Design and Preclinical Development of a Phage Product for the Treatment of Antibiotic-Resistant Staphylococcus aureus Infections


Abstract
:1. Introduction
- Obligately lytic, to avoid specialized transduction of bacterial genes, and maximize chances for bacterial killing;
- Not known, by empirical testing and/or inference from genomics, to be prone to generalized transduction; and,
- Fully sequenced, to avoid phages with known antibiotic resistance or bacterial virulence genes, and to help assess other lifestyle traits.
- Have broad activity against the target pathogen but not other species, to maximize potential utility and minimize off-target effects; and,
- Be capable of complementation, in which resistant mutants arising to one phage are sensitive to another phage.
2. Methods
2.1. Bacteriophages, Source and Propagation
2.2. Bacteria
2.3. Phage Sensitivity Assays
2.4. Frequency of Resistance and Complementation
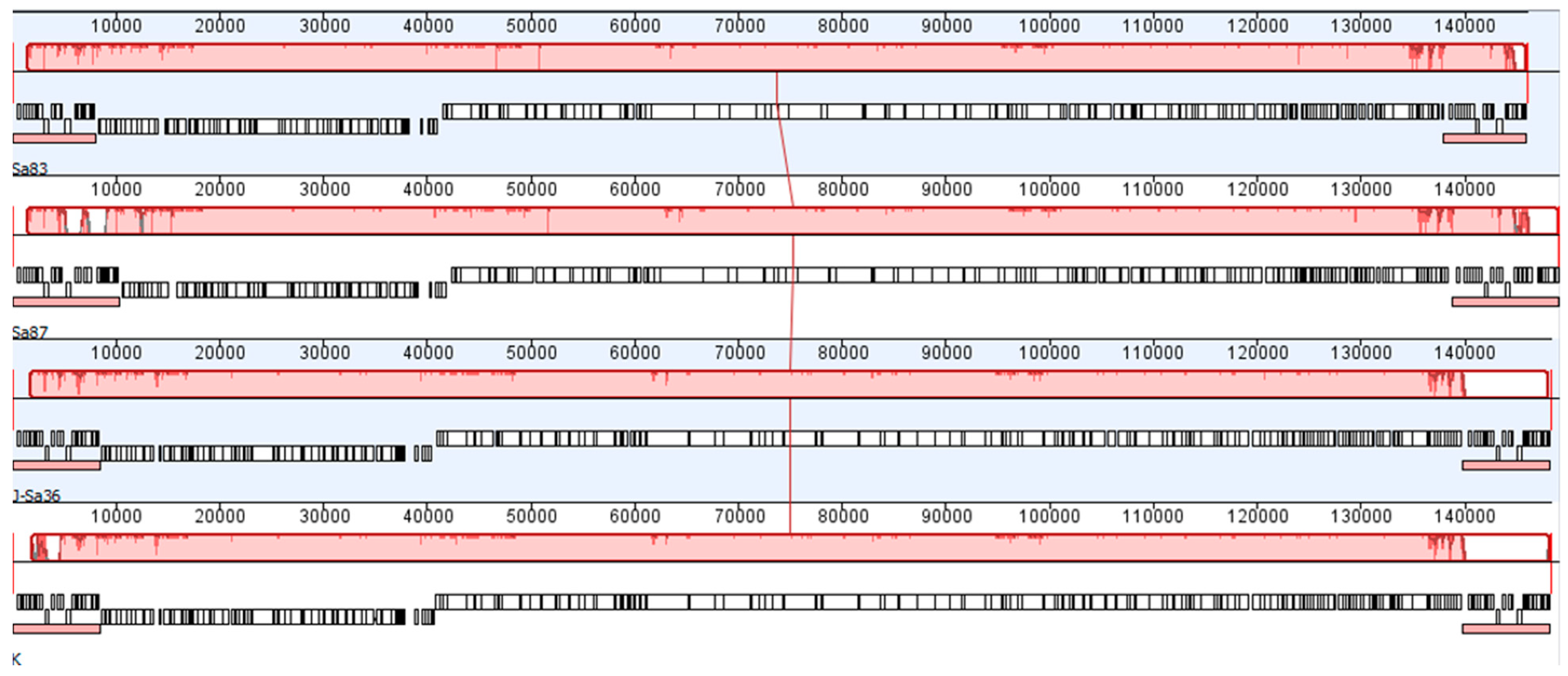
2.5. Genome Sequencing and Analysis
2.6. Animal Studies
2.7. Animal Welfare
3. Results
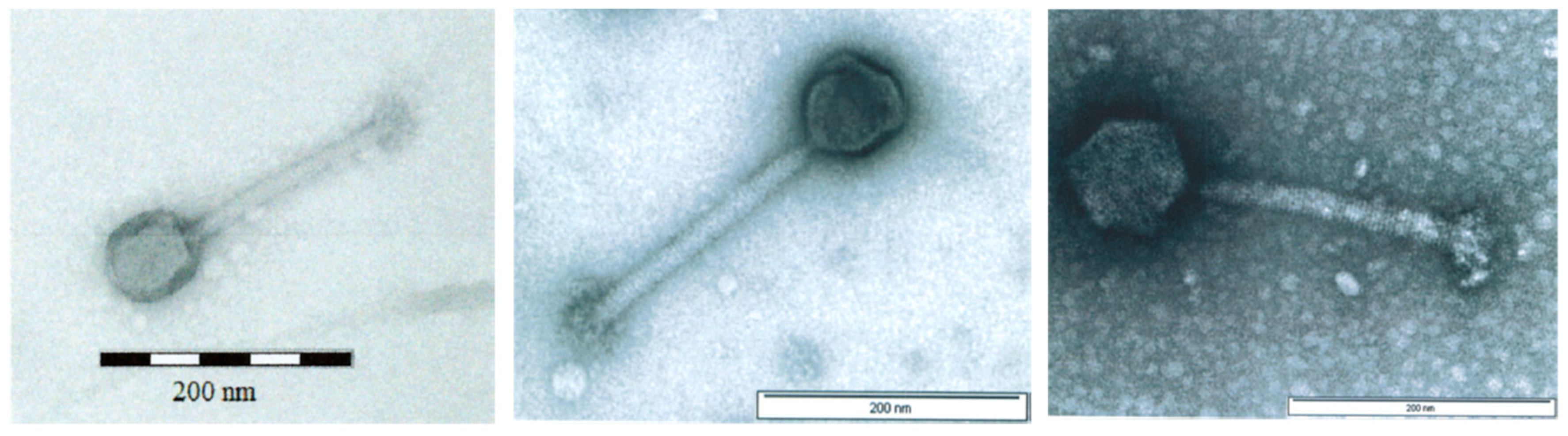
3.1. Physicochemical Characteristics of AB-SA01 Component Phages
3.2. In Vitro Activity of AB-SA01
3.3. Frequency of Resistance and Complementation
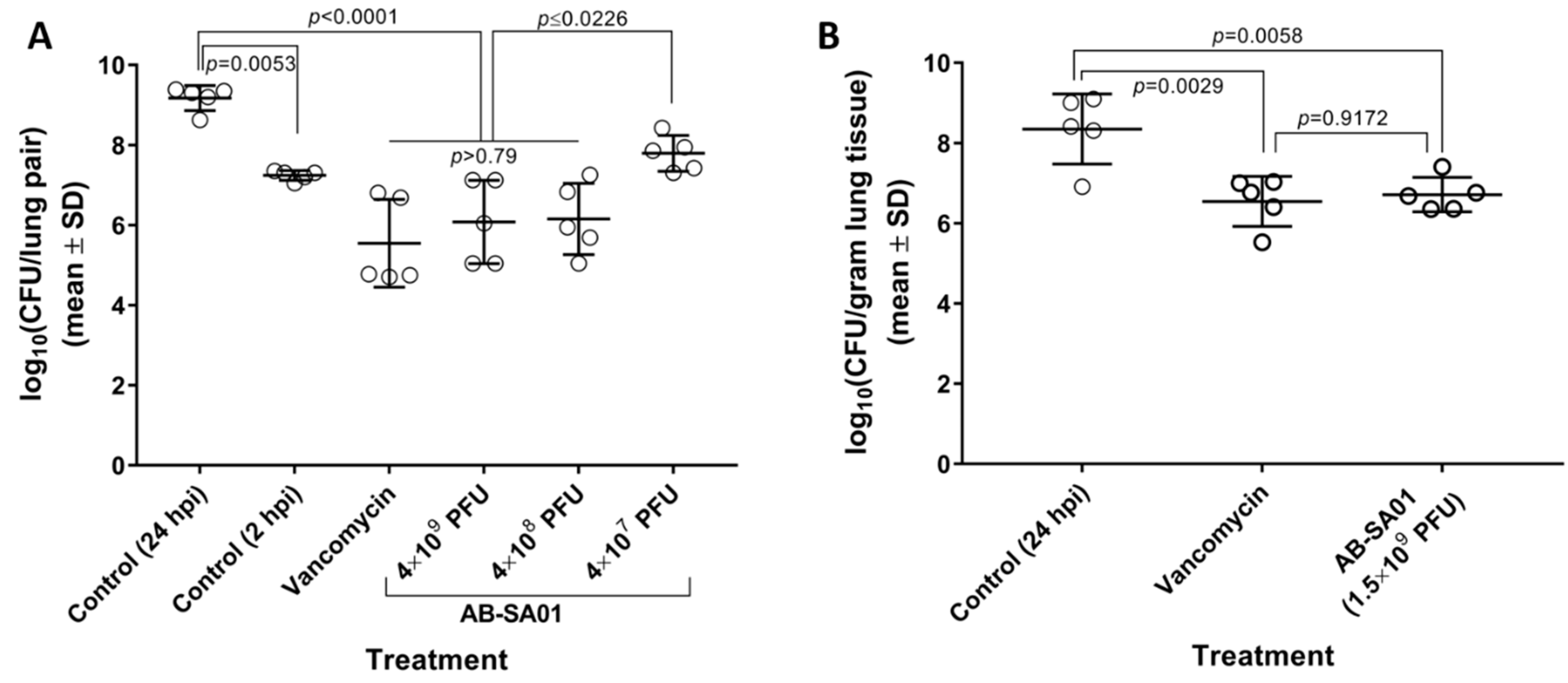
3.4. In Vivo Activity of AB-SA01
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Summers, W.C. The strange history of phage therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, J. Tackling Drug-Resistance Infections Globally: Final Report and Reccomendations; Review on Antimicrobial Resistance: London, UK, 2016. [Google Scholar]
- Rice, L.B. Federal funding for the study of antimicrobial resistance in nosocomial pathogens: No ESKAPE. J. Infect. Dis. 2008, 197, 1079–1081. [Google Scholar] [CrossRef] [PubMed]
- Stefani, S.; Chung, D.R.; Lindsay, J.A.; Friedrich, A.W.; Kearns, A.M.; Westh, H.; Mackenzie, F.M. Methicillin-resistant Staphylococcus aureus (MRSA): Global epidemiology and harmonisation of typing methods. Int. J. Antimicrob. Agents 2012, 39, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Otter, J.A.; French, G.L. Molecular epidemiology of community-associated meticillin-resistant Staphylococcus aureus in Europe. Lancet Infect. Dis. 2010, 10, 227–239. [Google Scholar] [CrossRef]
