Naturally Occurring Mutations within HBV Surface Promoter II Sequences Affect Transcription Activity, HBsAg and HBV DNA Levels in HBeAg-Positive Chronic Hepatitis B Patients


Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Laboratory Tests for Patient Samples
2.3. HBV DNA Amplification, Sequencing, and Sequence Analysis
2.4. Plasmids
2.5. Cell Cultures, Transfection and Luciferase Assay
2.6. Detection of HBsAg, HBV DNA, HBV Total RNAs and Pregenomic RNA in Cell Culture System
2.7. Statistical Analyses
2.8. GenBank Accession Numbers
3. Results
3.1. Patient Characteristics
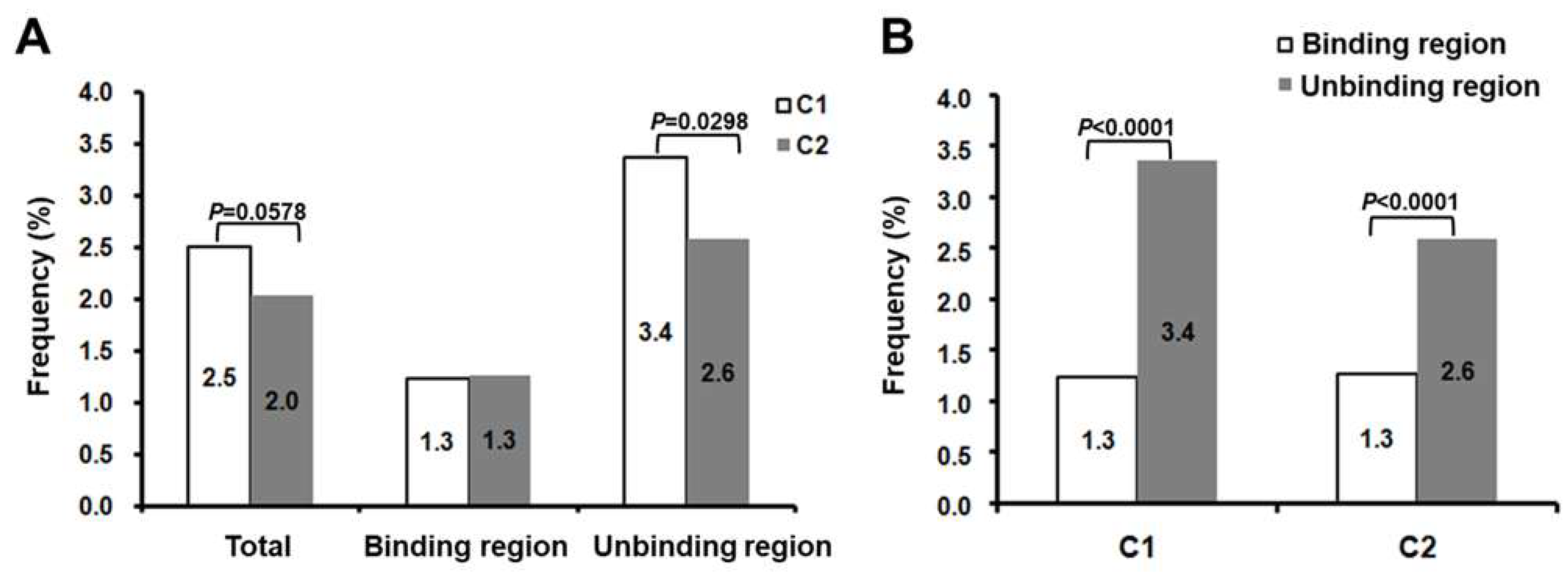
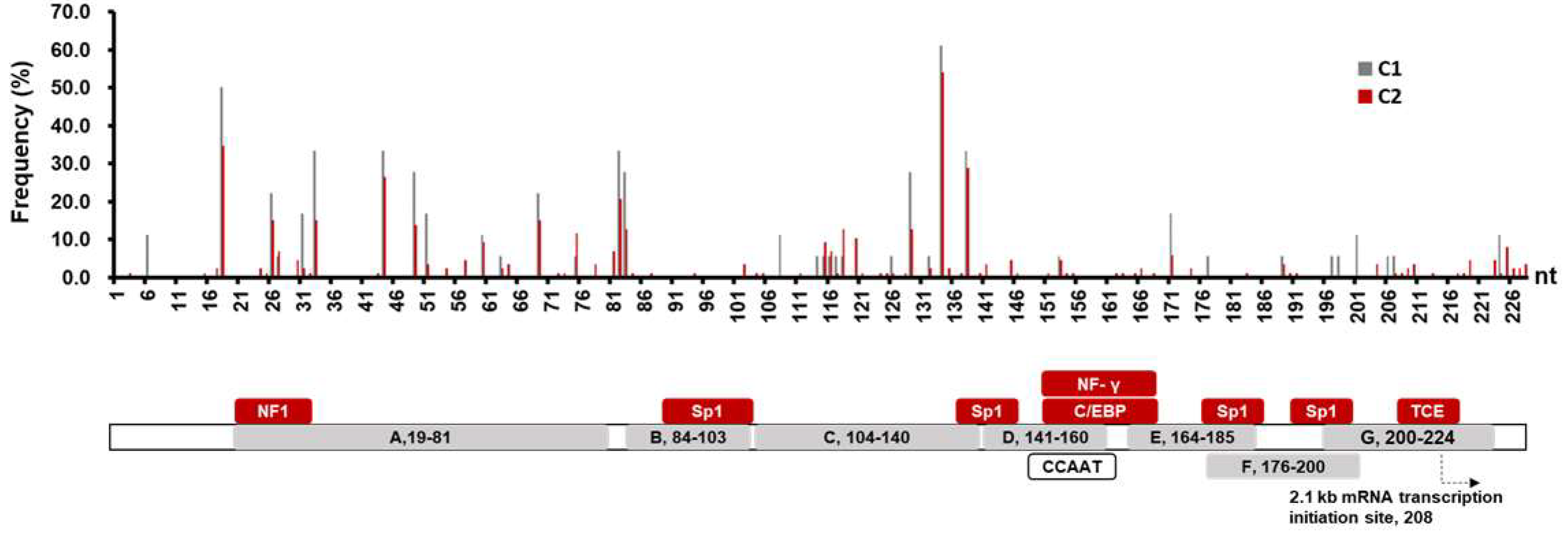
3.2. Comparison of SPII Mutations in C1 and C2 Sequences
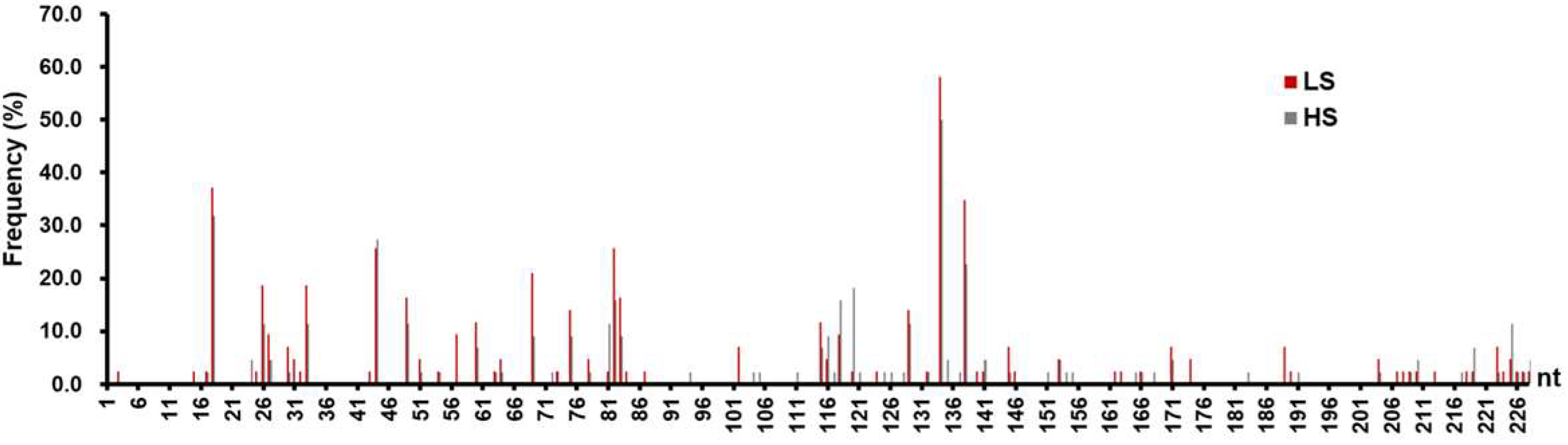
3.3. Mutation Analysis on the C2 SPII Sequences
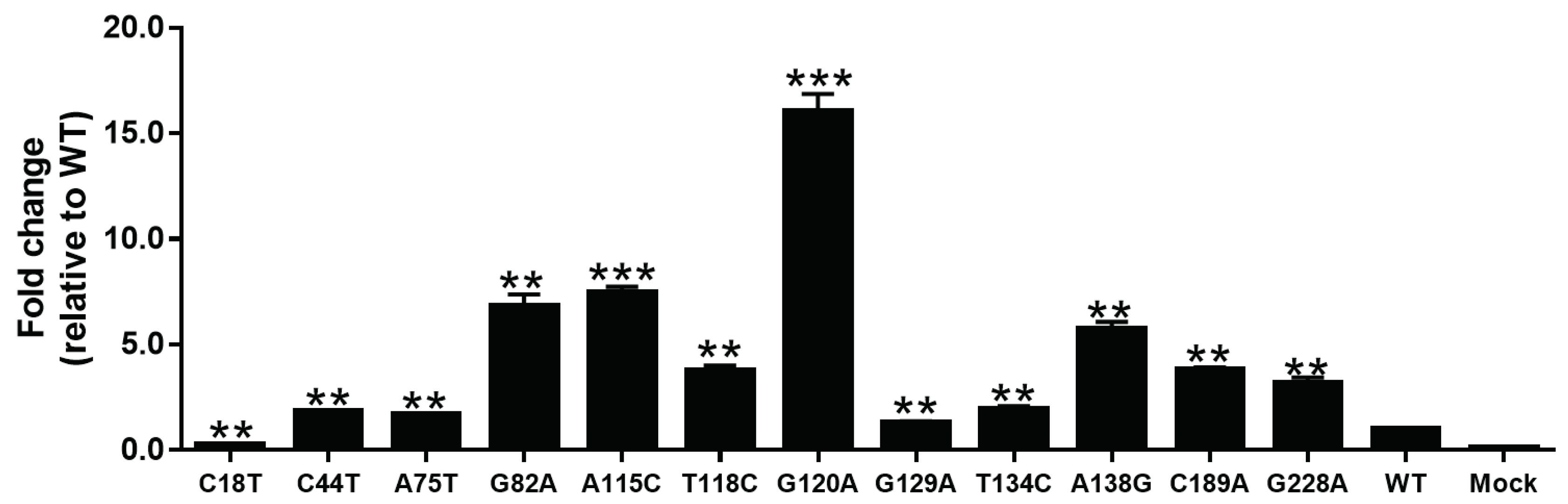
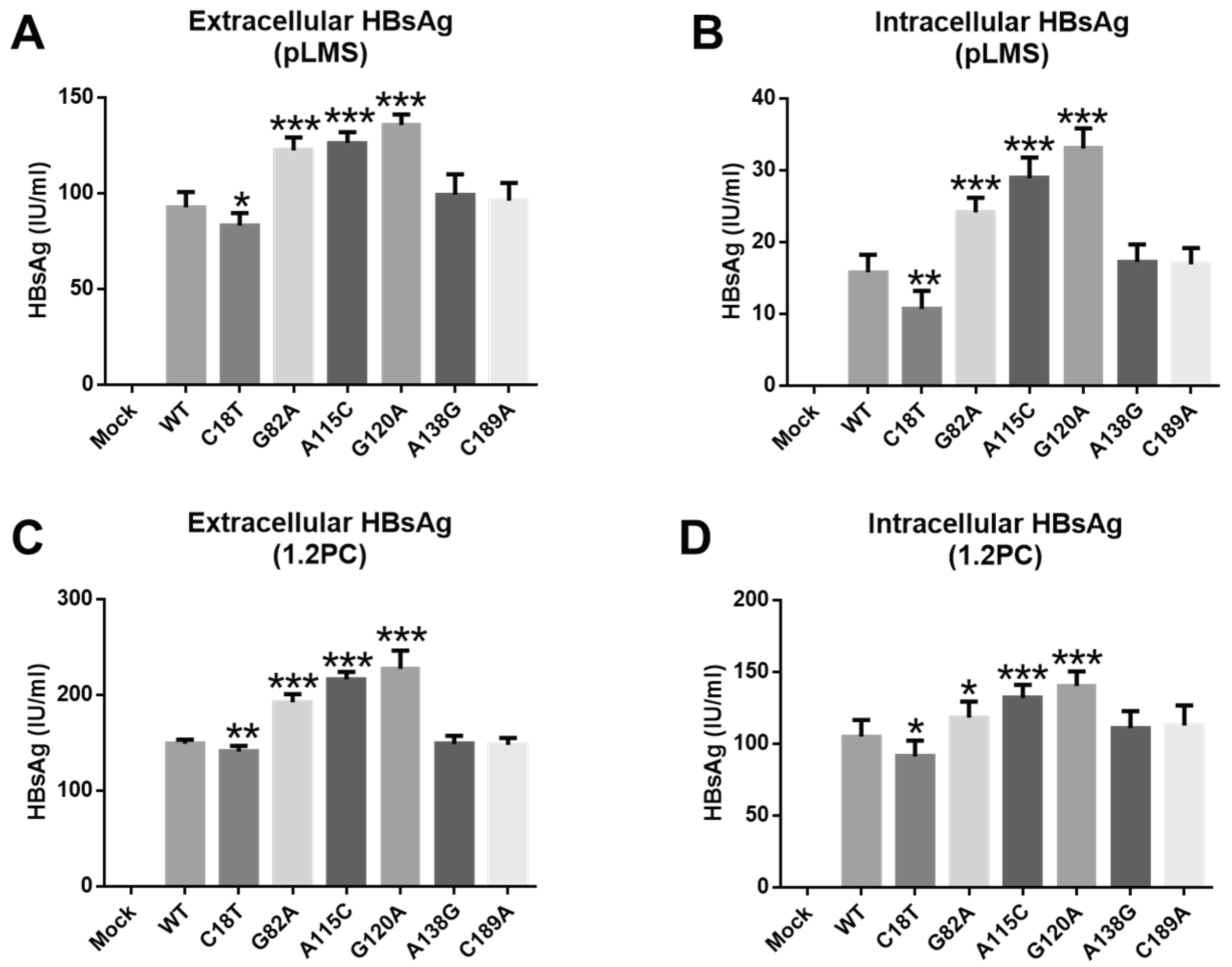
3.4. The Influence of SPII Mutations on Promoter Activity
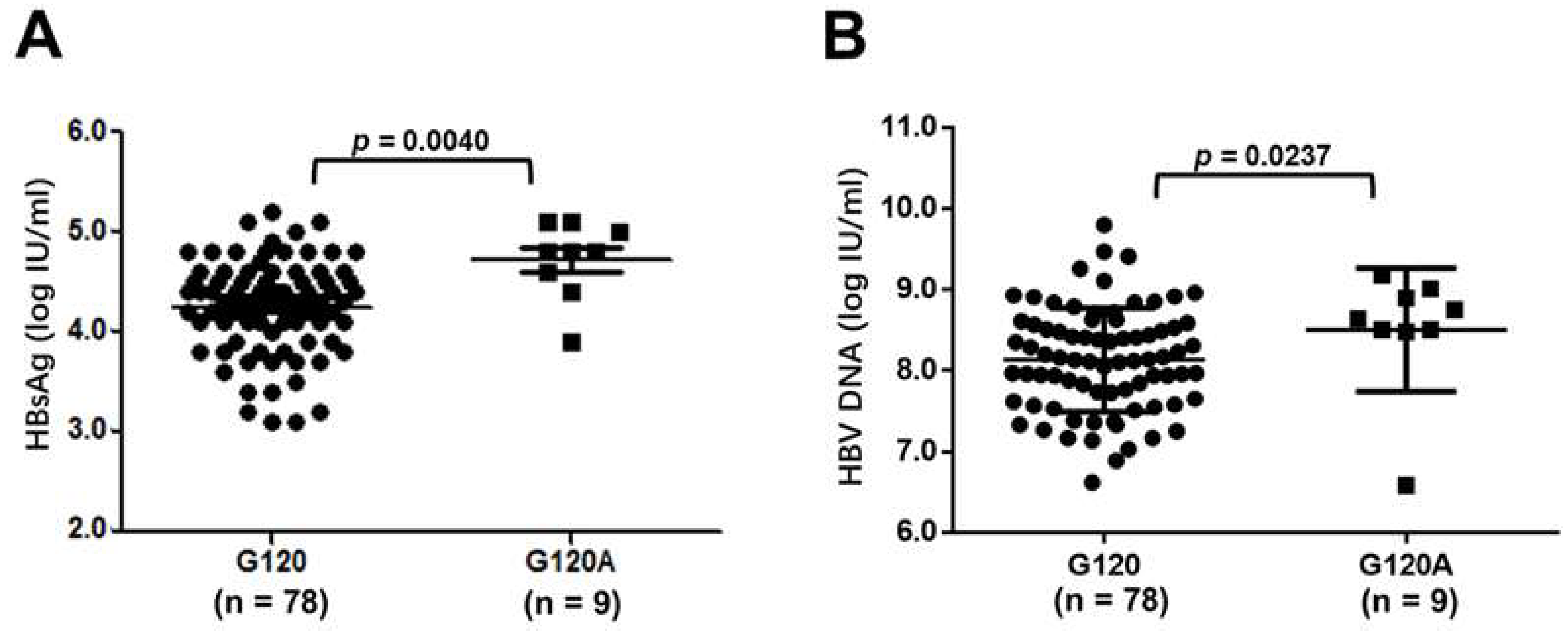
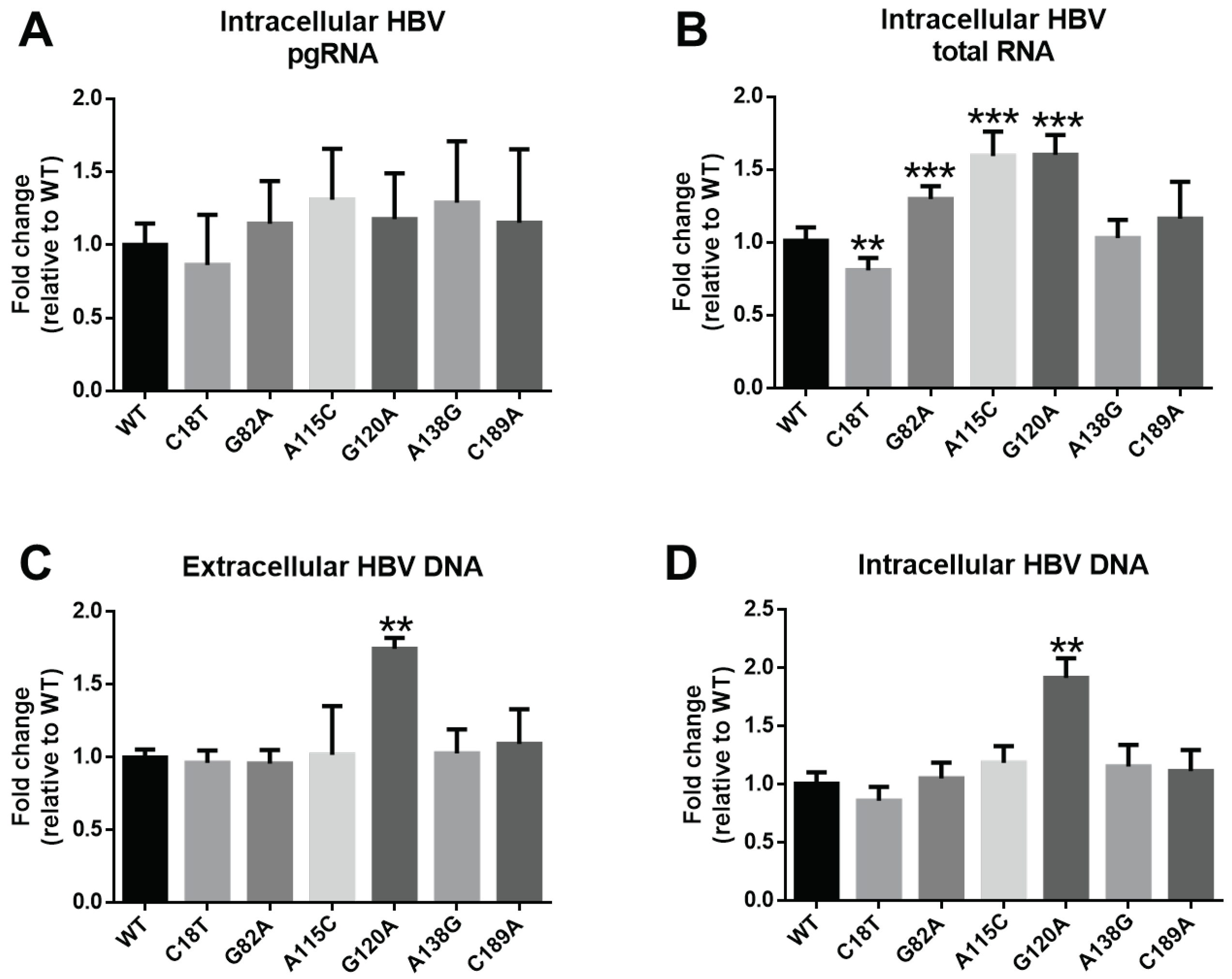
3.5. Influence of SPII Mutations on HBsAg Levels and HBV Replication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stasi, C.; Silvestri, C.; Voller, F. Emerging trends in epidemiology of hepatitis B Virus Infection. J. Clin. Transl. Hepatol. 