Establishment of Primary Transgenic Human Airway Epithelial Cell Cultures to Study Respiratory Virus–Host Interactions

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Human Airway Epithelial Cell (hAEC) Culture
2.3. Lentiviral Vectors
2.4. shRNA Ligation
2.5. Lentiviral Particle Production
2.6. Lentiviral Transduction
2.7. Flow Cytometry
2.8. FACS Sorting
2.9. Immunofluorescence
2.10. Cell Viability
2.11. Virus Infection
2.12. GFP Knockdown
2.13. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.14. Data Presentation
3. Results
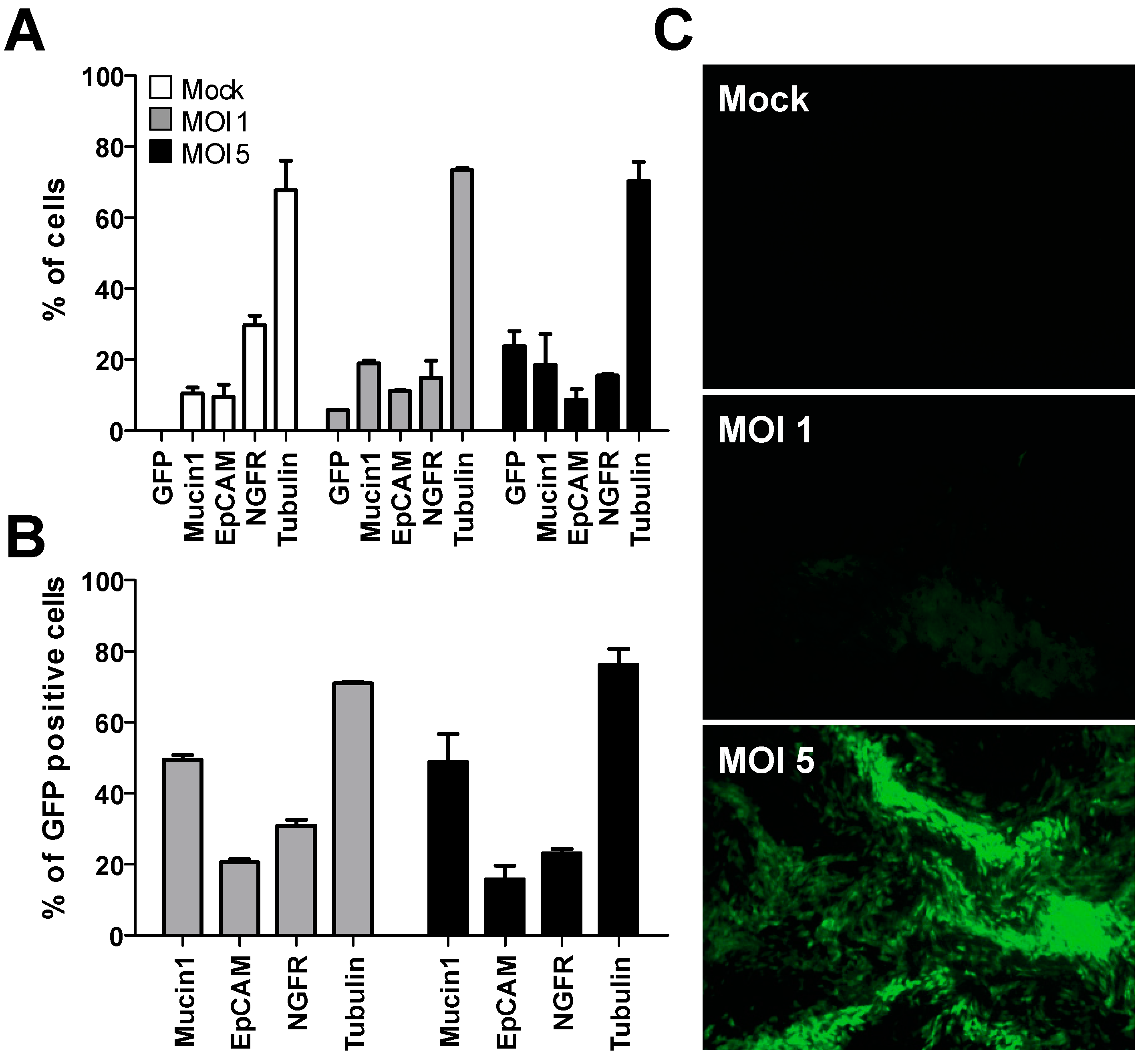
3.1. Transduction of Undifferentiated Primary Human Bronchial Cells
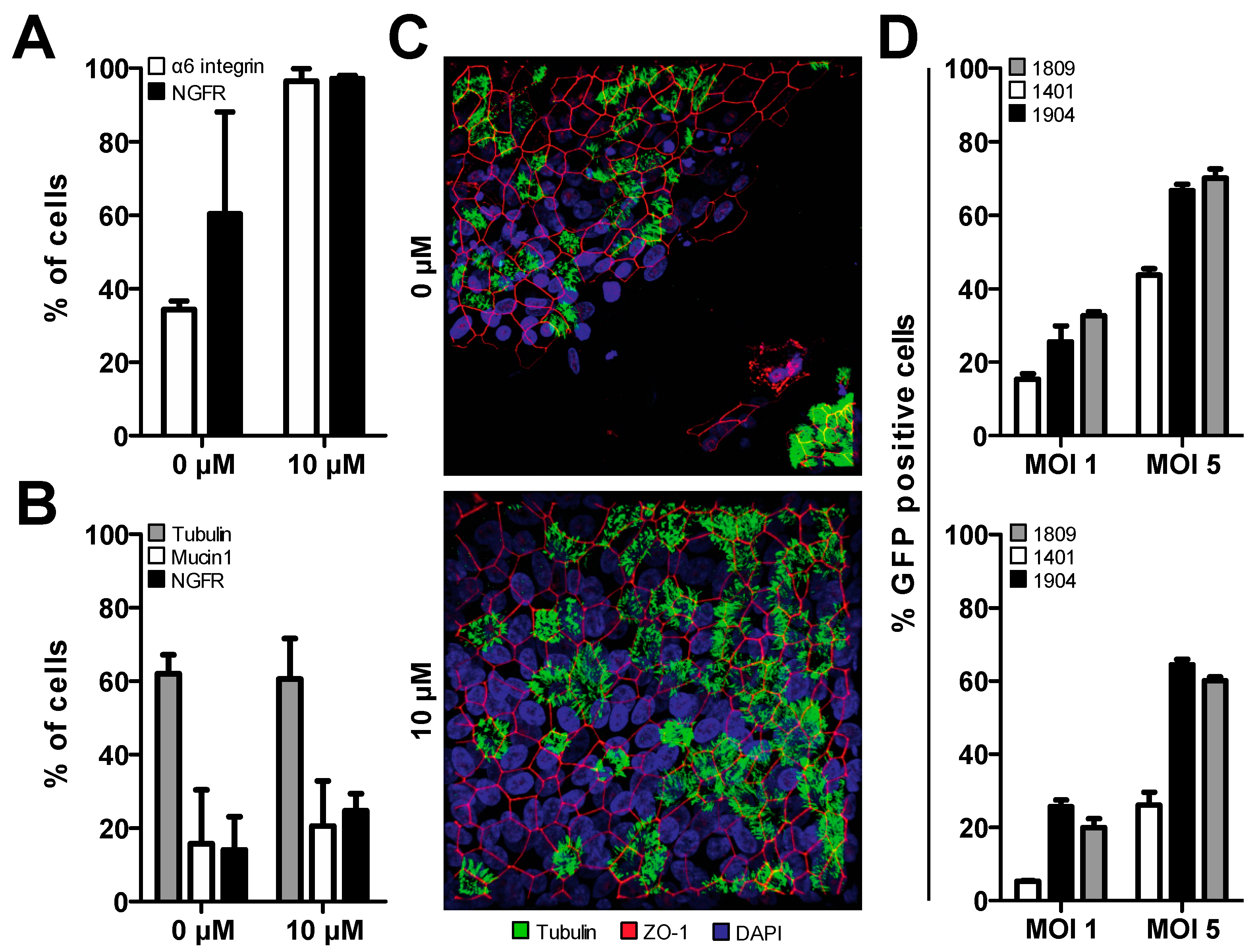
3.2. Treatment of Primary Cells with Y-27632 Prolongs Basal Cell Phenotype
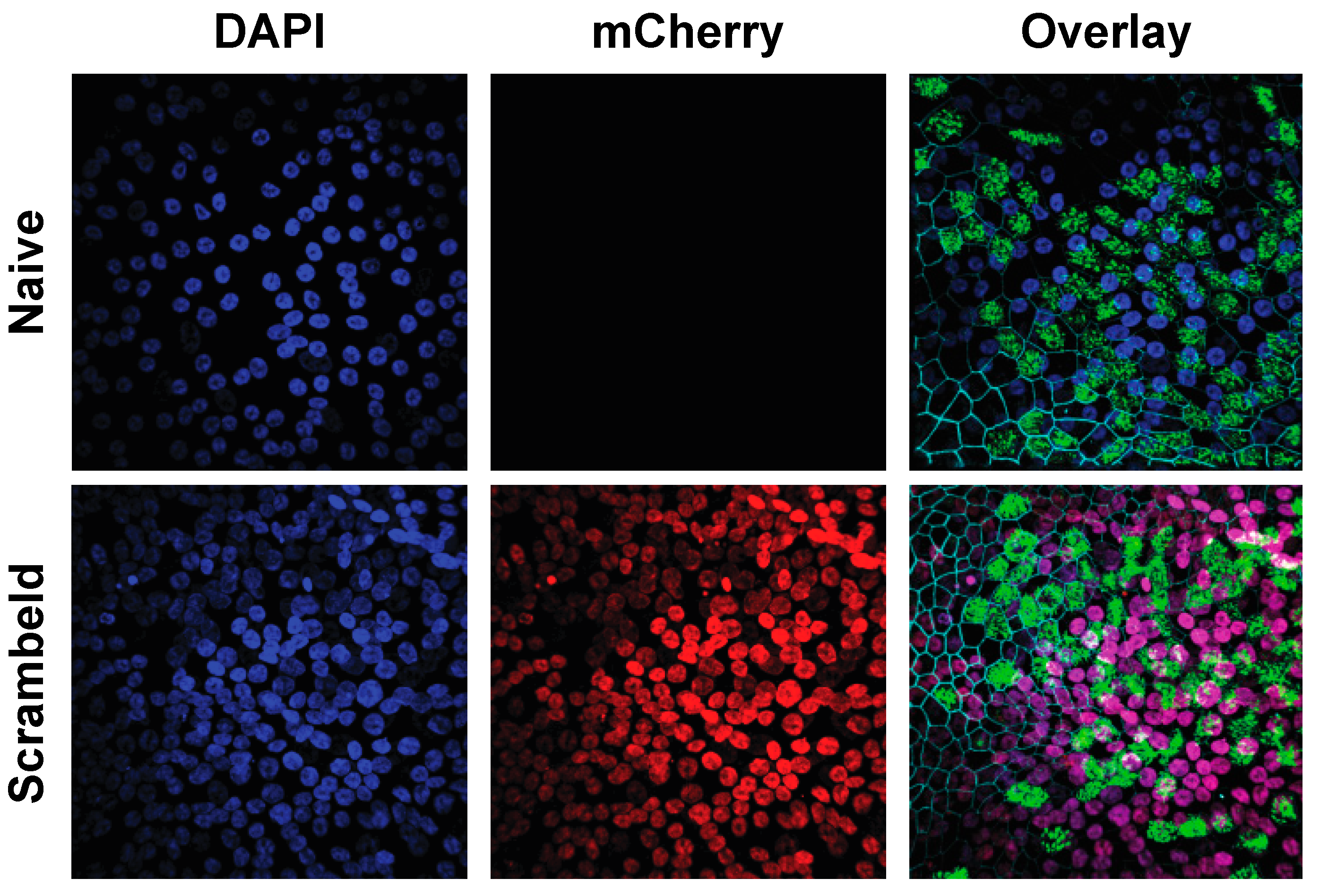
3.3. Establishment of Homogenously Transgenic hAEC Cultures
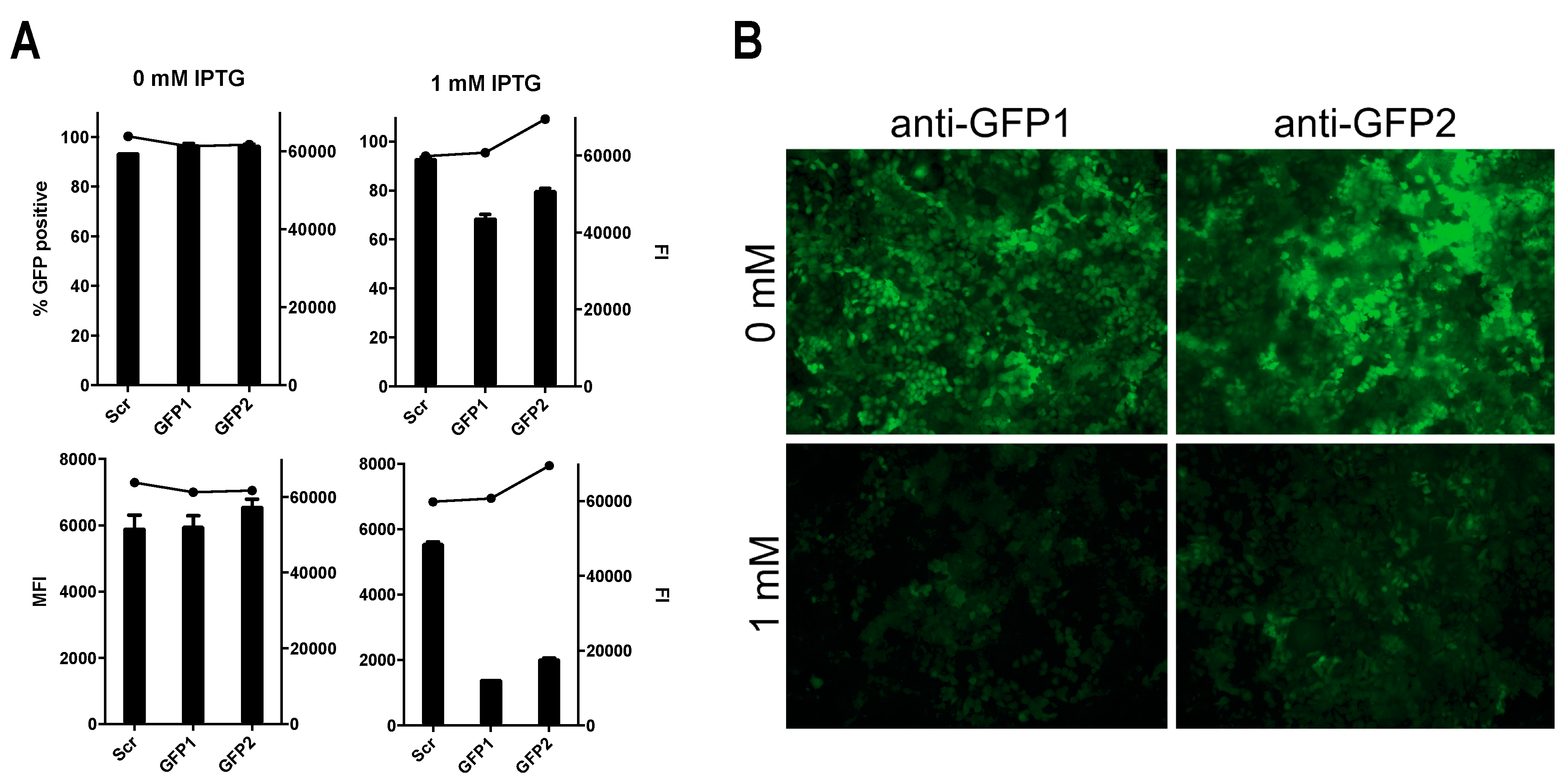
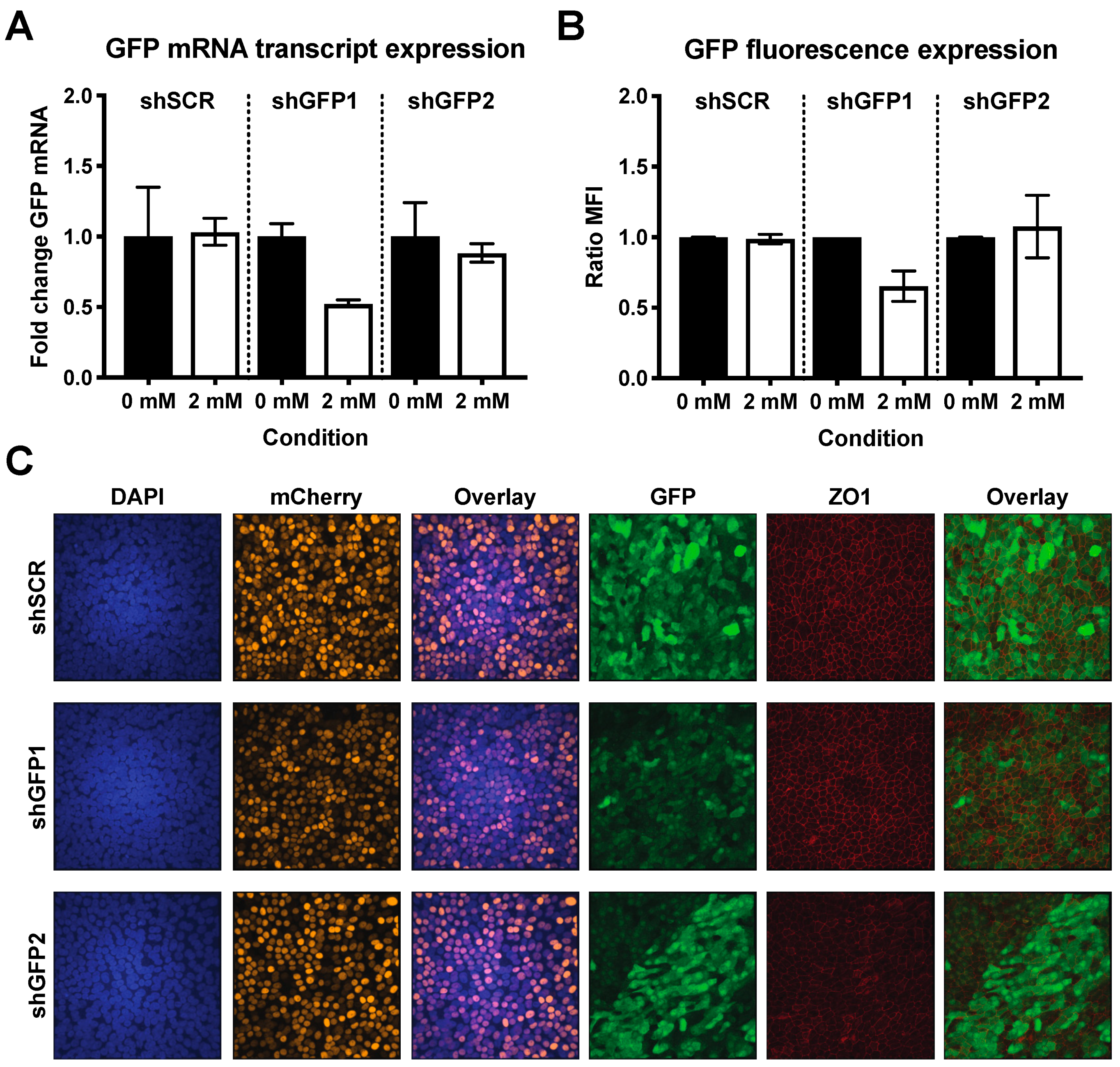
3.4. Successful Induction of Isopropyl β-D-1-Thiogalactopyranoside (IPTG)-Controlled shRNA Expression
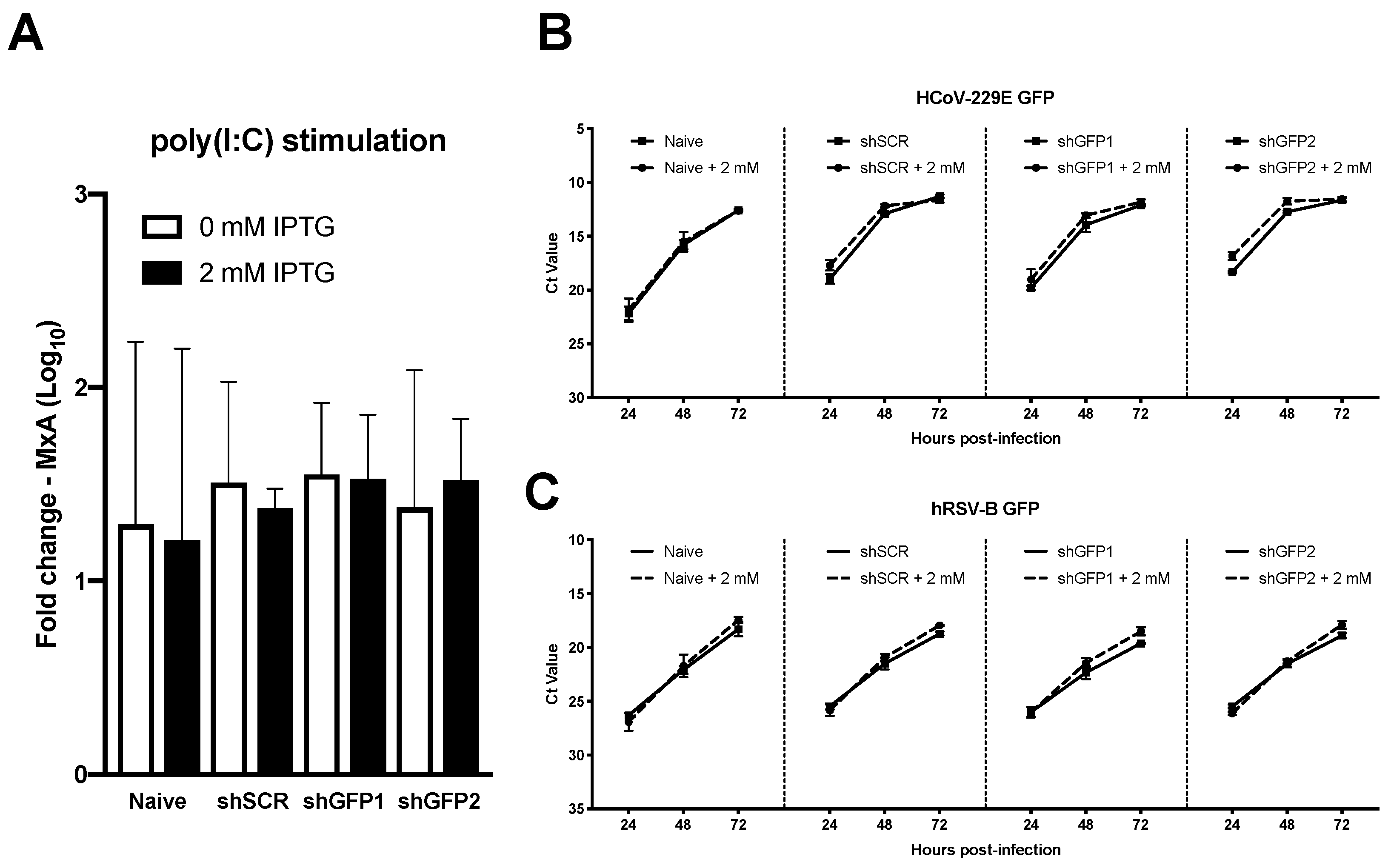
3.5. Transgenesis of Primary hAEC Cultures Does Not Affect Host Innate Immune Response or Viral Replication
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rackley, C.R.; Stripp, B.R. Building and maintaining the epithelium of the lung. J. Clin. Investig. 2012, 122, 2724–2730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randell, S.H.; Boucher, R.C. Effective mucus clearance is essential for respiratory health. Am. J. Respir. Cell Mol. Biol. 2006, 35, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Rock, J.R.; Onaitis, M.W.; Rawlins, E.L.; Lu, Y.; Clark, C.P.; Xue, Y.; Randell, S.H.; Hogan, B.L.M. Basal cells as stem cells of the mouse trachea and human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 12771–12775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pezzulo, A.A.; Starner, T.D.; Scheetz, T.E.; Traver, G.L.; Tilley, A.E.; Harvey, B.