- WHO. Prioritization of Pathogens to Guide Discovery, Research and Development Of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- CDC. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2013. [Google Scholar]
- Kallberg, C.; Ardal, C.; Salvesen Blix, H.; Klein, E.; Martinez, E.M.; Lindbaek, M.; Outterson, K.; Rottingen, J.A.; Laxminarayan, R. Introduction and geographic availability of new antibiotics approved between 1999 and 2014. PLoS ONE 2018, 13, e0205166. [Google Scholar] [CrossRef] [PubMed]
- Senior, K. FDA approves first drug in new class of antibiotics. Lancet 2000, 355, 1523. [Google Scholar] [CrossRef]
- Ikeda-Dantsuji, Y.; Hanaki, H.; Nakae, T.; Takesue, Y.; Tomono, K.; Honda, J.; Yanagihara, K.; Mikamo, H.; Fukuchi, K.; Kaku, M.; et al. Emergence of Linezolid-Resistant Mutants in a Susceptible-Cell Population of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2011, 55, 2466–2468. [Google Scholar] [CrossRef] [PubMed]
- Morales, G.; Picazo, J.J.; Baos, E.; Candel, F.J.; Arribi, A.; Peláez, B.; Andrade, R.; de la Torre, M.-Á.; Fereres, J.; Sánchez-García, M. Resistance to Linezolid Is Mediated by the cfr Gene in the First Report of an Outbreak of Linezolid-Resistant Staphylococcus aureus. Clin. Infect. Dis. 2010, 50, 821–825. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.S.; Schneider, T.; Sahl, H.-G. Mechanisms of daptomycin resistance in Staphylococcus aureus: Role of the cell membrane and cell wall. Ann. N. Y. Acad. Sci. 2013, 1277, 139–158. [Google Scholar] [CrossRef]
- Dortet, L.; Anguel, N.; Fortineau, N.; Richard, C.; Nordmann, P. In vivo acquired daptomycin resistance during treatment of methicillin-resistant Staphylococcus aureus endocarditis. Int. J. Infect. Dis. 2013, 17, e1076–e1077. [Google Scholar] [CrossRef]
- Stein, G.E.; Babinchak, T. Tigecycline: An update. Diagn. Microbiol. Infect. Dis. 2013, 75, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Long, S.W.; Olsen, R.J.; Mehta, S.C.; Palzkill, T.; Cernoch, P.L.; Perez, K.K.; Musick, W.L.; Rosato, A.E.; Musser, J.M. PBP2a mutations causing high-level ceftaroline resistance in clinical methicillin-resistant Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 2014, 58, 6668–6674. [Google Scholar] [CrossRef] [PubMed]
