CRISPR-Cas Targeting of Host Genes as an Antiviral Strategy

Abstract
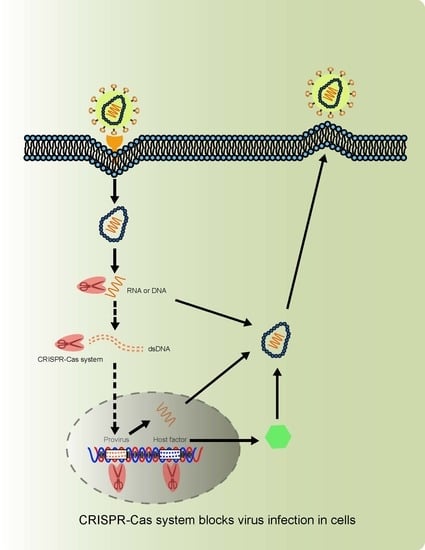
:
1. Introduction
1.1. The Development of CRISPR-Cas System
1.1.1. Canonical CRISPR-SpCas9
1.1.2. CRISPR Interference (CRISPRi)
1.1.3. CRISPR Activation (CRISPRa)
1.1.4. Non Canonical CRISPR-Cas System-SaCas9, Cpf1, FnCas9, C2c1/2/3 and CRISPR-Cas13
SaCas9
Cpf1
FnCas9
Class 2 Candidate 1/2/3 (C2c1/2/3)
CRISPR-Cas13
1.2. Virus-Host Interactions and Strategies Blocking Virus Infection
1.3. CRISPR-Cas System and Human Virus Restriction
1.3.1. Human Immunodeficiency Virus (HIV)
CRISPR-SpCas9 Targeting Host Factors and HIV-1 Genome
CRISPR-SaCas9 Targeting Host Factors and HIV-1 Genome
CRISPRa Targeting HIV-1 LTR and Host Factors
1.3.2. Hepatitis B Virus (HBV)
1.3.3. Human Papillomavirus (HPV)
1.3.4. Epstein–Barr Virus (EBV)
1.4. CRISPR and Human RNA Viruses
1.4.1. Hepatitis C Virus (HCV)
1.4.2. Other RNA Virus: Dengue Virus (DENV) and Zika Virus (ZIKV), West Nile Virus (WNV), Influenza Virus, Ebola Virus (EBOV), SARS-CoV, MERS-CoV
1.5. CRISPR and Plant Viruses
2. Challenge of Using CRISPR-Cas System Targets Host Factors to Protest against Virus Infection in Humans
2.1. Virus Mutation and Host Immune System
2.2. Off Target and Side Effect
2.3. Delivery Method
2.4. Clinical Trials
3. Conclusions and Future Directions
Acknowledgments
Conflicts of Interest
References
- Brouns, S.J.; Jore, M.M.; Lundgren, M.; Westra, E.R.; Slijkhuis, R.J.; Snijders, A.P.; Dickman, M.J.; Makarova, K.S.; Koonin, E.V.; van der Oost, J. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 2008, 321, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Garneau, J.E.; Dupuis, M.E.; Villion, M.; Romero, D.A.; Barrangou, R.; Boyaval, P.; Fremaux, C.; Horvath, P.; Magadan, A.H.; Moineau, S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 2010, 468, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Horvath, P. A decade of discovery: CRISPR functions and applications. Nat. Microbiol. 2017, 2, 17092. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Marraffini, L.A. CRISPR-Cas systems: Prokaryotes upgrade to adaptive immunity. Mol. Cell 2014, 54, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelsen, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhu, S.; Cai, C.; Yuan, P.; Li, C.; Huang, Y.; Wei, W. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 2014, 509, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Ran, F.A.; Hsu, P.D.; Konermann, S.; Shehata, S.I.; Dohmae, N.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 2014, 156, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Zhou, K.; Ma, L.; Gressel, S.; Doudna, J.A. STRUCTURAL BIOLOGY. A Cas9-guide RNA complex preorganized for target DNA recognition. Science 2015, 348, 1477–1481. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Linder, S.J.; Cascio, V.M.; Fu, Y.; Ho, Q.H.; Joung, J.K. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 2013, 10, 977–979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W.; et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, A.K.; Muruve, D.A. Immunity to adeno-associated virus vectors in animals and humans: A continued challenge. Gene Ther. 2008, 15, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Mingozzi, F.; High, K.A. Immune responses to AAV vectors: Overcoming barriers to successful gene therapy. Blood 2013, 122, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, R.; Bella, R.; Yin, C.; Otte, J.; Ferrante, P.; Gendelman, H.E.; Li, H.; Booze, R.; Gordon, J.; Hu, W.; et al. Excision of HIV-1 DNA by gene editing: A proof-of-concept in vivo study. Gene Ther. 2016, 23, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Kaya, H.; Mikami, M.; Endo, A.; Endo, M.; Toki, S. Highly specific targeted mutagenesis in plants using Staphylococcus aureus Cas9. Sci. Rep. 2016, 6, 26871. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Chen, C.; Han, Y.; Chen, Z.; Lu, X.; Liang, F.; Li, S.; Qin, W.; Lin, S. Expanding CRISPR/Cas9 Genome Editing Capacity in Zebrafish Using SaCas9. G3 2016, 6, 2517–2521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liang, P.; Ding, C.; Zhang, Z.; Zhou, J.; Xie, X.; Huang, R.; Sun, Y.; Sun, H.; Zhang, J.; et al. Efficient Production of Gene-Modified Mice using Staphylococcus aureus Cas9. Sci. Rep. 2016, 6, 32565. [Google Scholar] [CrossRef] [PubMed]
- Nishimasu, H.; Cong, L.; Yan, W.X.; Ran, F.A.; Zetsche, B.; Li, Y.; Kurabayashi, A.; Ishitani, R.; Zhang, F.; Nureki, O. Crystal Structure of Staphylococcus aureus Cas9. Cell 2015, 162, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Kim, J.; Hur, J.K.; Been, K.W.; Yoon, S.H.; Kim, J.S. Genome-wide analysis reveals specificities of Cpf1 endonucleases in human cells. Nat. Biotechnol. 2016, 34, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Nishimasu, H.; Zetsche, B.; Hirano, H.; Slaymaker, I.M.; Li, Y.; Fedorova, I.; Nakane, T.; Makarova, K.S.; Koonin, E.V.; et al. Crystal Structure of Cpf1 in Complex with Guide RNA and Target DNA. Cell 2016, 165, 949–962. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Heidenreich, M.; Mohanraju, P.; Fedorova, I.; Kneppers, J.; DeGennaro, E.M.; Winblad, N.; Choudhury, S.R.; Abudayyeh, O.O.; Gootenberg, J.S.; et al. Multiplex gene editing by CRISPR-Cpf1 using a single crRNA array. Nat. Biotechnol. 2017, 35, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Endo, A.; Masafumi, M.; Kaya, H.; Toki, S. Efficient targeted mutagenesis of rice and tobacco genomes using Cpf1 from Francisella novicida. Sci. Rep. 2016, 6, 38169. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Lowder, L.G.; Zhang, T.; Malzahn, A.A.; Zheng, X.; Voytas, D.F.; Zhong, Z.; Chen, Y.; Ren, Q.; Li, Q.; et al. A CRISPR-Cpf1 system for efficient genome editing and transcriptional repression in plants. Nat. Plants 2017, 3, 17018. [Google Scholar] [CrossRef] [PubMed]