-G.; Crystal, R.G.; McCray, P.B.; Zabner, J. The air-liquid interface and use of primary cell cultures are important to recapitulate the transcriptional profile of in vivo airway epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L25–L31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jong, P.M.; van Sterkenburg, M.A.J.A.; Kempenaar, J.A.; Dijkman, J.H.; Ponec, M. Serial culturing of human bronchial epithelial cells derived from biopsies. Vitr. Cell. Dev. Biol. Anim. J. Soc. Vitr. Biol. 1993, 29, 379–387. [Google Scholar] [CrossRef]
- De Jong, P.M.; van Sterkenburg, M.A.; Hesseling, S.C.; Kempenaar, J.A.; Mulder, A.A.; Mommaas, A.M.; Dijkman, J.H.; Ponec, M. Ciliogenesis in human bronchial epithelial cells cultured at the air-liquid interface. Am. J. Respir. Cell. Mol. Biol. 1994, 10. [Google Scholar] [CrossRef] [PubMed]
- Matrosovich, M.N.; Matrosovich, T.Y.; Gray, T.; Roberts, N.A.; Klenk, H.D. Human and avian influenza viruses target different cell types in cultures of human airway epithelium. Proc. Natl. Acad. Sci. USA 2004, 101, 4620–4624. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Bukreyev, A.; Thompson, C.I.; Watson, B.; Peeples, M.E.; Collins, P.L.; Pickles, R.J. Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium. J. Virol. 2005, 79, 1113–1124. [Google Scholar] [CrossRef]
- Sims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe acute respiratory syndrome coronavirus infection of human ciliated airway epithelia: Role of ciliated cells in viral spread in the conducting airways of the lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar] [CrossRef]