- Karlowsky, J.A.; Nichol, K.; Zhanel, G.G. Telavancin: Mechanisms of action, in vitro activity, and mechanisms of resistance. Clin. Infect. Dis 2015, 61 (Suppl. 2), S58–S68. [Google Scholar] [CrossRef]
- Jones, R.N.; Moeck, G.; Arhin, F.F.; Dudley, M.N.; Rhomberg, P.R.; Mendes, R.E. Results from Oritavancin Resistance Surveillance Programs (2011 to 2014): Clarification for Using Vancomycin as a Surrogate To Infer Oritavancin Susceptibility. Antimicrob. Agents Chemother. 2016, 60, 3174–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morroni, G.; Brenciani, A.; Brescini, L.; Fioriti, S.; Simoni, S.; Pocognoli, A.; Mingoia, M.; Giovanetti, E.; Barchiesi, F.; Giacometti, A.; et al. A high rate of ceftobiprole resistance among clinical MRSA from a hospital in central Italy. Antimicrob. Agents Chemother. 2018, 62, e01663-1810. [Google Scholar] [CrossRef] [PubMed]
- Werth, B.J.; Jain, R.; Hahn, A.; Cummings, L.; Weaver, T.; Waalkes, A.; Sengupta, D.; Salipante, S.J.; Rakita, R.M.; Butler-Wu, S.M. Emergence of dalbavancin non-susceptible, vancomycin-intermediate Staphylococcus aureus (VISA) after treatment of MRSA central line-associated bloodstream infection with a dalbavancin- and vancomycin-containing regimen. Clin. Microbiol. Infect. 2018, 24, e421–e429. [Google Scholar] [CrossRef] [PubMed]
- Molina-Manso, D.; del Prado, G.; Ortiz-Perez, A.; Manrubia-Cobo, M.; Gomez-Barrena, E.; Cordero-Ampuero, J.; Esteban, J. In vitro susceptibility to antibiotics of staphylococci in biofilms isolated from orthopaedic infections. Int. J. Antimicrob. Agents 2013, 41, 521–523. [Google Scholar] [CrossRef] [PubMed]
- Czaplewski, L.; Bax, R.; Clokie, M.; Dawson, M.; Fairhead, H.; Fischetti, V.A.; Foster, S.; Gilmore, B.F.; Hancock, R.E.; Harper, D.; et al. Alternatives to antibiotics-a pipeline portfolio review. Lancet Infect. Dis. 2016, 16, 239–251. [Google Scholar] [CrossRef]
- O’Neill, A.J.; Cove, J.H.; Chopra, I. Mutation frequencies for resistance to fusidic acid and rifampicin in Staphylococcus aureus. J. Antimicrob. Chemother. 2001, 47, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, S.; Washington, DC, USA. FDA’s Strategic Approach for Combating Antimicrobial Rsistance. Personal Communication, 2018. [Google Scholar]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [Green Version]