2017, 5, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Sandhu, P.; Haque, M.; Humphries-Bickley, T.; Ravi, S.; Song, J. Hepatitis B virus immunopathology, model systems, and current therapies. Front. Immunol. 2017, 8, 436. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Liu, K. Complete and incomplete hepatitis B virus particles: Formation, function, and application. Viruses 2017, 9, 56. [Google Scholar] [CrossRef]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; König, A. Molecular virology of hepatitis B virus and targets for antiviral intervention. Intervirology 2014, 57, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Trépo, C.; Chan, H.L.; Lok, A. Hepatitis B virus infection. Lancet 2014, 384, 2053–2063. [Google Scholar] [CrossRef]
- Martinot-Peignoux, M.; Asselah, T.; Marcellin, P. HBsAg quantification to optimize treatment monitoring in chronic hepatitis B patients. Liver Int. 2015, 35 (Suppl. 1), 82–90. [Google Scholar] [CrossRef]
- Martinot-Peignoux, M.; Lapalus, M.; Asselah, T.; Marcellin, P. HBsAg quantification: Useful for monitoring natural history and treatment outcome. Liver Int. 2014, 34 (Suppl. 1), 97–107. [Google Scholar] [CrossRef]
- Su, M.Z.; Xiang, K.H.; Li, Y.; Li, Y.T.; Deng, J.; Xu, X.Z.; Yan, L.; Zhuang, H.; Li, T. Higher detection rates of amino acid substitutions in HBV reverse transcriptase/surface protein overlapping sequence is correlated with lower serum HBV DNA and HBsAg levels in HBeAg-positive chronic hepatitis B patients with subgenotype B2. Infect. Genet. Evol. 2016, 40, 275–281. [Google Scholar] [CrossRef]
- Moolla, N.; Kew, M.; Arbuthnot, P. Regulatory elements of hepatitis B virus transcription. J. Viral Hepat. 2002, 9, 323–331. [Google Scholar] [CrossRef]
- Shi, K.Q.; Chen, X.; Xu, Y. Regulatory mechanisms for HBV replication. Int. J. Clin. Exp. Med. 2017, 10, 215–223. [Google Scholar]
- Bock, C.T.; Kubicka, S.; Manns, M.P.; Trautwein, C. Two control elements in the hepatitis B virus S-promoter are important for full promoter activity mediated by CCAAT-binding factor. Hepatology 1999, 29, 1236–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De-Medina, T.; Faktor, O.; Shaul, Y. The S promoter of hepatitis B virus is regulated by positive and negative elements. Mol. Cell. Biol. 1988, 8, 2449–2455. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Ren, X.; Liu, Y.; Li, X.; Bai, S.; Zhong, Y.; Wang, L.; Mao, P.; Wang, H.; Xin, S.; et al. Association of hepatitis B virus mutations in basal core promoter and precore regions with severity of liver disease: An investigation of 793 Chinese patients with mild and severe chronic hepatitis B and acute-on-chronic liver failure. J. Gastroenterol. 2011, 46, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Lindh, M.; Hannoun, C.; Dhillon, A.P.; Norkrans, G.; Horal, P. Core promoter mutations and genotypes in relation to viral replication and liver damage in East Asian hepatitis B virus carriers. J. Infect. Dis. 1999, 179, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Meier-Stephenson, V.; Bremner, W.T.R.; Dalton, C.S.; van Marle, G.; Coffin, C.S.; Patel, T.R. Comprehensive analysis of hepatitis B virus promoter region mutations. Viruses 2018, 10, 603. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Piwon, N.; Will, H. Wild-type levels of pregenomic RNA and replication but reduced pre-C RNA and e-antigen synthesis of hepatitis B virus with C (1653) --> T, A (1762) --> T and G (1764) --> A mutations in the core promoter. J. Gen. Virol. 1998, 79 Pt 2, 375–380. [Google Scholar] [CrossRef]