- Dijkman, R.; Koekkoek, S.M.; Molenkamp, R.; Schildgen, O.; van der Hoek, L. Human bocavirus can be cultured in differentiated human airway epithelial cells. J. Virol. 2009, 83, 7739–7748. [Google Scholar] [CrossRef]
- Pyrc, K.; Sims, A.C.; Dijkman, R.; Jebbink, M.; Long, C.; Deming, D.; Donaldson, E.; Vabret, A.; Baric, R.; Van Der Hoek, L.; et al. Culturing the unculturable: Human coronavirus HKU1 infects, replicates, and produces progeny virions in human ciliated airway epithelial cell cultures. J. Virol. 2010, 84. [Google Scholar] [CrossRef] [PubMed]
- Kindler, E.; Jonsdottir, H.R.; Muth, D.; Hamming, O.J.; Hartmann, R.; Rodriguez, R.; Geffers, R.; Fouchier, R.A.M.; Drosten, C.; Muller, M.A.; et al. Efficient replication of the novel human betacoronavirus EMC on primary human epithelium highlights its zoonotic potential. MBio 2013, 4, e00611-12. [Google Scholar] [CrossRef]
- Dijkman, R.; Jebbink, M.F.; Koekkoek, S.M.; Deijs, M.; Jónsdóttir, H.R.; Molenkamp, R.; Ieven, M.; Goossens, H.; Thiel, V.; van der Hoek, L. Isolation and Characterization of Current human coronavirus strains in primary human epithelial cell cultures reveal differences in target cell tropism. J. Virol. 2013, 87. [Google Scholar] [CrossRef]
- Tseng, C.-T.K.; Huang, C.; Newman, P.; Wang, N.; Narayanan, K.; Watts, D.M.; Makino, S.; Packard, M.M.; Zaki, S.R.; Chan, T.-S.; et al. Severe Acute Respiratory Syndrome Coronavirus Infection of Mice Transgenic for the Human Angiotensin-Converting Enzyme 2 Virus Receptor. J. Virol. 2007, 81, 1162–1173. [Google Scholar] [CrossRef] [Green Version]
- Barnard, D.L. Animal models for the study of influenza pathogenesis and therapy. Antiviral Res. 2009, 82, A110–A122. [Google Scholar] [CrossRef] [PubMed]