- Gorski, A.; Dabrowska, K.; Switala-Jelen, K.; Nowaczyk, M.; Weber-Dabrowska, B.; Boratynski, J.; Wietrzyk, J.; Opolski, A. New insights into the possible role of bacteriophages in host defense and disease. Med. Immunol. 2003, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage Transcytosis Provides a Mechanism To Cross Epithelial Cell Layers. mBio 2017, 8, e01874–e01817. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.J.; Hyman, P. Phage choice, isolation, and preparation for phage therapy. Curr. Pharm. Biotechnol. 2010, 11, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef] [PubMed]
- Casey, E.; van Sinderen, D.; Mahony, J. In Vitro Characteristics of Phages to Guide ‘Real Life’ Phage Therapy Suitability. Viruses 2018, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Reindel, R.; Fiore, C.R. Phage Therapy: Considerations and Challenges for Development. Clin. Infect. Dis. 2017, 64, 1589–1590. [Google Scholar] [CrossRef] [PubMed]
- Carlton, R.M. Phage therapy: Past history and future prospects. Arch. Immunol. Ther. Exp. 1999, 47, 267–274. [Google Scholar]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clinic. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages using the small drop plaque assay system. Methods mol. Biol. 2009, 501, 81–85. [Google Scholar] [PubMed]
- O’Flynn, G.; Ross, R.P.; Fitzgerald, G.F.; Coffey, A. Evaluation of a cocktail of three bacteriophages for biocontrol of Escherichia coli O157:H7. Appl. Environ. Microbiol. 2004, 70, 3417–3424. [Google Scholar] [CrossRef]
- Soding, J. Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed]
- Kadurugamuwa, J.L.; Sin, L.V.; Yu, J.; Francis, K.P.; Kimura, R.; Purchio, T.; Contag, P.R. Rapid Direct Method for Monitoring Antibiotics in a Mouse Model of Bacterial Biofilm Infection. Antimicrob. Agents Chemother. 2003, 47, 3130–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlson, K. Working with bacteriophages: Common techniques and methodological approaches. In Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Tattevin, P.; Diep, B.A.; Jula, M.; Perdreau-Remington, F. Long-term follow-up of methicillin-resistant Staphylococcus aureus molecular epidemiology after emergence of clone USA300 in San Francisco jail populations. J. Clin. Microbiol. 2008, 46, 4056–4057. [Google Scholar] [CrossRef]
