, N.; Shiraishi, T.; Maejima, K.; Hashimoto, M.; Miura, C.; Neriya, Y.; Namba, S. A dual strategy for the suppression of host antiviral silencing: Two distinct suppressors for viral replication and viral movement encoded by potato virus M. J. Virol. 2011, 85, 10269–10278. [Google Scholar] [CrossRef] [PubMed]
- Gramstat, A.; Courtpozanis, A.; Rohde, W. The 12 kDa protein of potato virus M displays properties of a nucleic acid-binding regulatory protein. FEBS Lett. 1990, 276, 34–38. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Yasunaga, T.; Miyata, T. Secondary structure of MS2 phage RNA and bias in code word usage. Nucleic Acids Res. 1979, 7, 2073–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Tuohy, T.M.F.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Cowe, E. Synonymous codon usage inSaccharomyces cerevisiae. Yeast 1991, 7, 657–678. [Google Scholar] [CrossRef]
- Sueoka, N. Directional mutation pressure and neutral molecular evolution. Proc. Natl. Acad. Sci. USA 1988, 85, 2653–2657. [Google Scholar] [CrossRef]
- Comeron, J.M.; Aguadé, M. An Evaluation of measures of synonymous codon usage bias. J. Mol. Evol. 1998, 47, 268–274. [Google Scholar] [CrossRef]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Kyte, J.; Doolittle, R.F. A simple method for displaying the hydropathic character of a protein. J. Mol. Biol. 1982, 157, 105–132. [Google Scholar] [CrossRef] [Green Version]
- Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 1999, 238, 53–58. [Google Scholar] [CrossRef]
- Duret, L.; Mouchiroud, D. Expression pattern and, surprisingly, gene length shape codon usage in Caenorhabditis, Drosophila, and Arabidopsis. Proc. Natl. Acad. Sci. USA 1999, 96, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Fuglsang, A. Accounting for background nucleotide composition when measuring codon usage bias: Brilliant idea, difficult in practice. Mol. Biol. Evol. 2006, 23, 1345–1347. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.R.; Papamichail, D.; Skiena, S.; Futcher, B.; Wimmer, E.; Mueller, S. Virus attenuation by genome-scale changes in codon pair bias. Science 2008, 320, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Warthi, G. Genetic Code Optimization for Cotranslational Protein Folding: Codon Directional Asymmetry Correlates with Antiparallel Betasheets, tRNA Synthetase Classes. Comput. Struct. Biotechnol. J. 2017, 15, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H.; Pollock, D.D. The ambush hypothesis: Hidden stop codons prevent off-frame gene reading. DNA Cell Biol. 2004, 23, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. The ambush hypothesis at the whole-organism level: Off frame, ‘hidden’ stops in vertebrate mitochondrial genes increase developmental stability. Comput. Biol. Chem. 2010, 34, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Localized Context-Dependent Effects of the “Ambush” Hypothesis: More Off-Frame Stop Codons Downstream of Shifty