- Gray, T.E.; Guzman, K.; Davis, C.W.; Abdullah, L.H.; Nettesheim, P. Mucociliary differentiation of serially passaged normal human tracheobronchial epithelial cells. Am. J. Respir. Cell. Mol. Biol. 1996, 14, 104–112. [Google Scholar] [CrossRef]
- Liu, X.; Ory, V.; Chapman, S.; Yuan, H.; Albanese, C.; Kallakury, B.; Timofeeva, O.A.; Nealon, C.; Dakic, A.; Simic, V.; et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am. J. Pathol. 2012, 180, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Suprynowicz, F.A.; Upadhyay, G.; Krawczyk, E.; Kramer, S.C.; Hebert, J.D.; Liu, X.; Yuan, H.; Cheluvaraju, C.; Clapp, P.W.; Boucher, R.C., Jr.; et al. Conditionally reprogrammed cells represent a stem-like state of adult epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, 20035–20040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsdottir, H.R.; Dijkman, R. Characterization of human coronaviruses on well-differentiated human airway epithelial cell cultures. Methods Mol. Biol. 2015, 1282, 73–87. [Google Scholar]
- Paddison, P.J.; Caudy, A.A.; Bernstein, E.; Hannon, G.J.; Conklin, D.S. Short hairpin RNAs (shRNAs) induce sequence-specific silencing in mammalian cells. Genes Dev. 2002, 16, 948–958. [Google Scholar] [CrossRef] [Green Version]
- Taxman, D.J.; Moore, C.B.; Guthrie, E.H.; Huang, M.T.-H. Short Hairpin RNA (shRNA): Design, Delivery, and Assessment of Gene Knockdown. In Methods in Molecular Biology; Clifton, N.J., Ed.; Springer: Berlin, Germany, 2010; Volume 629, pp. 139–156. [Google Scholar]
- Mao, Y.; Yan, R.; Li, A.; Zhang, Y.; Li, J.; Du, H.; Chen, B.; Wei, W.; Zhang, Y.; Sumners, C.; et al. Lentiviral Vectors Mediate Long-Term and High Efficiency Transgene Expression in HEK 293T cells. Int. J. Med. Sci. 2015, 12, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Everman, J.L.; Rios, C.; Seibold, M.A. Primary Airway Epithelial Cell Gene Editing Using CRISPR-Cas9. In Methods in Molecular Biology; Clifton, N.J., Ed.; Springer: Berlin, Germany, 2018; Volume 1706, pp. 267–292. [Google Scholar]