- Diep, B.A.; Carleton, H.A.; Chang, R.F.; Sensabaugh, G.F.; Perdreau-Remington, F. Roles of 34 virulence genes in the evolution of hospital- and community-associated strains of methicillin-resistant Staphylococcus aureus. J. Infect. Dis. 2006, 193, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Chambers, H.F.; Graber, C.J.; Szumowski, J.D.; Miller, L.G.; Han, L.L.; Chen, J.H.; Lin, F.; Lin, J.; Phan, T.H.; et al. Emergence of multidrug-resistant, community-associated, methicillin-resistant Staphylococcus aureus clone USA300 in men who have sex with men. Ann. Intern. Med. 2008, 148, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F.; et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Guimin, Z.; Yin, Z.; Paramasivan, S.; Richter, K.; Morales, S.; Wormald, P.J.; Vreugde, S. Bacteriophage effectively kills multidrug resistant Staphylococcus aureus clinical isolates from chronic rhinosinusitis patients. Int. Forum Allergy Rhinol. 2018, 8, 406–414. [Google Scholar]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef]
- Silverman, J.A.; Oliver, N.; Andrew, T.; Li, T. Resistance Studies with Daptomycin. Antimicrob. Agents Chemother. 2001, 45, 1799–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zurenko, G.E.; Yagi, B.H.; Schaadt, R.D.; Allison, J.W.; Kilburn, J.O.; Glickman, S.E.; Hutchinson, D.K.; Barbachyn, M.R.; Brickner, S.J. In vitro activities of U-100592 and U-100766, novel oxazolidinone antibacterial agents. Antimicrob. Agents Chemother. 1996, 40, 839–845. [Google Scholar] [CrossRef] [PubMed]
- De Sordi, L.; Khanna, V.; Debarbieux, L. The Gut Microbiota Facilitates Drifts in the Genetic Diversity and Infectivity of Bacterial Viruses. Cell Host Microbe 2017, 22, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Buckling, A.; Rainey, P.B. Antagonistic coevolution between a bacterium and a bacteriophage. Proc. Biol. Sci. 2002, 269, 931–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabijan, A.; Ho, J.; Lin, R.C.Y.; Maddocks, S.; Gilbey, T.; Sandaradura, I.; Chan, S.; Morales, S.; Venturini, C.; Branston, S.; et al. Safety and tolerability of bacteriophage therapy in the treatment of severe Staphylococcus aureus bacteremia. Manuscript in preparation.