- Ungerer, J.; Pakrasi, H.B. Cpf1 Is A Versatile Tool for CRISPR Genome Editing across Diverse Species of Cyanobacteria. Sci. Rep. 2016, 6, 39681. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Cheong, S.A.; Lee, J.G.; Lee, S.W.; Lee, M.S.; Baek, I.J.; Sung, Y.H. Generation of knockout mice by Cpf1-mediated gene targeting. Nat. Biotechnol. 2016, 34, 808–810. [Google Scholar] [CrossRef] [PubMed]
- Fonfara, I.; Richter, H.; Bratovic, M.; Le Rhun, A.; Charpentier, E. The CRISPR-associated DNA-cleaving enzyme Cpf1 also processes precursor CRISPR RNA. Nature 2016, 532, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Saroj, S.D.; Llewellyn, A.C.; Tzeng, Y.L.; Weiss, D.S. A CRISPR/Cas system mediates bacterial innate immune evasion and virulence. Nature 2013, 497, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Price, A.A.; Sampson, T.R.; Ratner, H.K.; Grakoui, A.; Weiss, D.S. Cas9-mediated targeting of viral RNA in eukaryotic cells. Proc. Natl. Acad. Sci. USA 2015, 112, 6164–6169. [Google Scholar] [CrossRef] [PubMed]
- Shmakov, S.; Abudayyeh, O.O.; Makarova, K.S.; Wolf, Y.I.; Gootenberg, J.S.; Semenova, E.; Minakhin, L.; Joung, J.; Konermann, S.; Severinov, K.; et al. Discovery and Functional Characterization of Diverse Class 2 CRISPR-Cas Systems. Mol. Cell 2015, 60, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef] [PubMed]
- East-Seletsky, A.; O’Connell, M.R.; Knight, S.C.; Burstein, D.; Cate, J.H.; Tjian, R.; Doudna, J.A. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 2016, 538, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, X.; Wang, J.; Wang, M.; Chen, P.; Yin, M.; Li, J.; Sheng, G.; Wang, Y. Two Distant Catalytic Sites Are Responsible for C2c2 RNase Activities. Cell 2017, 168, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, P.; Wang, M.; Li, X.; Wang, J.; Yin, M.; Wang, Y. C2c1-sgRNA Complex Structure Reveals RNA-Guided DNA Cleavage Mechanism. Mol. Cell 2017, 65, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gao, P.; Rajashankar, K.R.; Patel, D.J. PAM-Dependent Target DNA Recognition and Cleavage by C2c1 CRISPR-Cas Endonuclease. Cell 2016, 167, 1814–1828. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA targeting with CRISPR-Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with CRISPR-Cas13. Science 2017. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Zheng, Y.H.; Lovsin, N.; Peterlin, B.M. Newly identified host factors modulate HIV replication. Immunol. Lett. 2005, 97, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [PubMed]
- Kim, S.Y.; Byrn, R.; Groopman, J.; Baltimore, D. Temporal aspects of DNA and RNA synthesis during human immunodeficiency virus infection: Evidence for differential gene expression. J. Virol. 1989, 63, 3708–3713. [Google Scholar] [PubMed]
- Pomerantz, R.J.; Trono, D.; Feinberg, M.B.; Baltimore, D. Cells nonproductively infected with HIV-1 exhibit an aberrant pattern of viral RNA expression: A molecular model for latency. Cell 1990, 61, 1271–1276. [Google Scholar] [CrossRef]
- Update on acquired immune deficiency syndrome (AIDS)—United States. Morb. Mortal. Wkly. Rep. 1982, 31, 507–508, 513–514.
- Bayer, R.; Levine, C.; Murray, T.H. Guidelines for Confidentiality in Research on AIDS. AIDS Res. 1983, 1, 275–297. [Google Scholar] [CrossRef] [PubMed]
- Palella, F.J., Jr.; Delaney, K.M.; Moorman, A.C.; Loveless, M.O.; Fuhrer, J.; Satten, G.A.; Aschman, D.J.; Holmberg, S.D. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N. Engl. J. Med. 1998, 338, 853–860. [Google Scholar] [CrossRef] [PubMed]
- Durand, C.M.; Blankson, J.N.; Siliciano, R.F. Developing strategies for HIV-1 eradication. Trends Immunol. 2012, 33, 554–562. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, J.D.; Siliciano, R.F. HIV-1 eradication strategies: Design and assessment. Curr. Opin. HIV AIDS 2013, 8, 318–325. [Google Scholar] [CrossRef] [PubMed]