- Munye, M.M.; Shoemark, A.; Hirst, R.A.; Delhove, J.M.; Sharp, T.V.; McKay, T.R.; O’Callaghan, C.; Baines, D.L.; Howe, S.J.; Hart, S.L. BMI-1 extends proliferative potential of human bronchial epithelial cells while retaining their mucociliary differentiation capacity. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L258–L267. [Google Scholar] [CrossRef]
- Sinn, P.L.; Hwang, B.-Y.; Li, N.; Ortiz, J.L.S.; Shirazi, E.; Parekh, K.R.; Cooney, A.L.; Schaffer, D.V.; McCray, P.B. Novel GP64 envelope variants for improved delivery to human airway epithelial cells. Gene Ther. 2017, 24, 674–679. [Google Scholar] [CrossRef]
- Eekels, J.J.; Geerts, D.; Jeeninga, R.E.; Berkhout, B. Long-term inhibition of HIV-1 replication with RNA interference against cellular co-factors. Antivir. Res. 2011, 89, 43–53. [Google Scholar] [CrossRef]
- GPP Web Portal. Available online: https://www.broadinstitute.org/rnai/public/) (accessed on 12 August 2019).
- Cervantes-Barragan, L.; Zust, R.; Maier, R.; Sierro, S.; Janda, J.; Levy, F.; Speiser, D.; Romero, P.; Rohrlich, P.S.; Ludewig, B.; et al. Dendritic cell-specific antigen delivery by coronavirus vaccine vectors induces long-lasting protective antiviral and antitumor immunity. MBio 2010, 1, e00171-10. [Google Scholar] [CrossRef]