Codons. DNA Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Do anticodons of misacylated tRNAs preferentially mismatch codons coding for the misloaded amino acid? BMC Mol. Biol. 2010, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Error compensation of tRNA misacylation by codon-anticodon mismatch prevents translational amino acid misinsertion. Comput. Biol. Chem. 2011, 35, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, H. Coding constraints modulate chemically spontaneous mutational replication gradients in mitochondrial genomes. Curr. Genom. 2012, 13, 37–54. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, R.M.; Seligmann, H. Cryptic tRNAs in chaetognath mitochondrial genomes. Comput. Biol. Chem. 2016, 62, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, D.; Tao, J. Analysis of codon usage patterns in Herbaceous Peony (Paeonia lactiflora Pall.) based on transcriptome data. Genes 2015, 6, 1125–1139. [Google Scholar] [CrossRef] [PubMed]
- Je, M.; Kim, H.; Son, H.S. Analysis of the codon usage pattern of the RdRP gene of mycovirus infecting. Aspergillus spp. Virol. J. 2019, 16, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Wang, R.; Zhang, C.; Wang, S.; He, W.; Zhang, J.; Liu, J.; Cai, Y.; Zhou, J.; Su, S. Genetic and evolutionary analysis of emerging H3N2 canine influenza virus. Emerg. Microbes Infect. 2018, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, G.; Wang, H.; Wang, S.; Xing, G.; Zhang, C.; Zhang, W.; Liu, J.; Zhang, J.; Su, S.; Zhou, J. Insights into the genetic and host adaptability of emerging porcine circovirus 3. Virulence 2018, 9, 1301–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Z.; Wang, R.; Zhang, L.; Shen, B.; Wang, N.; Xu, Q.; He, W.; He, W.; Li, G.; Su, S. Evolutionary changes of the novel Influenza D virus hemagglutinin-esterase fusion gene revealed by the codon usage pattern. Virulence 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Van Hemert, F.; van der Kuyl, A.C.; Berkhout, B. Impact of the biased nucleotide composition of viral RNA genomes on RNA structure and codon usage. J. Gen. Virol. 2016, 97, 2608–2619. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Chen, W.; Yasaka, R.; Chen, C.; Chen, X. Temporal analysis and adaptive evolution of the global population of potato virus M. Infect. Genet. Evol. 2019, 73, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Rybicki, E.P. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Sawyer, S.A. GENECONV: A Computer Package for the Statistical Detection of Gene Conversion; Department of Mathematics, Washington University in Louis: Washington, WA, USA, 1999. [Google Scholar]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef] [PubMed]
- Smith, J. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-Scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Page, R.D.M. Tree View: An application to display phylogenetic trees on personal computers. Bioinformatics 1996, 12, 357–358. [Google Scholar] [CrossRef] [Green Version]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Steel, M. The phylogenetic handbook: A practical approach to phylogenetic analysis and hypothesis testing edited by Lemey, P., Salemi, M., and Vandamme, A.M. Biometrics 2010, 66, 324–325. [Google Scholar] [CrossRef]