- Blas-Garcia, A.; Apostolova, N.; Esplugues, J.V. Oxidative stress and mitochondrial impairment after treatment with anti-HIV drugs: Clinical implications. Curr. Pharm. Des. 2011, 17, 4076–4086. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5 Delta 32/Delta 32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef] [PubMed]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Muessig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kuecherer, C.; Blau, O.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantaion. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Delta32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ye, C.; Liu, J.; Zhang, D.; Kimata, J.T.; Zhou, P. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS ONE 2014, 9, e115987. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Guan, X.; Du, T.; Jin, W.; Wu, B.; Liu, Y.; Wang, P.; Hu, B.; Griffin, G.E.; Shattock, R.J.; et al. Inhibition of HIV-1 infection of primary CD4+ T-cells by gene editing of CCR5 using adenovirus-delivered CRISPR/Cas9. J. Gener. Virol. 2015, 96, 2381–2393. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Ferreira, L.M.; Collins, R.; Meissner, T.B.; Boutwell, C.L.; Friesen, M.; Vrbanac, V.; Garrison, B.S.; Stortchevoi, A.; Bryder, D.; et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 2014, 15, 643–652. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yang, H.; Gao, Y.; Chen, Z.; Xie, L.; Liu, Y.; Liu, Y.; Wang, X.; Li, H.; Lai, W.; et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol. Ther. J. Am. Soc. Gene Ther. 2017, 25, 1782–1789. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.K.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; DiMarzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Sheridan, K.E.; Ceradini, D.; Choe, S.; Landau, N.R. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J. Exp. Med. 1997, 185, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Toma, J.; Stawiski, E.; Fransen, S.; Wrin, T.; Parkin, N.; Whitcomb, J.M.; Coakley, E.; Hecht, F.M.; Deeks, S.G.; et al. Characterization of human immunodeficiency virus type 1 populations containing CXCR4-using variants from recently infected individuals. AIDS Res. Hum. Retroviruses 2009, 25, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Hou, P.; Chen, S.; Wang, S.; Yu, X.; Chen, Y.; Jiang, M.; Zhuang, K.; Ho, W.; Hou, W.; Huang, J.; et al. Genome editing of CXCR4 by CRISPR/cas9 confers cells resistant to HIV-1 infection. Sci. Rep. 2015, 5, 15577. [Google Scholar] [CrossRef] [PubMed]
- Hultquist, J.F.; Schumann, K.; Woo, J.M.; Manganaro, L.; McGregor, M.J.; Doudna, J.; Simon, V.; Krogan, N.J.; Marson, A. A Cas9 Ribonucleoprotein Platform for Functional Genetic Studies of HIV-Host Interactions in Primary Human T Cells. Cell Rep. 2016, 17, 1438–1452. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Yao, Y.; Xiao, H.; Li, J.; Liu, Q.; Yang, Y.; Adah, D.; Lu, J.; Zhao, S.; Qin, L.; et al. Simultaneous knockout of CXCR4 and CCR5 genes in CD4+ T cells via CRISPR/Cas9 confers resistance to both X4- and R5-tropic HIV-1 infection. Hum. Gene Ther. 2017, 29, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, S.; Jin, X.; Wang, Q.; Yang, K.; Li, C.; Xiao, Q.; Hou, P.; Liu, S.; Wu, S.; et al. Genome editing of the HIV co-receptors CCR5 and CXCR4 by CRISPR-Cas9 protects CD4+ T cells from HIV-1 infection. Cell Biosci. 2017, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, 2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Lei, R.; Le Duff, Y.; Li, J.; Guo, F.; Wainberg, M.A.; Liang, C. The CRISPR/Cas9 system inactivates latent HIV-1 proviral DNA. Retrovirology 2015, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, 6413. [Google Scholar] [CrossRef] [PubMed]
- Maertens, G.; Cherepanov, P.; Pluymers, W.; Busschots, K.; de Clercq, E.; Debyser, Z.; Engelborghs, Y. LEDGF/p75 is essential for nuclear and chromosomal targeting of HIV-1 integrase in human cells. J. Biol. Chem. 2003, 278, 33528–33539. [Google Scholar] [CrossRef] [PubMed]
- Christ, F.; Debyser, Z. The LEDGF/p75 integrase interaction, a novel target for anti-HIV therapy. Virology 2013, 435, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Fadel, H.J.; Morrison, J.H.; Saenz, D.T.; Fuchs, J.R.; Kvaratskhelia, M.; Ekker, S.C.; Poeschla, E.M. TALEN knockout of the PSIP1 gene in human cells: Analyses of HIV-1 replication and allosteric integrase inhibitor mechanism. J. Virol. 2014, 88, 9704–9717. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef] [PubMed]
- Schaller, T.; Ocwieja, K.E.; Rasaiyaah, J.; Price, A.J.; Brady, T.L.; Roth, S.L.; Hue, S.; Fletcher, A.J.; Lee, K.; KewalRamani, V.N.; et al. HIV-1 capsid-cyclophilin interactions determine nuclear import pathway, integration targeting and replication efficiency. PLoS Pathog. 2011, 7, e1002439. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [PubMed]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Konig, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; De Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Gori, J.L.; Hsu, P.D.; Maeder, M.L.; Shen, S.; Welstead, G.G.; Bumcrot, D. Delivery and Specificity of CRISPR-Cas9 Genome Editing Technologies for Human Gene Therapy. Hum. Gene Ther. 2015, 26, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Sather, B.D.; Romano Ibarra, G.S.; Sommer, K.; Curinga, G.; Hale, M.; Khan, I.F.; Singh, S.; Song, Y.; Gwiazda, K.; Sahni, J.; et al. Efficient modification of CCR5 in primary human hematopoietic cells using a megaTAL nuclease and AAV donor template. Sci. Transl. Med. 2015, 7, 307ra156. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; DeClercq, J.J.; Hayward, S.B.; Li, P.W.; Shivak, D.A.; Gregory, P.D.; Lee, G.; Holmes, M.C. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 2016, 44, e30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Exline, C.M.; DeClercq, J.J.; Llewellyn, G.N.; Hayward, S.B.; Li, P.W.; Shivak, D.A.; Surosky, R.T.; Gregory, P.D.; Holmes, M.C.; et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015, 33, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Chen, S.; Xiao, Q.; Liu, Z.; Liu, S.; Hou, P.; Zhou, L.; Hou, W.; Ho, W.; Li, C.; et al. Genome modification of CXCR4 by Staphylococcus aureus Cas9 renders cells resistance to HIV-1 infection. Retrovirology 2017, 14, 51. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.; Zhang, T.; Qu, X.; Zhang, Y.; Putatunda, R.; Xiao, X.; Li, F.; Xiao, W.; Zhao, H.; Dai, S.; et al. In Vivo Excision of HIV-1 Provirus by saCas9 and Multiplex Single-Guide RNAs in Animal Models. Mol. Ther. 2017, 25, 1168–1186. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; Di Mascio, M.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The challenge of finding a cure for HIV infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, C.; Zhang, T.; Li, F.; Yang, W.; Kaminski, R.; Fagan, P.R.; Putatunda, R.; Young, W.B.; Khalili, K.; et al. CRISPR/gRNA-directed synergistic activation mediator (SAM) induces specific, persistent and robust reactivation of the HIV-1 latent reservoirs. Sci. Rep. 2015, 5, 16277. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.M.; Lazar, D.C.; Scott, T.A.; Hart, J.R.; Takahashi, M.; Burnett, J.C.; Planelles, V.; Morris, K.V.; Weinberg, M.S. Potent and Targeted Activation of Latent HIV-1 Using the CRISPR/dCas9 Activator Complex. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Limsirichai, P.; Gaj, T.; Schaffer, D.V. CRISPR-mediated Activation of Latent HIV-1 Expression. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Jiang, Z.; Lu, P.; Ma, L.; Li, C.; Pan, H.; Fu, Z.; Qu, X.; Wang, P.; Deng, J.; et al. Specific Reactivation of Latent HIV-1 by dCas9-SunTag-VP64-mediated Guide RNA Targeting the HIV-1 Promoter. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 508–521. [Google Scholar] [CrossRef] [PubMed]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, X.; Stremlau, M.; Lee, M.; Sodroski, J. Removal of arginine 332 allows human TRIM5alpha to bind human immunodeficiency virus capsids and to restrict infection. J. Virol. 2006, 80, 6738–6744. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Akkina, R. Human immunodeficiency virus type 1 restriction by human-rhesus chimeric tripartite motif 5alpha (TRIM 5alpha) in CD34(+) cell-derived macrophages in vitro and in T cells in vivo in severe combined immunodeficient (SCID-hu) mice transplanted with human fetal tissue. Hum. Gene Ther. 2008, 19, 217–228. [Google Scholar] [PubMed]
- Pham, Q.T.; Bouchard, A.; Grutter, M.G.; Berthoux, L. Generation of human TRIM5alpha mutants with high HIV-1 restriction activity. Gene Ther. 2010, 17, 859–871. [Google Scholar] [CrossRef] [PubMed]
- Neagu, M.R.; Ziegler, P.; Pertel, T.; Strambio-de-Castillia, C.; Grutter, C.; Martinetti, G.; Mazzucchelli, L.; Grutter, M.; Manz, M.G.; Luban, J. Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J. Clin. Investig. 2009, 119, 3035–3047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Svarovskaia, E.S.; Barr, R.; Zhang, Y.; Khan, M.A.; Strebel, K.; Pathak, V.K. A single amino acid substitution in human APOBEC3G antiretroviral enzyme confers resistance to HIV-1 virion infectivity factor-induced depletion. Proc. Natl. Acad. Sci. USA 2004, 101, 5652–5657. [Google Scholar] [CrossRef] [PubMed]
- Schrofelbauer, B.; Chen, D.; Landau, N.R. A single amino acid of APOBEC3G controls its species-specific interaction with virion infectivity factor (Vif). Proc. Natl. Acad. Sci. USA 2004, 101, 3927–3932. [Google Scholar] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Hue, S.; Schaller, T.; Verschoor, E.; Pillay, D.; Towers, G.J. Mutation of a single residue renders human tetherin resistant to HIV-1 Vpu-mediated depletion. PLoS Pathog. 2009, 5, e1000443. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4(+) T-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef] [PubMed]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Wu, L. SAMHD1: A new contributor to HIV-1 restriction in resting CD4+ T-cells. Retrovirology 2012, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Baldauf, H.M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4(+) T cells. Nat. Med. 2012, 18, 1682–1687. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [PubMed]