- Lemon, K.; Nguyen, D.T.; Ludlow, M.; Rennick, L.J.; Yuksel, S.; van Amerongen, G.; McQuaid, S.; Rima, B.K.; de Swart, R.L.; Duprex, W.P. Recombinant subgroup B human respiratory syncytial virus expressing enhanced green fluorescent protein efficiently replicates in primary human cells and is virulent in cotton rats. J. Virol. 2015, 89, 2849–2856. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Vijgen, L.; Keyaerts, E.; Moes, E.; Maes, P.; Duson, G.; Van Ranst, M. Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J. Clin. Microbiol. 2005, 43, 5452–5456. [Google Scholar] [CrossRef]
- Van de Pol, A.C.; van Loon, A.M.; Wolfs, T.F.; Jansen, N.J.; Nijhuis, M.; Breteler, E.K.; Schuurman, R.; Rossen, J.W. Increased detection of respiratory syncytial virus, influenza viruses, parainfluenza viruses, and adenoviruses with real-time PCR in samples from patients with respiratory symptoms. J. Clin. Microbiol. 2007, 45, 2260–2262. [Google Scholar] [CrossRef]
- Jonsdottir, H.R.; Thiel, V.; Dijkman, R. Lentiviral transduction of well-differentiated hAEC cultures. Institute of Virology and Immunology: Bern, BE, Switzerland, 2012; (unpublished). [Google Scholar]
- Kremer, K.L.; Dunning, K.R.; Parsons, D.W.; Anson, D.S. Gene delivery to airway epithelial cells in vivo: A direct comparison of apical and basolateral transduction strategies using pseudotyped lentivirus vectors. J. Gene Med. 2007, 9, 362–368. [Google Scholar] [CrossRef]
- Jonsdottir, H.R.; Thiel, V.; Dijkman, R. Suspension Transduction of undifferentiated hAEC cultures. Institute of Virology and Immunolog: Bern, BE, Switzerland, 2012; (unpublished). [Google Scholar]
- Jonsdottir, H.R.; Thiel, V.; Dijkman, R. Influence of Y-27632 on epithelial structure and morphology of well-differentiated hAEC cultures. Institute of Virology and Immunology: Bern, BE, Switzerland, 2013; (Unpublished). [Google Scholar]
