The Involvement of Histone H3 Acetylation in Bovine Herpesvirus 1 Replication in MDBK Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Antibodies and Reagents
2.3. Cytotoxicity Assays by Trypan-Blue Exclusion Test
2.4. Western Blotting Analysis
2.5. Immunoprecipitation (IP) Assay
2.6. Virus Replication Inhibition Assay
2.7. Quantification of mRNA by qRT-PCR
3. Results
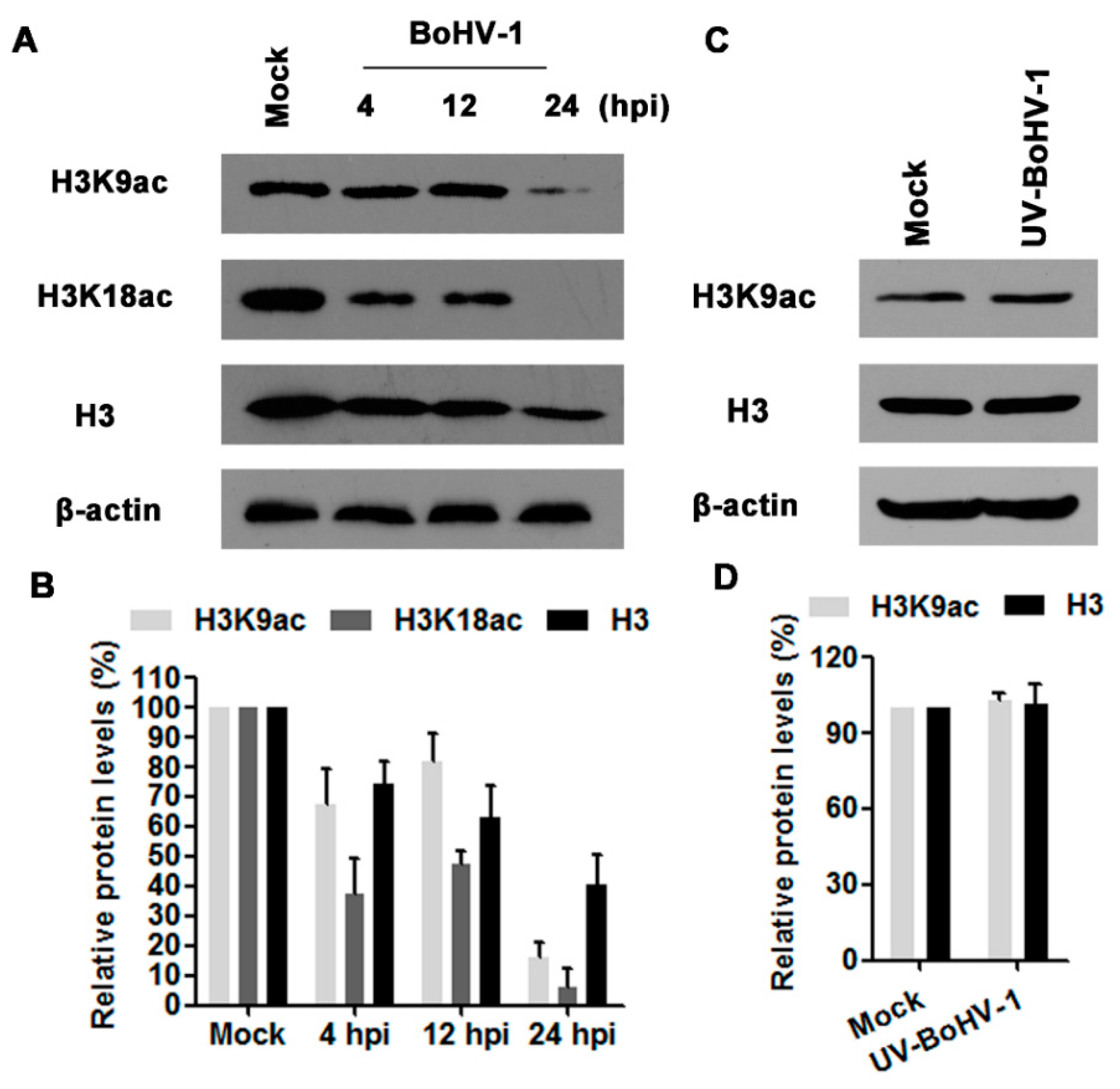
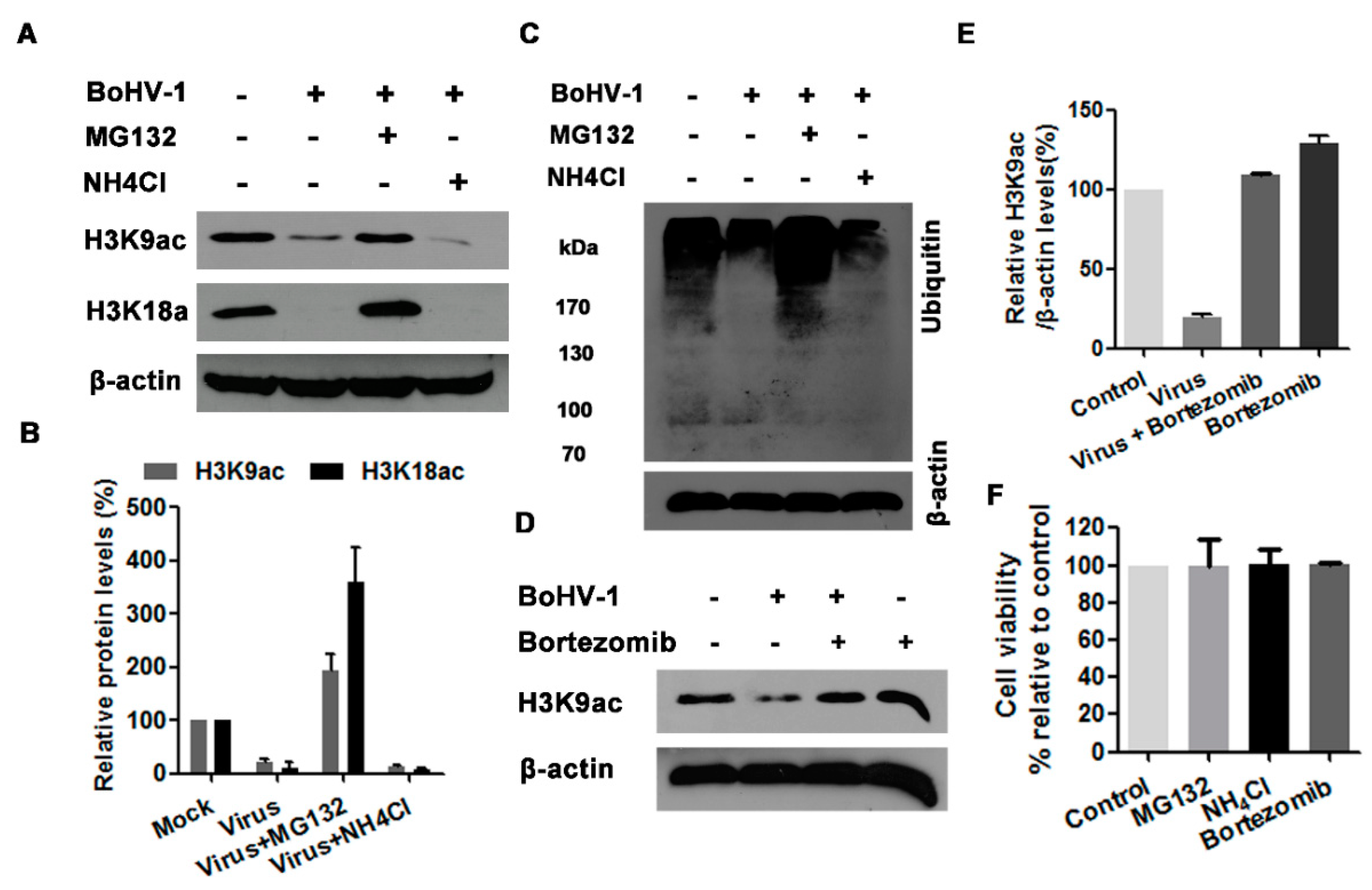
3.1. BoHV-1 Infection of MDBK Cells Decreases Histone H3 Acetylation
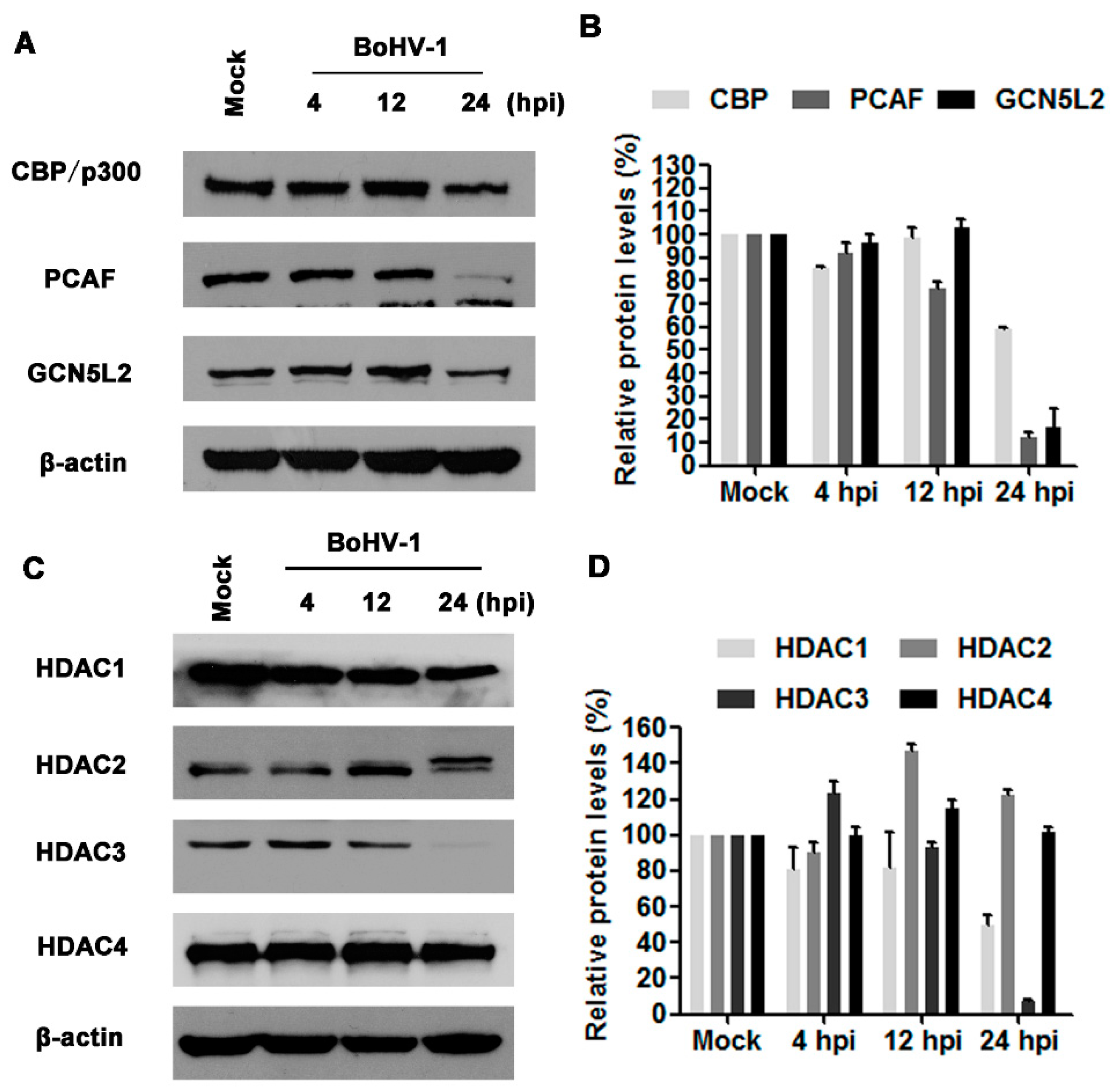
3.2. BoHV-1 Infection Differentially Affects the Expression of HATs and HDACs
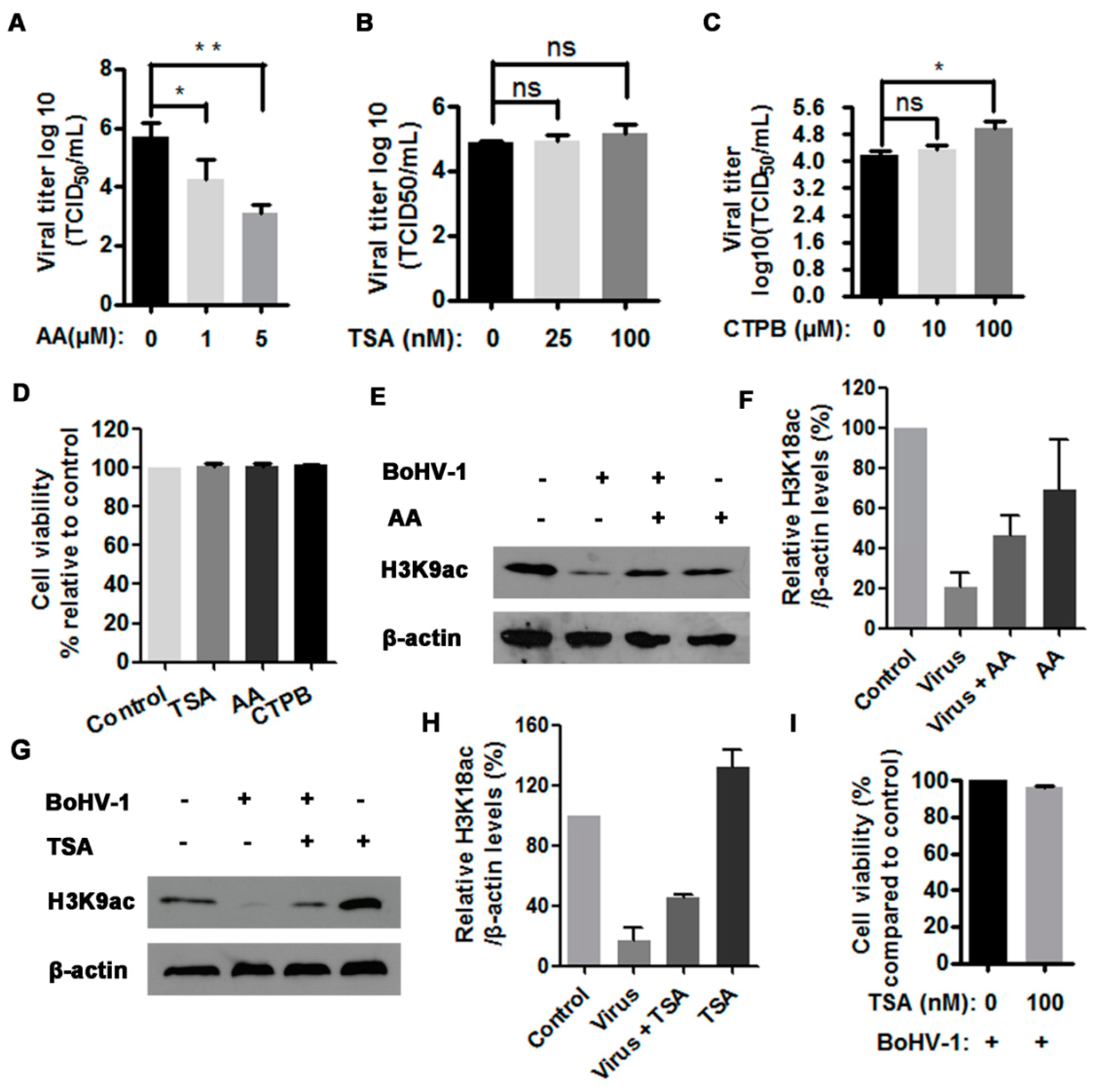
3.3. The HAT Inhibitor Limits BoHV-1 Replication
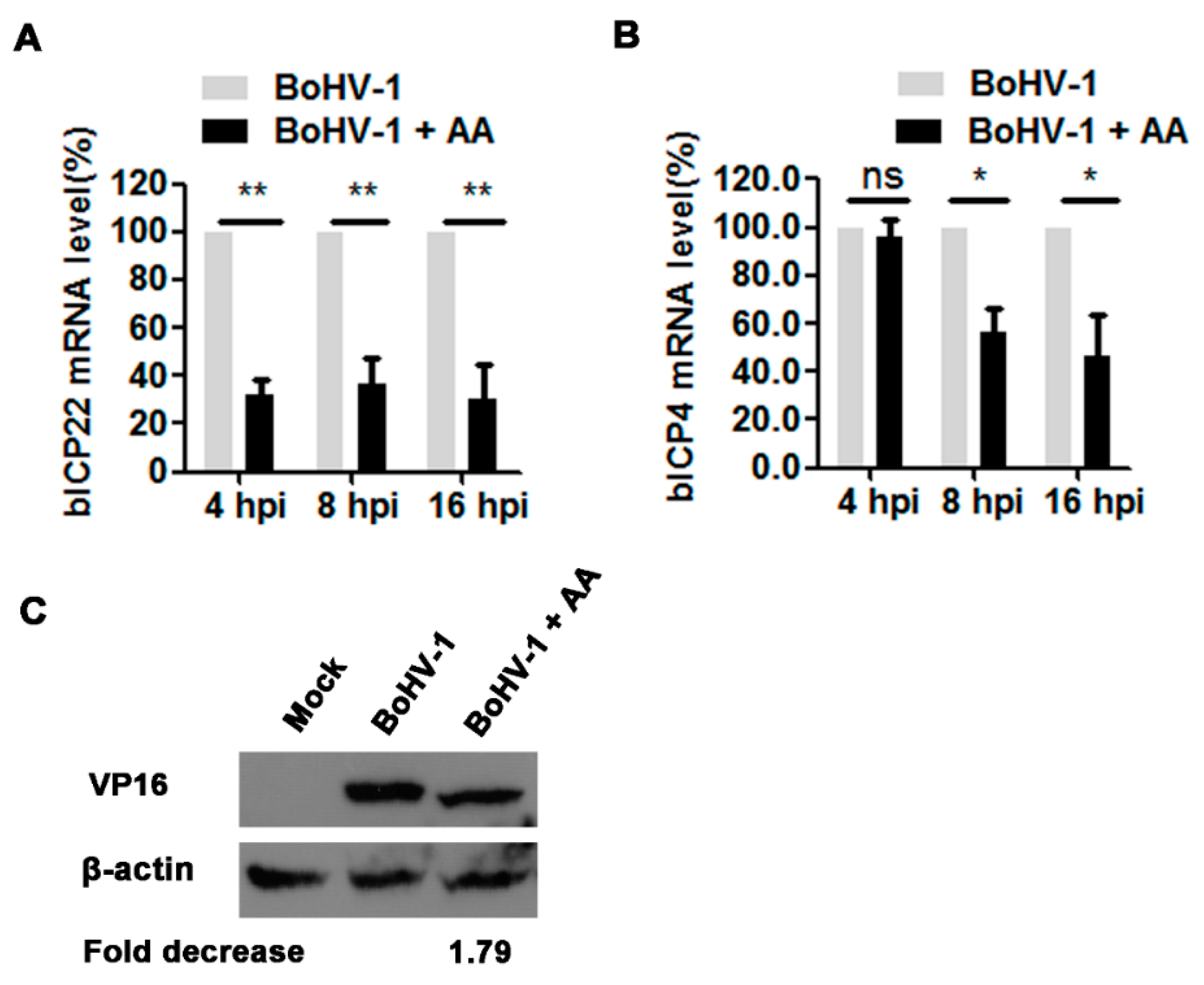
3.4. The HAT Inhibitor Affects Viral Gene Expression
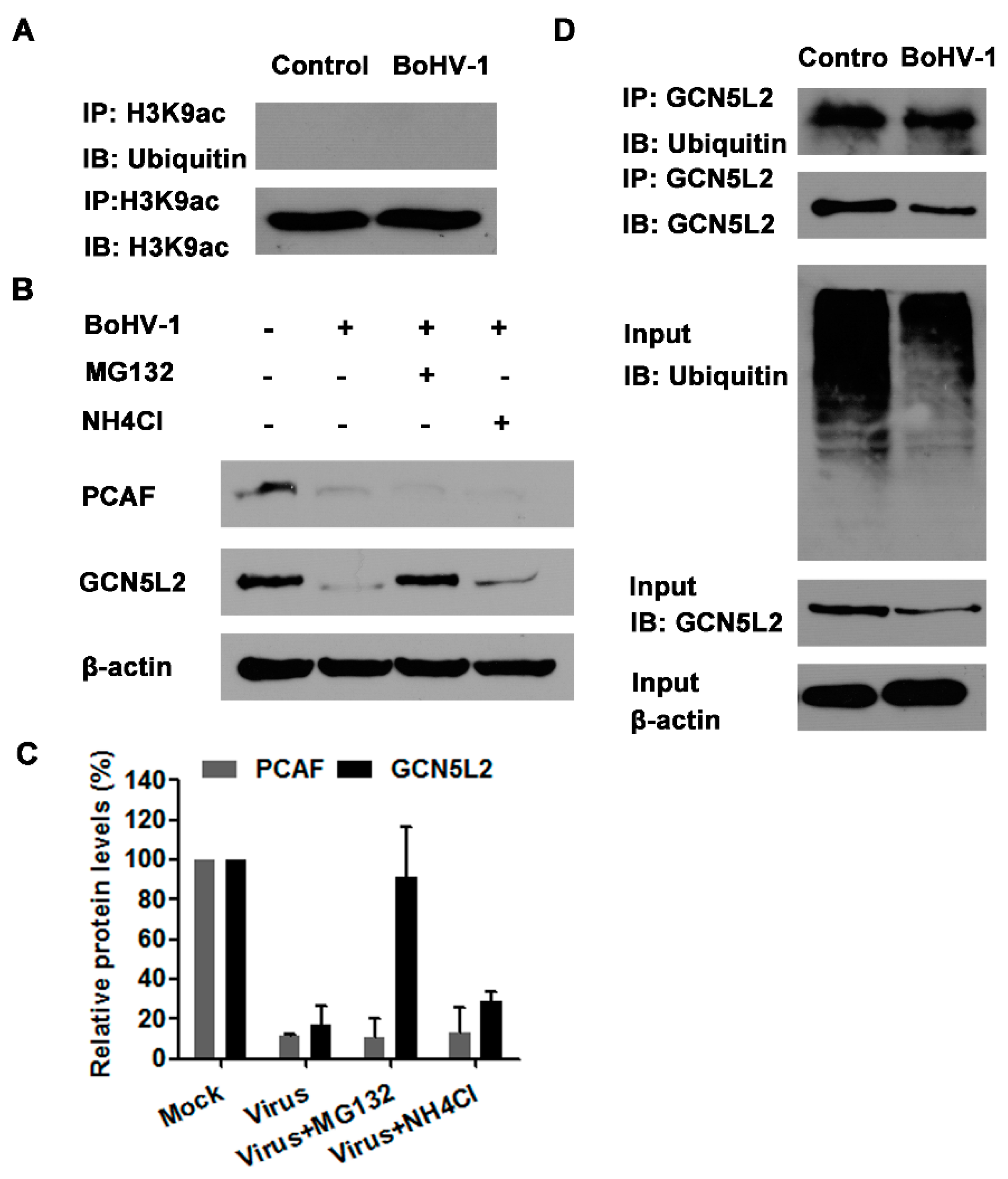
3.5. The Proteasome Pathway—Mediated GCN5L2 Degradation Is Potentially Involved in BoHV-1 Infection-Decreased Histone H3 Acetylation
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tikoo, S.K.; Campos, M.; Babiuk, L.A. Bovine herpesvirus 1 (BHV-1): Biology, pathogenesis, and control. Adv. Virus Res. 1995, 45, 191–223. [Google Scholar] [PubMed]
- Santos-Rosa, H.; Schneider, R.; Bannister, A.J.; Sherriff, J.; Bernstein, B.E.; Emre, N.C.; Schreiber, S.L.; Mellor, J.; Kouzarides, T. Active genes are tri-methylated at K4 of histone H3. Nature 2002, 419, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Risalde, M.A.; Molina, V.; Sanchez-Cordon, P.J.; Pedrera, M.; Panadero, R.; Romero-Palomo, F.; Gomez-Villamandos, J.C. Response of proinflammatory and anti-inflammatory cytokines in calves with subclinical bovine viral diarrhea challenged with bovine herpesvirus-1. Vet. Immunol. Immunopathol. 2011, 144, 135–143. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, C.; Zhang, D.; Ma, Y.; Ding, X.; Zhu, G. BHV-1 induced oxidative stress contributes to mitochondrial dysfunction in MDBK cells. Vet. Res. 2016, 47, 47. [Google Scholar] [CrossRef] [PubMed]
- Jones, C. Regulation of innate immune responses by bovine herpesvirus 1 and infected cell protein 0 (BICP0). Viruses 2009, 1, 255–275. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 2007, 8, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Neibergs, H.L.; Seabury, C.M.; Wojtowicz, A.J.; Wang, Z.; Scraggs, E.; Kiser, J.N.; Neupane, M.; Womack, J.E.; Van Eenennaam, A.; Hagevoort, G.R.; et al. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned holstein calves. BMC Genom. 2014, 15, 1164. [Google Scholar] [CrossRef] [PubMed]
- Fulton, R.W.; d’Offay, J.M.; Landis, C.; Miles, D.G.; Smith, R.A.; Saliki, J.T.; Ridpath, J.F.; Confer, A.W.; Neill, J.D.; Eberle, R.; et al. Detection and characterization of viruses as field and vaccine strains in feedlot cattle with bovine respiratory disease. Vaccine 2016, 34, 3478–3492. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, S.; Workman, J.L. Histone exchange, chromatin structure and the regulation of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.; Mallampalli, R.K. Regulation of histone modifying enzymes by the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 694–702. [Google Scholar] [CrossRef] [PubMed]
- Bonisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizzen, C.A.; Allis, C.D. Linking histone acetylation to transcriptional regulation. Cell. Mol. Life Sci. 1998, 54, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Agalioti, T.; Chen, G.; Thanos, D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002, 111, 381–392. [Google Scholar] [CrossRef]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef] [PubMed]
- Herrera, F.J.; Triezenberg, S.J. Vp16-dependent association of chromatin-modifying coactivators and underrepresentation of histones at immediate-early gene promoters during herpes simplex virus infection. J. Virol. 2004, 78, 9689–9696. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus icp0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Mantyla, E.; Salokas, K.; Oittinen, M.; Aho, V.; Mantysaari, P.; Palmujoki, L.; Kalliolinna, O.; Ihalainen, T.O.; Niskanen, E.A.; Timonen, J.; et al. Promoter-targeted histone acetylation of chromatinized parvoviral genome is essential for the progress of infection. J. Virol. 2016, 90, 4059–4066. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Fukuyama, S.; Sakai-Tagawa, Y.; Takashita, E.; Shoemaker, J.E.; Kawaoka, Y. C646, a novel p300/creb-binding protein-specific inhibitor of histone acetyltransferase, attenuates influenza a virus infection. Antimicrob. Agents Chemother. 2015, 60, 1902–1906. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Harms, J.S.; Splitter, G.A. Bovine herpesvirus 1 tegument protein vp22 interacts with histones, and the carboxyl terminus of VP22 is required for nuclear localization. J. Virol. 2001, 75, 8251–8258. [Google Scholar] [CrossRef] [PubMed]
- Seal, B.S.; Martinez, J.D.; Hall, M.R.; St Jeor, S.C. Occurrence of bovine herpesvirus-1 DNA in nucleosomes and chromatin of bovine herpesvirus-1-infected cells: Identification of a virion-associated protein in chromatin of infected cells. Arch. Virol. 1988, 99, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Barber, K.A.; Daugherty, H.C.; Ander, S.E.; Jefferson, V.A.; Shack, L.A.; Pechan, T.; Nanduri, B.; Meyer, F. Protein composition of the bovine herpesvirus 1.1 virion. Vet. Sci. 2017, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yuan, C.; Ding, X.; Jones, C.; Zhu, G. The role of phospholipase c signaling in bovine herpesvirus 1 infection. Vet. Res. 2017, 48, 45. [Google Scholar] [CrossRef] [PubMed]
- Misra, V.; Bratanich, A.C.; Carpenter, D.; O’Hare, P. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the bhv alpha gene trans-inducing factor. J. Virol. 1994, 68, 4898–4909. [Google Scholar] [PubMed]