- Goujon, C.; Moncorge, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hue, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Voit, R.A.; McMahon, M.A.; Sawyer, S.L.; Porteus, M.H. Generation of an HIV resistant T-cell line by targeted “stacking” of restriction factors. Mol. Ther. J. Am. Soc. Gene Ther. 2013, 21, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Bogerd, H.P.; Kornepati, A.V.; Marshall, J.B.; Kennedy, E.M.; Cullen, B.R. Specific induction of endogenous viral restriction factors using CRISPR/Cas-derived transcriptional activators. Proc. Natl. Acad. Sci. USA 2015, 112, E7249–E7256. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y.; Wu, Y.; Gottlinger, H.G. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Rosa, A.; Chande, A.; Ziglio, S.; de Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Zhang, K.; Li, J. Application of CRISPR/Cas9 Technology to HBV. Int. J. Mol. Sci. 2015, 16, 26077–26086. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Lu, M.; Yang, D. CRISPR/Cas9-based tools for targeted genome editing and replication control of HBV. Virol. Sin. 2015, 30, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antivir. Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Luth, S.; Buchholz, F.; Schulze zur Wiesch, J.; et al. CRISPR/Cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Wang, S.; Yang, L.; Liang, Y.; Huang, Y.; Liu, H.; Li, P.; Yang, C.; Yang, X.; et al. Removal of Integrated Hepatitis B Virus DNA Using CRISPR-Cas9. Front. Cell. Infect. Microbiol. 2017, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gener. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Pankowicz, F.P.; Jarrett, K.E.; Lagor, W.R.; Bissig, K.D. CRISPR/Cas9: At the cutting edge of hepatology. Gut 2017, 66, 1329–1340. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting Hepatitis B Virus With CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Complete Spectrum of CRISPR/Cas9-induced Mutations on HBV cccDNA. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Zhang, R.; Ding, S.; Zhang, T.; Yuan, Q.; Guan, G.; Chen, X.; Zhang, T.; Zhuang, H.; et al. The gRNA-miRNA-gRNA Ternary Cassette Combining CRISPR/Cas9 with RNAi Approach Strongly Inhibits Hepatitis B Virus Replication. Theranostics 2017, 7, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.W.; Liu, S.; Zhang, R.Y.; Ding, S.L.; Xie, X.M.; Long, L.; Chen, X.M.; Zhuang, H.; Lu, F.M. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J. Gastroenterol. 2015, 21, 9554–9565. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xie, K.; Xu, Y.; Wang, L.; Chen, K.; Zhang, L.; Fang, J. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Sluder, A.; Daito, T.; Matsunaga, S.; Ryo, A.; Nagamori, S.; Iwamoto, M.; Nakajima, S.; Tsukuda, S.; Borroto-Esoda, K. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter NTCP. Hepatology 2014, 59, 1726–1737. [Google Scholar] [CrossRef] [PubMed]
- Nkongolo, S.; Ni, Y.; Lempp, F.A.; Kaufman, C.; Lindner, T.; Essernobis, K.; Lohmann, V.; Mier, W.; Mehrle, S.; Urban, S. Cyclosporin A inhibits hepatitis B and hepatitis D virus entry by cyclophilin-independent interference with the NTCP receptor. J. Hepatol. 2014, 60, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Cannie, I.; Urban, S. Efficient inhibition of hepatitis B virus infection by acylated peptides derived from the large viral surface protein. J. Virol. 2005, 79, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.; Dandri, M.; Mier, W.; Lãtgehetmann, M.; Volz, T.; von Weizsäcker, F.; Haberkorn, U.; Fischer, L.; Pollok, J.M.; Erbes, B. Prevention of hepatitis B virus infection in vivo by entry inhibitors derived from the large envelope protein. Nat. Biotechnol. 2008, 26, 335–341. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lütgehetmann, M. The entry inhibitor Myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Hoh, A.; Heeg, M.; Ni, Y.; Schuch, A.; Binder, B.; Hennecke, N.; Blum, H.E.; Nassal, M.; Protzer, U.; Hofmann, M. Hepatitis B Virus-Infected HepG2hNTCP Cells Serve as a Novel Immunological Tool To Analyze the Antiviral Efficacy of CD8+ T Cells In Vitro. J. Virol. 2015, 89, 7433–7438. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Retzlaff, M.; Nassal, M.; Beck, J. Chaperone activation of the hepadnaviral reverse transcriptase for template RNA binding is established by the Hsp70 and stimulated by the Hsp90 system. Nucleic Acids Res. 2007, 35, 6124–6136. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.P.; Liu, F.; He, H.W.; Han, Y.X.; Peng, Z.G.; Li, B.W.; You, X.F.; Song, D.Q.; Li, Z.R.; Yu, L.Y. Heat Stress Cognate 70 Host Protein as a Potential Drug Target against Drug Resistance in Hepatitis B Virus. Antimicrob. Agents Chemother. 2010, 54, 2070–2077. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.B.; Wang, Y.F.; Zhang, Y.; Zheng, L.Y.; Yang, X.A.; Wang, N.; Jiang, J.H.; Ma, F.; Yin, D.T.; Sun, C.Y. In vitro activity of cepharanthine hydrochloride against clinical wild-type and lamivudine-resistant hepatitis B virus isolates. Eur. J. Pharmacol. 2012, 683, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.Y.; Quan, X.; Yi, Y.S.; Jung, G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology 2011, 410, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Seeger, C. Hsp90 is Required for the Activity of a Hepatitis B Virus Reverse Transcriptase. Proc. Natl. Acad. Sci. USA 1996, 93, 1060–1064. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Miao, X.; Qi, Z.; Zeng, W.; Liang, J.; Liang, Z. Hepatitis B virus X protein upregulates HSP90alpha expression via activation of c-Myc in human hepatocarcinoma cell line, HepG2. Virol. J. 2010, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Königer, C.; Wingert, I.; Marsmann, M.; Rösler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Mcallister, R.; Boregowda, R.; Ji, A.S.; Ledesma, F.C.; Caldecott, K.W.; Seeger, C.; Hu, J. Does Tyrosyl DNA Phosphodiesterase-2 Play a Role in Hepatitis B Virus Genome Repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed]