- Slopek, S.; Weber-Dabrowska, B.; Dabrowski, M.; Kucharewicz-Krukowska, A. Results of bacteriophage treatment of suppurative bacterial infections in the years 1981-1986. Arch. Immunol. Ther. Exp. 1987, 35, 569–583. [Google Scholar]
- Weber-Dabrowska, B.; Mulczyk, M.; Gorski, A. Bacteriophage therapy of bacterial infections: An update of our institute’s experience. Arch. Immunol. Ther. Exp. 2000, 48, 547–551. [Google Scholar]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the Eliava Institute. Med. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef]
- Wright, A.; Hawkins, C.H.; Anggard, E.E.; Harper, D.R. A controlled clinical trial of a therapeutic bacteriophage preparation in chronic otitis due to antibiotic-resistant Pseudomonas aeruginosa; a preliminary report of efficacy. Clin. Otolaryngol. 2009, 34, 349–357. [Google Scholar] [CrossRef]
- Chanishvili, N. Phage therapy–history from Twort and d’Herelle through Soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar]
- Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Fortuna, W.; Letkiewicz, S.; Szufnarowski, K.; Pawelczyk, Z.; Rogoz, P.; Klak, M.; Wojtasik, E.; et al. Clinical aspects of phage therapy. Adv. Virus Res. 2012, 83, 73–121. [Google Scholar]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Jennes, S.; Merabishvili, M.; Soentjens, P.; Pang, K.W.; Rose, T.; Keersebilck, E.; Soete, O.; François, P.-M.; Teodorescu, S.; Verween, G.; et al. Use of bacteriophages in the treatment of colistin-only-sensitive Pseudomonas aeruginosa septicaemia in a patient with acute kidney injury-a case report. Crit. Care 2017, 21, 129. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evolut. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed]
- Gorski, A.; Miedzybrodzki, R.; Borysowski, J.; Weber-Dabrowska, B.; Lobocka, M.; Fortuna, W.; Letkiewicz, S.; Zimecki, M.; Filby, G. Bacteriophage therapy for the treatment of infections. Curr. Opin. Investig. Drugs 2009, 10, 766–774. [Google Scholar] [PubMed]
- Chaudhry, W.N.; Concepcion-Acevedo, J.; Park, T.; Andleeb, S.; Bull, J.J.; Levin, B.R. Synergy and Order Effects of Antibiotics and Phages in Killing Pseudomonas aeruginosa Biofilms. PLoS ONE 2017, 12, e0168615. [Google Scholar] [CrossRef] [PubMed]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. PeerJ 2014, 2, e590. [Google Scholar] [CrossRef] [PubMed]
- Oechslin, F. Resistance Development to Bacteriophages Occurring during Bacteriophage Therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Di Santo, J.P.; Weitz, J.S.; Debarbieux, L. Synergy between the Host Immune System and Bacteriophage Is Essential for Successful Phage Therapy against an Acute Respiratory Pathogen. Cell Host Microbe 2017, 22, 38–47.e4. [Google Scholar] [CrossRef]
- Skerrett, S.J.; Liggitt, H.D.; Hajjar, A.M.; Wilson, C.B. Cutting edge: Myeloid differentiation factor 88 is essential for pulmonary host defense against Pseudomonas aeruginosa but not Staphylococcus aureus. J. Immunol. 2004, 172, 3377–3381. [Google Scholar] [CrossRef]
- Hirche, T.O.; Benabid, R.; Deslee, G.; Gangloff, S.; Achilefu, S.; Guenounou, M.; Lebargy, F.; Hancock, R.E.; Belaaouaj, A. Neutrophil elastase mediates innate host protection against Pseudomonas aeruginosa. J. Immunol. 2008, 181, 4945–4954. [Google Scholar] [CrossRef]