- Arason, A.J.; Jonsdottir, H.R.; Halldorsson, S.; Benediktsdottir, B.E.; Bergthorsson, J.T.; Ingthorsson, S.; Baldursson, O.; Sinha, S.; Gudjonsson, T.; Magnusson, M.K. deltaNp63 Has a Role in Maintaining Epithelial Integrity in Airway Epithelium. PLoS ONE 2014, 9, e88683. [Google Scholar] [CrossRef]
- Lauber, C.; Vieyres, G.; Terczynska-Dyla, E.; Anggakusuma; Dijkman, R.; Gad, H.H.; Akhtar, H.; Geffers, R.; Vondran, F.W.; Thiel, V.; et al. Transcriptome analysis reveals a classical interferon signature induced by IFNlambda4 in human primary cells. Genes Immun. 2015, 16, 414–421. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Mou, H.; Vinarsky, V.; Tata, P.R.; Brazauskas, K.; Choi, S.H.; Crooke, A.K.; Zhang, B.; Solomon, G.M.; Turner, B.; Bihler, H.; et al. Dual SMAD Signaling Inhibition Enables Long-Term Expansion of Diverse Epithelial Basal Cells. Cell Stem Cell 2016, 19, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Brody, S.L.; Kaiko, G.E. Harnessing TGF-β and BMP signaling for expansion of p63-positive epithelial stem cells. Stem Cell Investig. 2016, 3, 82. [Google Scholar] [CrossRef]
- Guyot, B.; Maguer-Satta, V. Blocking TGF-β and BMP SMAD-dependent cell differentiation is a master key to expand all kinds of epithelial stem cells. Stem Cell Investig. 2016, 3, 88. [Google Scholar] [CrossRef]
- Zhang, C.; Lee, H.J.; Shrivastava, A.; Wang, R.; McQuiston, T.J.; Challberg, S.S.; Pollok, B.A.; Wang, T. Long-Term In Vitro Expansion of Epithelial Stem Cells Enabled by Pharmacological Inhibition of PAK1-ROCK-Myosin II and TGF-β Signaling. Cell Rep. 2018, 25, 598–610.e5. [Google Scholar] [CrossRef]
- Liu, Y.P.; Schopman, N.C.T.; Berkhout, B. Dicer-independent processing of short hairpin RNAs. Nucleic Acids Res. 2013, 41, 3723–3733. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Wang, H.; Ji, Y.; Yang, J.; Xu, S.; Huang, X.; Wang, Z.; Qin, L.; Tien, P.; Zhou, X.; et al. The Nucleocapsid Protein of Coronaviruses Acts as a Viral Suppressor of RNA Silencing in Mammalian Cells. J. Virol. 2015, 89, 9029–9043. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1° Antibody | Target | Dilution | Host | Clone | Supplier |