- Sengupta, S.; Rehman, S.; Durgapal, H.; Acharya, S.K.; Panda, S.K. Role of surface promoter mutations in hepatitis B surface antigen production and secretion in occult hepatitis B virus infection. J. Med. Virol. 2007, 79, 220–228. [Google Scholar] [CrossRef]
- Biswas, B.; Kandpal, M.; Vivekanandan, P. A G-quadruplex motif in an envelope gene promoter regulates transcription and virion secretion in HBV genotype B. Nucleic Acids Res. 2017, 45, 11268–11280. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Cheng, J.; Sun, Y.; Chen, X.; Li, T.; Wang, H.; Jiang, J.; Chen, X.; Long, H.; Tang, H.; et al. Randomized, three-arm study to optimize lamivudine efficacy in hepatitis B e antigen-positive chronic hepatitis B patients. J. Gastroenterol. Hepatol. 2015, 30, 748–755. [Google Scholar] [CrossRef]
- Hou, J.; Wang, G.; Wang, F.; Cheng, J.; Ren, H.; Zhuang, H.; Sun, J.; Li, L.; Li, J.; Meng, Q.; et al. Guideline of prevention and treatment for chronic hepatitis B (2015 Update). J. Clin. Transl. Hepatol. 2017, 5, 297–318. [Google Scholar] [CrossRef] [PubMed]
- Xiang, K.; Michailidis, E.; Ding, H.; Peng, Y.; Su, M.; Li, Y.; Liu, X.; Dao Thi, V.L.; Wu, X.; Schneider, W.M.; et al. Effects of amino acid substitutions in hepatitis B virus surface protein on virion secretion, antigenicity, HBsAg and viral DNA. J. Hepatol. 