- Fiorito, F.; Marfe, G.; De Blasio, E.; Granato, G.E.; Tafani, M.; de Martino, L.; Montagnaro, S.; Florio, S.; Pagnini, U. 2,3,7,8-tetrachlorodibenzo-p-dioxin regulates bovine herpesvirus type 1 induced apoptosis by modulating BCL-2 family members. Apoptosis 2008, 13, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, F.; Iovane, V.; Cantiello, A.; Marullo, A.; de Martino, L.; Iovane, G. Mg-132 reduces virus release in bovine herpesvirus-1 infection. Sci. Rep. 2017, 7, 13306. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Jones, C. The high mobility group at-hook 1 protein stimulates bovine herpesvirus 1 productive infection. Virus Res. 2017, 238, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Smith, J.A.; Balachandran, S.; Perwitasari, O.; Proll, S.C.; Thomas, M.J.; Korth, M.J.; Barber, G.N.; Schiff, L.A.; Katze, M.G. The cellular protein p58ipk regulates influenza virus mRNA translation and replication through a PKR-mediated mechanism. J. Virol. 2007, 81, 2221–2230. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Hussain, M.; Galvin, H.D.; Husain, M. Histone deacetylase 2 is a component of influenza a virus-induced host antiviral response. Front. Microbiol. 2017, 8, 1315. [Google Scholar] [CrossRef] [PubMed]
- Ghizzoni, M.; Wu, J.; Gao, T.; Haisma, H.J.; Dekker, F.J.; George Zheng, Y. 6-alkylsalicylates are selective tip60 inhibitors and target the acetyl-coa binding site. Eur. J. Med. Chem. 2012, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiang, X.; Chen, S.; Price, B.D. Inhibition of histone acetyltransferase activity by anacardic acid sensitizes tumor cells to ionizing radiation. FEBS Lett. 2006, 580, 