|---|---|---|---|---|---|
| Anti-β-Tubulin | Cilia | 1:200 | Mouse | ONS 1A6 | Abcam |
| Anti-ZO-1 | Tight junctions | 1:200 | Goat | Ab99462 | |
| Anti-GFP | Green fluorescent protein (GFP) | 1:200 | Mouse | Ab1218 | |
| Anti-mCherry | mCherry | 1:200 | Chicken | Ab205402 | |
| Anti-ZO-1 | Tight Junctions | 1:200 | Rabbit | 61-7300 | Thermofisher |
| 2° Antibody | Target | Dilution | Host | Supplier |
|---|---|---|---|---|
| Alexa Fluor® 488 | Anti-mouse | 1:400 | Donkey | Jackson Immunoresearch |
| Cy3 | Anti-goat | 1:400 | ||
| Alexa Fluor® 647 | Anti-goat | 1:400 | ||
| Alexa Fluor® 594 | Anti-chicken | 1:400 | ||
| Alexa Fluor® 647 | Anti-rabbit | 1:400 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jonsdottir, H.R.; Marti, S.; Geerts, D.; Rodriguez, R.; Thiel, V.; Dijkman, R. Establishment of Primary Transgenic Human Airway Epithelial Cell Cultures to Study Respiratory Virus–Host Interactions. Viruses 2019, 11, 747. https://doi.org/10.3390/v11080747
Jonsdottir HR, Marti S, Geerts D, Rodriguez R, Thiel V, Dijkman R. Establishment of Primary Transgenic Human Airway Epithelial Cell Cultures to Study Respiratory Virus–Host Interactions. Viruses. 2019; 11(8):747. https://doi.org/10.3390/v11080747
Chicago/Turabian StyleJonsdottir, Hulda R., Sabrina Marti, Dirk Geerts, Regulo Rodriguez, Volker Thiel, and Ronald Dijkman. 2019. "Establishment of Primary Transgenic Human Airway Epithelial Cell Cultures to Study Respiratory Virus–Host Interactions" Viruses 11, no. 8: 747. https://doi.org/10.3390/v11080747