2017, 66, 288–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, K.H. The Study of the Influence and Mechanisms of Hepatitis B Surface Protein Substitution on Hepatitis B Surface Antigen Levels. Ph.D. Thesis, Peking University, Beijing, China, 2017. [Google Scholar]
- Hao, R.; Xiang, K.H.; Peng, Y.Q.; Hou, J.L.; Sun, J.; Li, Y.; Su, M.Z.; Yan, L.; Zhuang, H.; Li, T. Naturally occurring deletion/insertion mutations within HBV whole genome sequences in HBeAg-positive chronic hepatitis B patients are correlated with baseline serum HBsAg and HBeAg levels and might predict a shorter interval to HBeAg loss and seroconversion during antiviral treatment. Infect. Genet. Evol. 2015, 33, 261–268. [Google Scholar]
- Peng, Y.Q.; Liu, B.M.; Hou, J.L.; Sun, J.; Hao, R.; Xiang, K.H.; Yan, L.; Zhang, J.; Zhuang, H.; Li, T. Naturally occurring deletions/insertions in HBV core promoter tend to decrease in hepatitis B e antigen-positive chronic hepatitis B patients during antiviral therapy. Antivir. Ther. 2015, 20, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Ryu, C.J.; Hong, H.J. Hepatitis B virus preS1 functions as a transcriptional activation domain. J. Gen. Virol. 1997, 78 Pt 5, 1083–1086. [Google Scholar] [CrossRef]