4353–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, M.; Horinouchi, S.; Beppu, T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 1995, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Nagesh, P.T.; Husain, M. Influenza a virus dysregulates host histone deacetylase 1 that inhibits viral infection in lung epithelial cells. J. Virol. 2016, 90, 4614–4625. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Med. Res. Rev. 2001, 21, 245–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Chi, X.; Wei, H.; Chen, Y.; Chen, Z.; Huang, S.; Chen, J.L. Influenza A virus-induced degradation of eukaryotic translation initiation factor 4B contributes to viral replication by suppressing IFITM3 protein expression. J. Virol. 2014, 88, 8375–8385. [Google Scholar] [CrossRef] [PubMed]
- Gorisch, S.M.; Wachsmuth, M.; Toth, K.F.; Lichter, P.; Rippe, K. Histone acetylation increases chromatin accessibility. J. Cell Sci. 2005, 118, 5825–5834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, W.S.; Henry, K.W.; Schwartz, M.F.; Berger, S.L. Histone modification patterns during gene activation. Methods Enzymol. 2004, 377, 130–153. [Google Scholar] [PubMed]
- Hancock, M.H.; Cliffe, A.R.; Knipe, D.M.; Smiley, J.R. Herpes simplex virus VP16, but not ICP0, is required to reduce histone occupancy and enhance histone acetylation on viral genomes in U2OS osteosarcoma cells. J. Virol. 2010, 84, 1366–1375. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, C.; Luo, J.; Su, W.; Li, M.; Zhao, N.; Lyu, W.; Attaran, H.; He, Y.; Ding, H.; et al. Histone deacetylase 1 plays an acetylation-independent role in influenza A virus replication. Front. Immunol. 2017, 8, 1757. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barozzi, I.; Termanini, A.; Prosperini, E.; Recchiuti, A.; Dalli, J.; Mietton, F.; Matteoli, G.; Hiebert, S.; Natoli, G. Requirement for the histone deacetylase HDAC3 for the inflammatory gene expression program in macrophages. Proc. Natl. Acad. Sci. USA 2012, 109, E2865–2874. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, H.; Kaufmann, J.K.; Wang, P.Y.; Nguyen, T.; Speranza, M.C.; Kasai, K.; Okemoto, K.; Otsuki, A.; Nakano, I.; Fernandez, S.; et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J. Clin. Investig. 2015, 125, 4269–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Saira, K.; Zhou, Y.; Jones, C. The infected cell protein 0 encoded by bovine herpesvirus 1 (BICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J. Virol. 2007, 81, 3077–3086. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, N.; Jones, C. Regulation of promyelocytic leukemia (PML) protein levels and cell morphology by bovine herpesvirus 1 infected cell protein 0 (BICP0) and mutant BICP0 proteins that do not localize to the nucleus. Virus Res. 2011, 156, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Reits, E.A.; Ressing, M.E.; Lipinska, A.D.; Abele, R.; Koch, J.; Marcondes Rezende, M.; Admiraal, P.; van Leeuwen, D.; Bienkowska-Szewczyk, K.; et al. Varicelloviruses avoid T cell recognition by ul49.5-mediated inactivation of the transporter associated with antigen processing. Proc. Natl. Acad. Sci. USA 2005, 102, 5144–5149. [Google Scholar] [CrossRef] [PubMed]