- Gerton, J.L.; DeRisi, J.; Shroff, R.; Lichten, M.; Brown, P.O.; Petes, T.D. Global mapping of meiotic recombination hotspots and coldspots in the yeast Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2000, 97, 11383–11390. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.M.; Nasrullah, I.; Qamar, R.; Tong, Y. Evolution of codon usage in Zika virus genomes is host and vector specific. Emerg. Microbes Infect. 2016, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.; Zhao, J.; Xing, G.; Li, G.; Wang, R.; Wang, Z.; Zhang, C.; Franzo, G.; Su, S.; Zhou, J. Genetic analysis and evolutionary changes of porcine Circovirus 2. Mol. Phylogenet. Evol. 2019, 106520. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, L.; He, W.; Zhang, X.; Wen, B.; Wang, C.; Xu, Q.; Li, G.; Zhou, J.; Veit, M.; et al. Genetic evolution and molecular selection of the HE gene of Influenza C virus. Viruses 2019, 11, 167. [Google Scholar] [CrossRef] [PubMed]
- Biswas, K.; Palchoudhury, S.; Chakraborty, P.; Bhattacharyya, U.; Ghosh, D.; Debnath, P.; Ramadugu, C.; Keremane, M.; Khetarpal, R.; Lee, R. Codon usage bias analysis of Citrus tristeza virus: Higher codon adaptation to citrus reticulata host. Viruses 2019, 11, 331. [Google Scholar] [CrossRef]
- Chakraborty, P.; Das, S.; Saha, B.; Sarkar, P.; Karmakar, A.; Saha, A.; Saha, D.; Saha, A. Phylogeny and synonymous codon usage pattern of Papaya ringspot virus coat protein gene in the sub-Himalayan region of north-east India. Can. J. Microbiol. 2015, 61, 555–564. [Google Scholar] [CrossRef]
- He, M.; Guan, S.Y.; He, C.Q. Evolution of rice stripe virus. Mol. Phylogenet. Evol. 2017, 109, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Liu, Q.; Fan, L.; Cui, X.; Zhou, X. Analysis of synonymous codon usage and evolution of begomoviruses. J. Zhejiang Univ. Sci. B 2008, 9, 667–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Wang, H.; Liu, S.; Zhang, B.; Wei, W. Analysis of synonymous codon usage bias of Zika virus and its adaption to the hosts. PLoS ONE 2016, 11, e0166260. [Google Scholar] [CrossRef] [PubMed]
- Khandia, R.; Singhal, S.; Kumar, U.; Ansari, A.; Tiwari, R.; Dhama, K.; Das, J.; Munjal, A.; Singh, R.K. Analysis of Nipah virus codon usage and adaptation to hosts. Front. Microbiol. 2019, 10, 886. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, A.J.; Fargette, D.; García-Arenal, F.; Gibbs, M.J. Time-the emerging dimension of plant virus studies. J. Gen. Virol. 2010, 91, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Torres-Pérez, F.; Palma, R.E.; Hjelle, B.; Holmes, E.C.; Cook, J.A. Spatial but not temporal co-divergence of a virus and its mammalian host. Mol. Ecol. 2011, 20, 4109–4122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irwin, N.R.; Bayerlová, M.; Missa, O.; Martínková, N. Complex patterns of host switching in New World arenaviruses. Mol. Ecol. 2012, 21, 4137–4150. [Google Scholar] [CrossRef] [PubMed]