- Barrera, A.; Guerra, B.; Notvall, L.; Lanford, R.E. Mapping of the hepatitis B virus pre-S1 domain involved in receptor recognition. J. Virol. 2005, 79, 9786–9798. [Google Scholar] [CrossRef]
- Chen, P.; Gan, Y.; Han, N.; Fang, W.; Li, J.; Zhao, F.; Hu, K.; Rayner, S. Computational evolutionary analysis of the overlapped surface (S) and polymerase (P) region in hepatitis B virus indicates the spacer domain in P is crucial for survival. PLoS ONE 2013, 8, e60098. [Google Scholar] [CrossRef]
- Lin, C.G.; Yang, S.J.; Hwang, W.L.; Su, T.S.; Lo, S.J. Demonstration of the presence of protease-cutting site in the spacer of hepatitis B viral Pol protein. J. Virol. Methods 1995, 51, 61–73. [Google Scholar] [CrossRef]
- Peiffer, K.H.; Kuhnhenn, L.; Jiang, B.; Mondorf, A.; Vermehren, J.; Knop, V.; Susser, S.; Walter, D.; Dietz, J.; Carra, G.; et al. Divergent preS Sequences in Virion-Associated Hepatitis B Virus Genomes and Subviral HBV Surface Antigen Particles From HBV e Antigen-Negative Patients. J. Infect. Dis. 2018, 218, 114–123. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Value (n = 106) |
|---|---|
| Age, median (range) | 31.5 (18–58) |