- Stapels, D.A.; Geisbrecht, B.V.; Rooijakkers, S.H. Neutrophil serine proteases in antibacterial defense. Curr. Opin. Microbiol. 2015, 23, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapels, D.A.; Ramyar, K.X.; Bischoff, M.; von Kockritz-Blickwede, M.; Milder, F.J.; Ruyken, M.; Eisenbeis, J.; McWhorter, W.J.; Herrmann, M.; van Kessel, K.P.; et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 13187–13192. [Google Scholar] [CrossRef] [PubMed]
- Ajuebor, J.; Buttimer, C.; Arroyo-Moreno, S.; Chanishvili, N.; Gabriel, E.M.; O’Mahony, J.; McAuliffe, O.; Neve, H.; Franz, C.; Coffey, A. Comparison of Staphylococcus Phage K with Close Phage Relatives Commonly Employed in Phage Therapeutics. Antibiotics 2018, 7, 37. [Google Scholar] [CrossRef] [PubMed]
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brussow, H. Metagenome analysis of Russian and Georgian Pyophage cocktails and a placebo-controlled safety trial of single phage versus phage cocktail in healthy Staphylococcus aureus carriers. Environ. Microbiol. 2018, 20, 3278–3293. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Hejnowicz, M.S.; Dabrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dabrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of staphylococcal Twort-like phages--potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [PubMed]
- Adriaenssens, E.M.; Wittmann, J.; Kuhn, J.H.; Turner, D.; Sullivan, M.B.; Dutilh, B.E.; Jang, H.B.; van Zyl, L.J.; Klumpp, J.; Lobocka, M.; et al. Taxonomy of prokaryotic viruses: 2017 update from the ICTV Bacterial and Archaeal Viruses Subcommittee. Arch. Virol. 2018, 163, 1125–1129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barylski, J.; Enault, F.; Dutilh, B.E.; Schuller, M.B.P.; Edwards, R.A.; Gillis, A.; Klumpp, J.; Knezevic, P.; Krupovic, M.; Kuhn, J.H.; et al. Analysis of Spounaviruses as a Case 3 Study for the Overdue Reclassification of 4 Tailed Bacteriophages. bioRxiv 2017. [Google Scholar] [CrossRef]
- Drilling, A.; Morales, S.; Jardeleza, C.; Vreugde, S.; Speck, P.; Wormald, P.J. Bacteriophage reduces biofilm of Staphylococcus aureus ex vivo isolates from chronic rhinosinusitis patients. Am. J. Rhinol. Allergy 2014, 28, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Drilling, A.; Morales, S.; Boase, S.; Jervis-Bardy, J.; James, C.; Jardeleza, C.; Tan, N.C.; Cleland, E.; Speck, P.; Vreugde, S.; et al. Safety and efficacy of topical bacteriophage and ethylenediaminetetraacetic acid treatment of Staphylococcus aureus infection in a sheep model of sinusitis. Int. Forum Allergy Rhinol. 2014, 4, 176–186. [Google Scholar] [CrossRef]
- Ooi MLD, A.J.; Morales, S.; Fong, S.; Moraitis, S.; Macias-Valle, L.; Vreugde, S.; Psaltis, A.; Wormald, P.-J. Phage Therapy for S. aureus Chronic Rhinosinusitis: A Phase 1 First-in-Human Study. Manuscript in preparation.




{kind=link}
{kind=link}
{kind=link}
| Panel Type | Phage | Percentage of Total Isolates Sensitive to Indicated Phage | % of MDR Isolates Sensitive to AB-SA01 | ||||
|---|---|---|---|---|---|---|---|
| Panel | Sa83 | Sa87 | J-Sa36 | AB-SA01 | |||
| Selection | AmpliPhi Reference Panel (n = 68) 1 | 85.2% | 86.8% | 76.4% | 94.1% | 94% (61/65) | |
| Prevalence 2 | 2013 Global Panel (n = 53) | 96.2% | 96.2% | 86.8% | 100% | 100% (38/38) | |
| 2015 Global Panel (n = 60) | 85.0% | 93.3% | 75.0% | 96.7% | 100% (28/28) | ||
| 2016 Global Panel (n = 60) | 80.0% | 83.3% | 63.3% | 88.3% | 94% (30/32) | ||
| Targeted | CDC VISA Panel (n = 14) | 64.3% | 64.3% | 64.3% | 64.3% | 69% (9/13) | |
| Regional USA300 Panel (n = 29) 3 | 100% | 100% | 100% | 100% | 100% (29/29) | ||
| Ghent CRS Panel (n = 90) | NT | NT | NT | 96.7% | Insufficient AST data | ||