- Hartmann-Stuhler, C.; Prange, R. Hepatitis B virus large envelope protein interacts with gamma2-adaptin, a clathrin adaptor-related protein. J. Virol. 2001, 75, 5343–5351. [Google Scholar] [CrossRef] [PubMed]
- Rost, M.; Mann, S.; Lambert, C.; Döring, T.; Thomé, N.; Prange, R. Gamma-adaptin, a novel ubiquitin-interacting adaptor, and Nedd4 ubiquitin ligase control hepatitis B virus maturation. J. Biol. Chem. 2006, 281, 29297. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhang, B.; Shi, Y.; Han, Z.; Zhang, Y.; Zhou, Y.; Zhang, W.; Niu, J.; Yu, X.F. Identification of BST-2/tetherin-induced hepatitis B virus restriction and hepatocyte-specific BST-2 inactivation. Sci. Rep. 2015, 5, 11736. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhao, X.; Cai, D.; Liu, Y.; Block, T.M.; Guo, J.T.; Guo, H. The Interferon-Inducible Protein Tetherin Inhibits Hepatitis B Virus Virion Secretion. J. Virol. 2015, 89, 9200–9212. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, K.; Matsunaga, S.; Watashi, K.; Sugiyama, M.; Kimura, H.; Yamamoto, N.; Mizokami, M.; Wakita, T.; Ryo, A. Molecular dissection of HBV evasion from restriction factor tetherin: A new perspective for antiviral cell therapy. Oncotarget 2015, 6, 21840. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, F.; Laimins, L.A. Human papillomaviruses: Targeting differentiating epithelial cells for malignant transformation. Oncogene 2003, 22, 5201–5207. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, I.; Mittal, S.; Banerjee, D.; Singh, P.; Dasgupta, S.; Chatterjee, S.; Biswas, J.; Panda, C.; Basu, P. Study of accuracy of colposcopy in VIA and HPV detection-based cervical cancer screening program. Aust. N. Z. J. Obstet. Gynaecol. 2014, 54, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Kjaer, S.K.; Sigurdsson, K.; Iversen, O.E.; Hernandez-Avila, M.; Wheeler, C.M.; Perez, G.; Brown, D.R.; Koutsky, L.A.; Tay, E.H.; et al. Impact of human papillomavirus (HPV)-6/11/16/18 vaccine on all HPV-associated genital diseases in young women. J. Natl. Cancer Inst. 2010, 102, 325–339. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Kornepati, A.V.; Goldstein, M.; Bogerd, H.P.; Poling, B.C.; Whisnant, A.W.; Kastan, M.B.; Cullen, B.R. Inactivation of the human papillomavirus E6 or E7 gene in cervical carcinoma cells by using a bacterial CRISPR/Cas RNA-guided endonuclease. J. Virol. 2014, 88, 11965–11972. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Takahashi, Y.; Narita, S.; Liu, Y.H.; Li, Y. In vitro and in vivo growth suppression of human papillomavirus 16-positive cervical cancer cells by CRISPR/Cas9. Biochem. Biophys. Res. Commun. 2014, 450, 1422–1426. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.S.; Stepp, W.H.; Stamos, J.D.; McBride, A.A. Host cell restriction factors that limit transcription and replication of human papillomavirus. Virus Res. 2017, 231, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Smith, N.; Wang, X.; Morgan, I.M. The Deacetylase SIRT1 Regulates the Replication Properties of Human Papillomavirus 16 E1 and E2. J. Virol. 2017, 91, e00102-17. [Google Scholar] [CrossRef] [PubMed]
- McKinney, C.C.; Kim, M.J.; Chen, D.; McBride, A.A. Brd4 Activates Early Viral Transcription upon Human Papillomavirus 18 Infection of Primary Keratinocytes. mBio 2016, 7, e01644-16. [Google Scholar] [CrossRef] [PubMed]
- Meuris, F.; Carthagena, L.; Jaracz-Ros, A.; Gaudin, F.; Cutolo, P.; Deback, C.; Xue, Y.; Thierry, F.; Doorbar, J.; Bachelerie, F. The CXCL12/CXCR4 Signaling Pathway: A New Susceptibility Factor in Human Papillomavirus Pathogenesis. PLoS Pathog. 2016, 12, e1006039. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Hong, S.; Maniar, K.P.; Cheng, S.; Jie, C.; Rademaker, A.W.; Krensky, A.M.; Clayberger, C. KLF13 regulates the differentiation-dependent human papillomavirus life cycle in keratinocytes through STAT5 and IL-8. Oncogene 2016, 35, 5565–5575. [Google Scholar] [CrossRef] [PubMed]
- Gunasekharan, V.K.; Li, Y.; Andrade, J.; Laimins, L.A. Post-Transcriptional Regulation of KLF4 by High-Risk Human Papillomaviruses Is Necessary for the Differentiation-Dependent Viral Life Cycle. PLoS Pathog. 2016, 12, e1005747. [Google Scholar] [CrossRef] [PubMed]
- DeSmet, M.; Kanginakudru, S.; Rietz, A.; Wu, W.H.; Roden, R.; Androphy, E.J. The Replicative Consequences of Papillomavirus E2 Protein Binding to the Origin Replication Factor ORC2. PLoS Pathog. 2016, 12, e1005934. [Google Scholar] [CrossRef] [PubMed]
- Peta, E.; Cappellesso, R.; Masi, G.; Sinigaglia, A.; Trevisan, M.; Grassi, A.; Di Camillo, B.; Vassarotto, E.; Fassina, A.; Palu, G.; et al. Down-regulation of microRNA-146a is associated with high-risk human papillomavirus infection and epidermal growth factor receptor overexpression in penile squamous cell carcinoma. Hum. Pathol. 2017, 61, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Tzellos, S.; Farrell, P.J. Epstein-barr virus sequence variation-biology and disease. Pathogens 2012, 1, 156–174. [Google Scholar] [CrossRef] [PubMed]