- Koppers-Lalic, D.; Rijsewijk, F.A.; Verschuren, S.B.; van Gaans-Van den Brink, J.A.; Neisig, A.; Ressing, M.E.; Neefjes, J.; Wiertz, E.J. The ul41-encoded virion host shutoff (VHS) protein and VHS-independent mechanisms are responsible for down-regulation of MHC class I molecules by bovine herpesvirus 1. J. Gen. Virol. 2001, 82, 2071–2081. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiang, Y.; Geiser, V.; Zhou, J.; Jones, C. Bovine herpesvirus 1 immediate-early protein (BICP0) interacts with the histone acetyltransferase p300, which stimulates productive infection and GC promoter activity. J. Gen. Virol. 2006, 87, 1843–1851. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, L.; Jiang, X.; Fu, X.; Qi, Y.; Zhu, G. The Involvement of Histone H3 Acetylation in Bovine Herpesvirus 1 Replication in MDBK Cells. Viruses 2018, 10, 525. https://doi.org/10.3390/v10100525
Zhu L, Jiang X, Fu X, Qi Y, Zhu G. The Involvement of Histone H3 Acetylation in Bovine Herpesvirus 1 Replication in MDBK Cells. Viruses. 2018; 10(10):525. https://doi.org/10.3390/v10100525
Chicago/Turabian StyleZhu, Liqian, Xinyi Jiang, Xiaotian Fu, Yanhua Qi, and Guoqiang Zhu. 2018. "The Involvement of Histone H3 Acetylation in Bovine Herpesvirus 1 Replication in MDBK Cells" Viruses 10, no. 10: 525. https://doi.org/10.3390/v10100525