- Rodelo-Urrego, M.; Pagán, I.; González-Jara, P.; Betancourt, M.; Moreno-Letelier, A.; Ayllón, M.A.; Fraile, A.; Piñero, D.; García-Arenal, F. Landscape heterogeneity shapes host-parasite interactions and results in apparent plant-virus codivergence. Mol. Ecol. 2013, 22, 2325–2340. [Google Scholar] [CrossRef]
- Puigbò, P.; Aragonès, L.; Garcia-Vallvé, S. RCDI/eRCDI: A web-server to estimate codon usage deoptimization. BMC Res. Notes 2010, 3, 87. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Codon | aa | CP | NABP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Potato | Tomato | Pepino | All | Potato | Tomato | Pepino | All | ||
| UUU | F | 0.89 | 0.63 | 0.65 | 0.73 | 1.98 | 1.8 | 1.96 | 1.96 |
| UUC | F | 1.11 * | 1.38 | 1.35 | 1.27 | 0.02 | 0.2 | 0.04 | 0.04 |
| UUA | L | 0.36 | 0.26 | 0.02 | 0.15 | 1.7 | 1.6 | 1.96 | 1.84 |
| UUG | L | 1.18 | 0.93 | 0.96 | 1.04 | 0.81 | 0.8 | 1.84 | 1.4 |
| CUU | L | 0.48 | 0.26 | 0.29 | 0.36 | 1.63 | 2.4 | 2.05 | 1.93 |
| CUC | L | 0.89 | 1.03 | 1 | 0.96 | 0.52 | 0.2 | 0 | 0.2 |
| CUA | L | 1 | 1.12 | 1.38 | 1.23 | 0.63 | 0.4 | 0.02 | 0.27 |
| CUG | L | 2.1 | 2.41 | 2.36 | 2.27 | 0.7 | 0.6 | 0.12 | 0.37 |
| AUU | I | 0.64 | 0.52 | 0.13 | 0.33 | 1.14 | 1.07 | 1.1 | 1.1 |
| AUC | I | 1.17 | 1.47 | 1.74 | 1.53 | 1.05 | 0.67 | 0.65 | 0.72 |
| AUA | I | 1.18 | 1.01 | 1.13 | 1.14 | 0.81 | 1.26 | 1.25 | 1.18 |
| GUU | V | 0.54 | 0.42 | 0.48 | 0.5 | 0.09 | 0 | 0 | 0.02 |
| GUC | V | 0.63 | 0.54 | 0.5 | 0.55 | 0.52 | 0.4 | 0.4 | 0.42 |
| GUA | V | 0.5 | 0.81 | 0.55 | 0.54 | 1.03 | 0.8 | 0.82 | 0.87 |
| GUG | V | 2.33 | 2.24 | 2.47 | 2.41 | 2.36 | 2.8 | 2.77 | 2.69 |
| UCU | S | 0.83 | 0.81 | 0.72 | 0.76 | 1.83 | 2.04 | 2.38 | 2.23 |
| UCC | S | 0.76 | 0.72 | 0.71 | 0.73 | 0.9 | 0.64 | 0.27 | 0.44 |
| UCA | S | 0.88 | 0.94 | 1.07 | 0.99 | 1.4 | 1.61 | 1.58 | 1.54 |
| UCG | S | 1.53 | 1.48 | 1.37 | 1.44 | 0.23 | 0 | 0.01 | 0.06 |
| AGU | S | 0.31 | 0.4 | 0.62 | 0.49 | 1.6 | 1.71 | 1.77 | 1.72 |
| AGC | S | 1.7 | 1.66 | 1.51 | 1.59 | 0.03 | 0 | 0 | 0.01 |
| CCU | P | 0.7 | 0.55 | 0.69 | 0.69 | 2.02 | 1.7 | 1.68 | 1.75 |
| CCC | P | 0.56 | 0.58 | 0.43 | 0.49 | 0.62 | 0.8 | 0.78 | 0.75 |
| CCA | P | 1.24 | 1.39 | 1.59 | 1.45 | 1.18 | 1.5 | 1.51 | 1.44 |
| CCG | P | 1.51 | 1.47 | 1.28 | 1.38 | 0.19 | 0 | 0.03 | 0.06 |
| ACU | T | 1.85 | 1.99 | 1.95 | 1.92 | 2.68 | 2.76 | 3.05 | 2.95 |
| ACC | T | 0.42 | 0.24 | 0.37 | 0.37 | 0.93 | 1.16 | 0.95 | 0.97 |
| ACA | T | 1.01 | 0.97 | 0.8 | 0.88 | 0.19 | 0 | 0 | 0.04 |
| ACG | T | 0.72 | 0.8 | 0.89 | 0.83 | 0.19 | 0.07 | 0 | 0.04 |
| GCU | A | 1.51 | 1.57 | 1.34 | 1.42 | 2.05 | 2.38 | 2.3 | 2.26 |
| GCC | A | 0.86 | 0.78 | 0.94 | 0.9 | 0.05 | 0.05 | 0.14 | 0.12 |
| GCA | A | 1.03 | 1.03 | 1.17 | 1.11 | 0.48 | 0.66 | 0.41 | 0.44 |
| GCG | A | 0.6 | 0.62 | 0.54 | 0.57 | 1.43 | 0.91 | 1.15 | 1.19 |