| NA | Expanded Access Requests (n = 27) 4 | 85.2% | 92.6% | 88.9% | 96.3% | Insufficient AST data | |
| Summary Values | Diversity Panels: Selection and Prevalence (n = 241) | 94.6% | - | ||||
| All Panels (n = 401) | 94.5% | 95% (n = 205) | |||||
| Bacteria | Number of Strains Tested | Number of Strains Productively Infected | ||||
|---|---|---|---|---|---|---|
| Order | Genus, Species | Sa83 | Sa87 | J-Sa36 | AB-SA01 | |
| Bacillales | Staphylococcus epidermidis | 5 | 2 | 2 | 2 | 2 |
| Lactobacillales | Streptococcus spp. | 3 | 0 | 0 | 0 | 0 |
| Corynebacteriales | Corynebacterium spp. | 4 | 0 | 0 | 0 | 0 |
| Micrococcales | Micrococcus luteus | 1 | 0 | 0 | 0 | 0 |
| Burkholderiales | Achromobacter xylosoxidans | 1 | 0 | 0 | 0 | 0 |
| Burkholderia cepacia | 1 | 0 | 0 | 0 | 0 | |
| Pseudomonales | Acinetobacter baumannii | 1 | 0 | 0 | 0 | 0 |
| Pseudomonas aeruginosa | 3 | 0 | 0 | 0 | 0 | |
| Pseudomonas oryzihabitans | 1 | 0 | 0 | 0 | 0 | |
| Enterobacteriales | Enterobacter cloacae | 1 | 0 | 0 | 0 | 0 |
| Escherichia coli | 1 | 0 | 0 | 0 | 0 | |
| Klebsiella pneumoniae | 1 | 0 | 0 | 0 | 0 | |
| Pantoea agglomerans | 1 | 0 | 0 | 0 | 0 | |
| Xanthamonadales | Stenotrophomonas maltophilia | 1 | 0 | 0 | 0 | 0 |
| Phage Used to Generate BIM | Bacterial Lawn | BIM Confirmation 1 | Test for Complementation | |||||
|---|---|---|---|---|---|---|---|---|
| Sa83 | Sa87 | J-Sa36 | Sa76 | Sa81 | J-Sa37 | |||
| Sa87 | parental | S | S | S | S | S | S | S |
| BIM 1 | I | S | - | S | S | S | R | |
| BIM 2 | I | S | - | S | S | S | R | |
| BIM 3 | NG 2 | - | - | - | - | - | - | |
| BIM 4 | I | S | - | S | S | S | R | |
| BIM 5 | I | S | - | I | S | S | R | |
| BIM 6 | I | S | - | S | S | S | R | |
| BIM 7 | I | S | - | S | S | S | R | |
| BIM 8 | I | S | - | I | S | S | R | |
| BIM 9 | I | S | - | I | S | S | R | |
| BIM 10 | I | S | - | I | S | S | R | |
| J-Sa36 | Parental | S | S | S | S | - | - | S |
| BIM 1 | I | S | I | - | - | - | R | |
| BIM 2 | I | I | I | - | - | - | R | |
| BIM 3 | I | I | S | - | - | - | S | |
| BIM 4 | I | I | I | - | - | - | S | |
| Phage | After 24 h Plate Incubation | After 48 h Plate Incubation | ||||
|---|---|---|---|---|---|---|
| Replicate 1 1 | Replicate 2 | Replicate 3 | Replicate 1 | Replicate 2 | Replicate 3 | |
| Sa83 | 1.1E-8 | 3.8E-9 | 5.0E-9 | 7.1E-9 | 3.8E-9 | 6.7E-9 |
| Sa87 | 2.0E-8 | 5.0E-9 | 5.0E-9 | 1.7E-8 | 5.0E-9 | 5.0E-9 |
| J-Sa36 | 2.9E-9 | 2.5E-9 | 1.2E-8 | 2.9E-9 | 1.3E-9 | 5.0E-9 |
| AB-SA01 2 | 1.4E-9 | 3.8E-9 | 3.3E-9 | 2.9E-9 | 0 3 | 3.3E-9 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lehman, S.M.; Mearns, G.; Rankin, D.; Cole, R.A.; Smrekar, F.; Branston, S.D.; Morales, S. Design and Preclinical Development of a Phage Product for the Treatment of Antibiotic-Resistant Staphylococcus aureus Infections. Viruses 2019, 11, 88. https://doi.org/10.3390/v11010088
Lehman SM, Mearns G, Rankin D, Cole RA, Smrekar F, Branston SD, Morales S. Design and Preclinical Development of a Phage Product for the Treatment of Antibiotic-Resistant Staphylococcus aureus Infections. Viruses. 2019; 11(1):88. https://doi.org/10.3390/v11010088
Chicago/Turabian StyleLehman, Susan M., Gillian Mearns, Deborah Rankin, Robert A. Cole, Frenk Smrekar, Steven D. Branston, and Sandra Morales. 2019. "Design and Preclinical Development of a Phage Product for the Treatment of Antibiotic-Resistant Staphylococcus aureus Infections" Viruses 11, no. 1: 88. https://doi.org/10.3390/v11010088