| Gender M/F (% M) | 80/26 (75.5) |
| HBV DNA (log IU/mL), median (range) | 8.1 (6.6–9.8) |
| HBsAg (log IU/mL), median (range) | 4.4 (3.1–5.2) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hao, R.; Xiang, K.; Shi, Y.; Zhao, D.; Tian, H.; Xu, B.; Zhu, Y.; Dong, H.; Ding, H.; Zhuang, H.; et al. Naturally Occurring Mutations within HBV Surface Promoter II Sequences Affect Transcription Activity, HBsAg and HBV DNA Levels in HBeAg-Positive Chronic Hepatitis B Patients. Viruses 2019, 11, 78. https://doi.org/10.3390/v11010078
Hao R, Xiang K, Shi Y, Zhao D, Tian H, Xu B, Zhu Y, Dong H, Ding H, Zhuang H, et al. Naturally Occurring Mutations within HBV Surface Promoter II Sequences Affect Transcription Activity, HBsAg and HBV DNA Levels in HBeAg-Positive Chronic Hepatitis B Patients. Viruses. 2019; 11(1):78. https://doi.org/10.3390/v11010078
Chicago/Turabian StyleHao, Ran, Kuanhui Xiang, Yan Shi, Dong Zhao, Huifang Tian, Baohong Xu, Yufang Zhu, Huan Dong, Hai Ding, Hui Zhuang, and et al. 2019. "Naturally Occurring Mutations within HBV Surface Promoter II Sequences Affect Transcription Activity, HBsAg and HBV DNA Levels in HBeAg-Positive Chronic Hepatitis B Patients" Viruses 11, no. 1: 78. https://doi.org/10.3390/v11010078