| UAU | Y | 0.99 | 1.08 | 1.01 | 1.01 | 1.13 | 0.86 | 0.84 | 0.91 |
| UAC | Y | 1.01 | 0.92 | 0.99 | 0.99 | 0.87 | 1.14 | 1.16 | 1.09 |
| CAU | H | 0.77 | 0.4 | 0.46 | 0.56 | 0.88 | 0.53 | 0.72 | 0.74 |
| CAC | H | 1.23 | 1.6 | 1.54 | 1.44 | 1.12 | 1.47 | 1.28 | 1.26 |
| CAA | Q | 0.74 | 0.58 | 0.44 | 0.56 | 1.37 | 2 | 2 | 1.89 |
| CAG | Q | 1.26 | 1.42 | 1.56 | 1.44 | 0.63 | 0 | 0 | 0.11 |
| AAU | N | 1.18 | 1.34 | 1.29 | 1.25 | 1.64 | 1.33 | 1.34 | 1.4 |
| AAC | N | 0.82 | 0.66 | 0.71 | 0.75 | 0.36 | 0.67 | 0.66 | 0.6 |
| AAA | K | 0.76 | 0.75 | 0.62 | 0.68 | 0.31 | 0.53 | 0.53 | 0.49 |
| AAG | K | 1.24 | 1.25 | 1.38 | 1.32 | 1.69 | 1.47 | 1.47 | 1.51 |
| GAU | D | 1.24 | 1.32 | 1.45 | 1.37 | 1.97 | 1.78 | 2 | 1.98 |
| GAC | D | 0.76 | 0.68 | 0.55 | 0.63 | 0.03 | 0.22 | 0 | 0.02 |
| GAA | E | 0.7 | 0.68 | 0.66 | 0.68 | 0.73 | 0.8 | 0.78 | 0.77 |
| GAG | E | 1.3 | 1.32 | 1.34 | 1.32 | 1.27 | 1.2 | 1.22 | 1.23 |
| UGU | C | 0.72 | 0.83 | 0.71 | 0.72 | 1.58 | 1.62 | 1.66 | 1.64 |
| UGC | C | 1.28 | 1.17 | 1.29 | 1.28 | 0.42 | 0.38 | 0.34 | 0.36 |
| CGU | R | 0.65 | 0.31 | 0.26 | 0.4 | 1.16 | 1.37 | 1.37 | 1.32 |
| CGC | R | 1.11 | 0.81 | 0.78 | 0.9 | 1.07 | 0.91 | 0.91 | 0.95 |
| CGA | R | 0.93 | 1.47 | 1.5 | 1.3 | 0.59 | 0.74 | 0.52 | 0.56 |
| CGG | R | 0.53 | 0.56 | 0.53 | 0.53 | 0.16 | 0 | 0 | 0.03 |
| AGA | R | 0.82 | 0.69 | 0.69 | 0.74 | 1.3 | 1.26 | 1.34 | 1.32 |
| AGG | R | 1.96 | 2.16 | 2.24 | 2.14 | 1.73 | 1.71 | 1.85 | 1.82 |
| GGU | G | 1.28 | 1.01 | 1.15 | 1.19 | 2.09 | 2.57 | 2.85 | 2.66 |
| GGC | G | 0.8 | 0.95 | 0.98 | 0.92 | 0.85 | 0.14 | 0.01 | 0.21 |
| GGA | G | 0.9 | 1.01 | 1 | 0.96 | 0.75 | 0.71 | 0.57 | 0.62 |
| GGG | G | 1.02 | 1.03 | 0.86 | 0.93 | 0.31 | 0.57 | 0.57 | 0.51 |
| Gene | ENC | GC3s | GC | Axis1 | Axis2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| r | P | r | P | r | P | r | P | r | P | ||
| CP | Gravy | 0.17691 * | 0.0309 | −0.00431 ns | 0.95835 | −0.01278 ns | 0.87702 | 0.14168 ns | 0.0848 | −0.29638 ** | 2.42 × 10−4 |
| Aromo | 0.27855 ** | 5.82 × 10−4 | −0.26752 ** | 9.73 × 10−4 | −0.23363 ** | 0.00414 | 0.39558 ** | 5.94 × 10−7 | −0.19631 * | 0.01642 | |
| NABP | Gravy | −0.26677 ** | 0.00298 | −0.48469 ** | 1.54 × 10−8 | −0.4726 ** | 3.88 × 10−8 | −0.62506 ** | 1.41 × 10−14 | −0.16583 ns | 0.06793 |
| Aromo | −0.15843 ns | 0.08135 | 0.21532 * | 0.01723 | 0.11425 ns | 0.21018 | 0.44691 ** | 2.47 × 10−7 | 0.30524 ** | 6.29 × 10−4 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, Z.; Gan, H.; Liang, X. Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts. Viruses 2019, 11, 752. https://doi.org/10.3390/v11080752
He Z, Gan H, Liang X. Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts. Viruses. 2019; 11(8):752. https://doi.org/10.3390/v11080752
Chicago/Turabian StyleHe, Zhen, Haifeng Gan, and Xinyan Liang. 2019. "Analysis of Synonymous Codon Usage Bias in Potato Virus M and Its Adaption to Hosts" Viruses 11, no. 8: 752. https://doi.org/10.3390/v11080752