- Raab-Traub, N. Novel mechanisms of EBV-induced oncogenesis. Curr. Opin. Virol. 2012, 2, 453–458. [Google Scholar] [CrossRef] [PubMed]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Ann. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef] [PubMed]
- Ok, C.Y.; Li, L.; Young, K.H. EBV-driven B-cell lymphoproliferative disorders: From biology, classification and differential diagnosis to clinical management. Exp. Mol. Med. 2015, 47, e132. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv. Virus Res. 2014, 88, 279–313. [Google Scholar] [PubMed]
- Wang, J.; Quake, S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Chan, C.P.; Wong, N.H.; Ho, C.H.; Ho, T.H.; Lei, T.; Deng, W.; Tsao, S.W.; Chen, H.; Kok, K.H.; et al. CRISPR/Cas9-mediated genome editing of Epstein-Barr virus in human cells. J. Gener. Virol. 2015, 96 Pt 3, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. CRISPR/Cas9-Mediated Genome Editing of Herpesviruses Limits Productive and Latent Infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. CRISPR/Cas9 Screens Reveal Epstein-Barr Virus-Transformed B Cell Host Dependency Factors. Cell Host Microbe 2017, 21, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Roehm, P.C.; Shekarabi, M.; Wollebo, H.S.; Bellizzi, A.; He, L.; Salkind, J.; Khalili, K. Inhibition of HSV-1 Replication by Gene Editing Strategy. Sci. Rep. 2016, 6, 23146. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed]
- Moradpour, D.; Penin, F.; Rice, C.M. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Strader, D.B.; Wright, T.; Thomas, D.L.; Seeff, L.B. American Association for the Study of Liver, D.; Diagnosis, management, and treatment of hepatitis C. Hepatology 2004, 39, 1147–1171. [Google Scholar] [CrossRef] [PubMed]
- Andreoni, M.; Giacometti, A.; Maida, I.; Meraviglia, P.; Ripamonti, D.; Sarmati, L. HIV-HCV co-infection: Epidemiology, pathogenesis and therapeutic implications. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1473–1483. [Google Scholar] [PubMed]
- Ren, Q.; Li, C.; Yuan, P.; Cai, C.; Zhang, L.; Luo, G.G.; Wei, W. A Dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci. Rep. 2015, 5, 8865. [Google Scholar] [CrossRef] [PubMed]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of hepatitis C virus RNA abundance by a liver-specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Doerflinger, M.; Forsyth, W.; Ebert, G.; Pellegrini, M.; Herold, M.J. CRISPR/Cas9-The ultimate weapon to battle infectious diseases? Cell. Microbiol. 2017, 19. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A CRISPR Activation Screen Identifies a Pan-avian Influenza Virus Inhibitory Host Factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.A.N.; Vetter, P.; Bausch, D.G.; Burgess, T.; Davey, R.T., Jr.; Fowler, R.; Hayden, F.G.; Jahrling, P.B.; Kalil, A.C.; Mayers, D.L.; et al. Ebola virus disease: An update on post-exposure prophylaxis. Lancet Infect. Dis. 2017. [Google Scholar] [CrossRef]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, 1206. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, S.B.; Bolay, F.; Kieh, M.; Grandits, G.; Badio, M.; Ballou, R.; Eckes, R.; Feinberg, M.; Follmann, D.; Grund, B.; et al. Phase 2 Placebo-Controlled Trial of Two Vaccines to Prevent Ebola in Liberia. N. Engl. J. Med. 2017, 377, 1438–1447. [Google Scholar] [CrossRef] [PubMed]
- Cote, M.; Misasi, J.; Ren, T.; Bruchez, A.; Lee, K.; Filone, C.M.; Hensley, L.; Li, Q.; Ory, D.; Chandran, K.; et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011, 477, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.H.; Obernosterer, G.; Raaben, M.; Herbert, A.S.; Deffieu, M.S.; Krishnan, A.; Ndungo, E.; Sandesara, R.G.; Carette, J.E.; Kuehne, A.I.; et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012, 31, 1947–1960. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef] [PubMed]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [PubMed]
- Younan, P.; Iampietro, M.; Nishida, A.; Ramanathan, P.; Santos, R.I.; Dutta, M.; Lubaki, N.M.; Koup, R.A.; Katze, M.G.; Bukreyev, A. Ebola Virus Binding to Tim-1 on T Lymphocytes Induces a Cytokine Storm. mBio 2017, 8, e00845-17. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Cai, H.; Pan, J.; Xiang, N.; Tien, P.; Ahola, T.; Guo, D. Functional screen reveals SARS coronavirus nonstructural protein nsp14 as a novel cap N7 methyltransferase. Proc. Natl. Acad. Sci. USA 2009, 106, 3484–3489. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.C.; Bailey, C.C.; Weyer, J.L.; Radoshitzky, S.R.; Becker, M.M.; Chiang, J.J.; Brass, A.L.; Ahmed, A.A.; Chi, X.; Dong, L.; et al. Distinct patterns of IFITM-mediated restriction of filoviruses, SARS coronavirus, and influenza A virus. PLoS Pathog. 2011, 7, e1001258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Muller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Zhang, Y.; Zhang, W.; Yuan, Y.; Bao, J.; et al. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Earnest, J.T.; Hantak, M.P.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. The tetraspanin CD9 facilitates MERS-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 2017, 13, e1006546. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, S.; Zafar, Y.; Briddon, R.W. Geminivirus disease complexes: The threat is spreading. Trends Plant Sci. 2006, 11, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Hanley-Bowdoin, L.; Bejarano, E.R.; Robertson, D.; Mansoor, S. Geminiviruses: Masters at redirecting and reprogramming plant processes. Nat. Rev. Microbiol. 2013, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.F.; Chen, K.C.; Cheng, Y.H.; Raja, J.A.; Huang, Y.L.; Chien, W.C.; Yeh, S.D. Generation of marker-free transgenic plants concurrently resistant to a DNA geminivirus and a RNA tospovirus. Sci. Rep. 2014, 4, 5717. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Navas-Castillo, J.; Moriones, E.; Hernandez-Zepeda, C.; Idris, A.; Brown, J.K.; Murilo Zerbini, F.; Martin, D.P. Establishment of three new genera in the family Geminiviridae: Becurtovirus, Eragrovirus and Turncurtovirus. Arch. Virol. 2014, 159, 2193–2203. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Zhang, H.; Zhang, Y.; Wang, Y.; Gao, C. Establishing a CRISPR-Cas-like immune system conferring DNA virus resistance in plants. Nat. Plants 2015, 1, 15144. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Abulfaraj, A.; Idris, A.; Ali, S.; Tashkandi, M.; Mahfouz, M.M. CRISPR/Cas9-mediated viral interference in plants. Genome Biol. 2015, 16, 238. [Google Scholar] [CrossRef] [PubMed]
- Ali, Z.; Ali, S.; Tashkandi, M.; Zaidi, S.S.; Mahfouz, M.M. CRISPR/Cas9-Mediated Immunity to Geminiviruses: Differential Interference and Evasion. Sci. Rep. 2016, 6, 26912. [Google Scholar] [CrossRef] [PubMed]
- Baltes, N.J.; Hummel, A.W.; Konecna, E.; Cegan, R.; Bruns, A.N.; Bisaro, D.M.; Voytas, D.F. Conferring resistance to geminiviruses with the CRISPR—Cas prokaryotic immune system. Nat. Plants 2015, 1, 15145. [Google Scholar] [CrossRef]
- Hutter, G.; Bodor, J.; Ledger, S.; Boyd, M.; Millington, M.; Tsie, M.; Symonds, G. CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses 2015, 7, 4186–4203. [Google Scholar] [CrossRef] [PubMed]
- Afkhami, S.; Yao, Y.; Xing, Z. Methods and clinical development of adenovirus-vectored vaccines against mucosal pathogens. Mol. Ther. Methods Clin. Dev. 2016, 3, 16030. [Google Scholar] [CrossRef] [PubMed]
- Wold, W.S.; Toth, K. Adenovirus vectors for gene therapy, vaccination and cancer gene therapy. Curr. Gene Ther. 2013, 13, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.X.; Cannon, P.M. The clinical applications of genome editing in HIV. Blood 2016, 127, 2546–2552. [Google Scholar] [CrossRef] [PubMed]
- Weatherley, D.A.V.; Boswell, M.T.; Rowland-Jones, S.L. Targeting TRIM5alpha in HIV Cure Strategies for the CRISPR-Cas9 Era. Front. Immunol. 2017, 8, 1616. [Google Scholar] [CrossRef] [PubMed]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed]


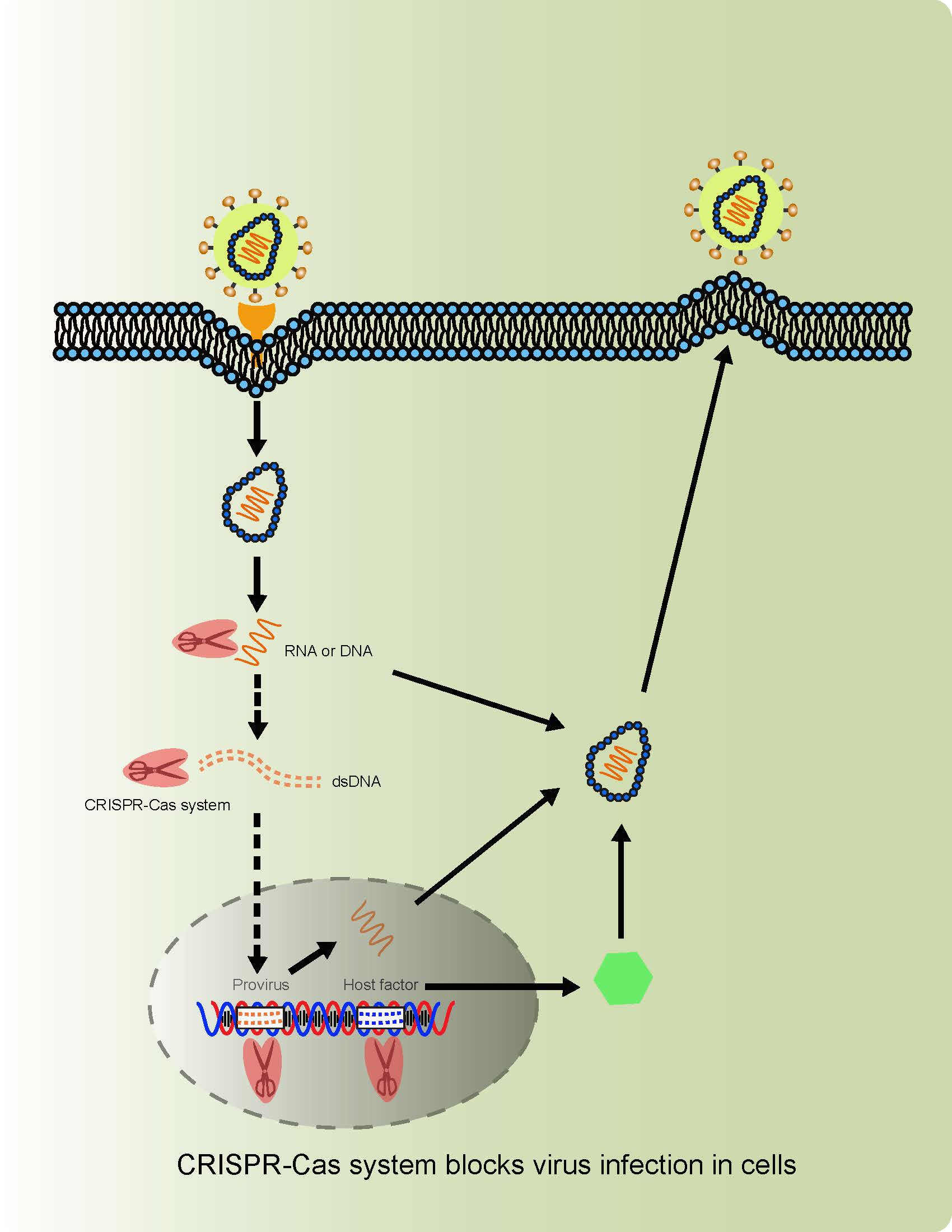{kind=link}
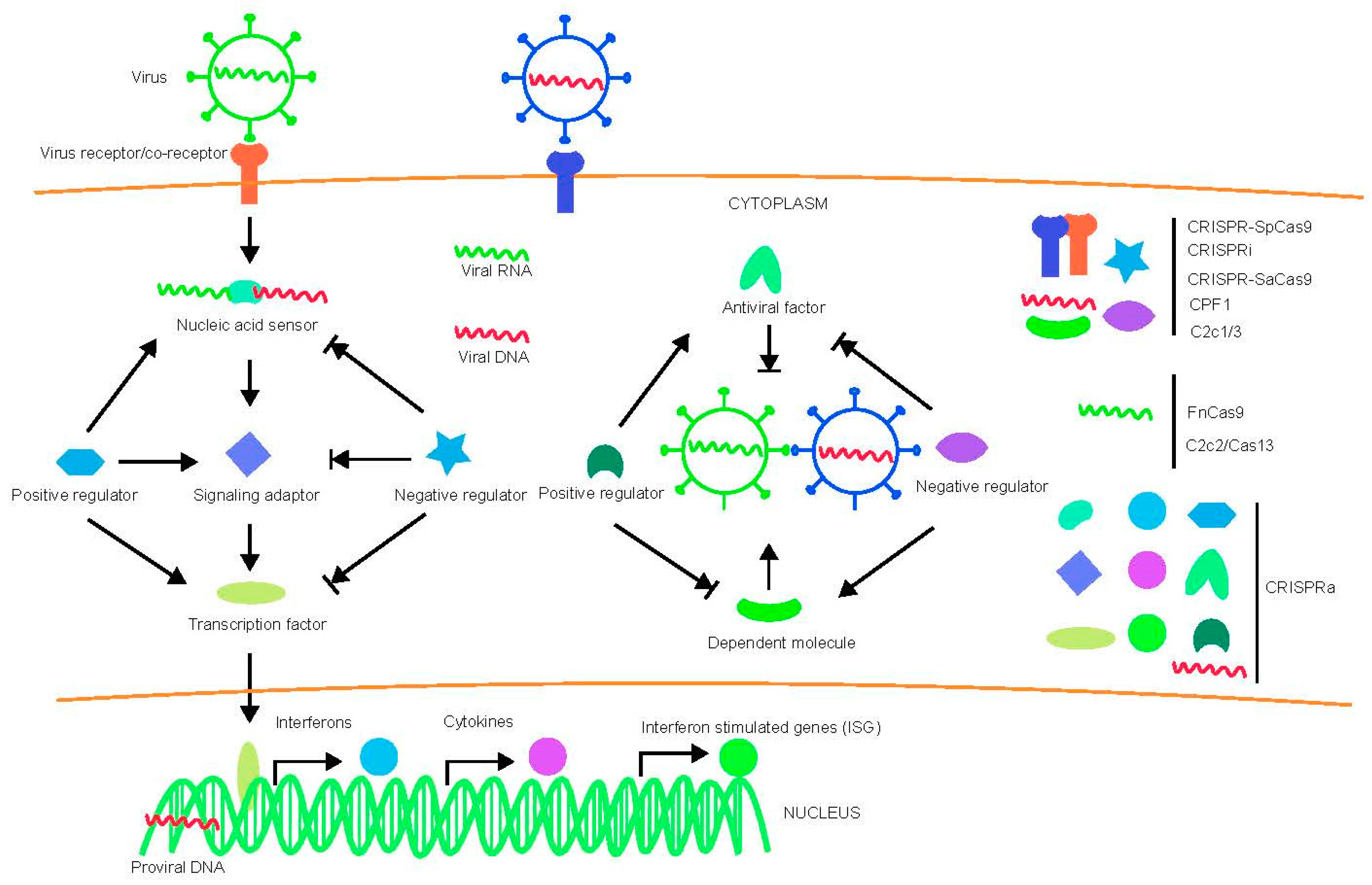{kind=link}
| CRISPR-Cas | DNA Cassette: Size | PAMs | Function | Targeting Virus | Virus Related Host Factor | Delivery Vehicle | Pros/Cons | |
|---|---|---|---|---|---|---|---|---|
| CRISPR-SpCas9 | Sp-Cas9-Flag-T2A-Puro:4641 bp | 5′-NGG-3′ | single-RNA-mediated DNA endonuclease | HIV, HBV, HPV, EBV, ZIKV, DENV | CCR5, CXCR4, LEDGF, TNPO3, BART, EBNA1, CLDN1, OCLN, CD81, AXL, EMC |
| Frequently used/Large protein size | |
| CRISPRi | Flag-NLS-dCas9-NLS-KRAB-T2A-Puro:5145 bp | 5′-NGG-3′ | single-RNA-mediated inhibition of mRNA transcription | - | - |
| Multiplex gene transcription inhibition/Large protein size, less application | |
| CRISPRa | dCas9-VP64 and MS2–p65–HSF1 | dCas9-VP64-Blast:4872 bp MS2-p65-HSF1-Hygro:2520 bp | 5′-NGG-3′ | single-RNA-mediated activation of mRNA transcription | HIV | IFNγ, APOBEC3G (A3G), APOBEC3G (A3G) |
| Multiplex gene activation/Relative low efficiency, Large protein size, less application |
| scFv-GCN4-sfGFP-VP64 and dCas9-24xGCN4-v4 | scFv-GCN4-sfGFP-VP64-GB1-NLS:2031 bp NLS-dCas9-24xGCN4_v4-NLS-P2A-BFP:6906 bp | Single-molecule imaging, transcriptional activation/Super large in size, less application | ||||||
| CRISPR-SaCas9 | NLS-SaCas9-NLS-3xHA:3411 bp | 5′-NNGRRT-3′ | single-RNA-mediated DNA endonuclease | HIV | CCR5, CXCR4 |
| Small in size/Relatively strict PAM, less application | |
| Cpf1 | AsCpf1:4056 bp LbCpf1:3819 bp FnCpf1:3900 bp | 5′-TTTN-3′ | single-crRNA-mediated DNA Endonuclease/RNase activity | - | - |
| a single and short crRNA, cohesive ends, Multiplex gene editing with tandemly arrayed crRNA/less application in cells | |
| FnCas9 | FnCas9:4887 bp | 5′-NGG-3′ | single-RNA-mediated PAM-independent inhibiting of translation of target RNA | HCV | - |
| RNA targeting, Less restrictive PAM/Large in size, less application | |
| C2c1/3 | AacC2c1:3387 bp | 5′-TTN-3′ | dual-RNA-guided DNA endonuclease | - | - |
| Small in size/requires a 111-nt sgRNA containing crRNA and tracrRNA, lower cleavage activity, no application in cells | |
| C2c2/Cas13 | LwCas13a-msfGFP-2A-Blast:4869 bp | depends on a non-G 3′ protospacer-flanking site (PFS) | single-effector endoRNase mediating ssRNA cleavage with a single crRNA guide | ZIKV, DENV | - |
| RNA targeting, nucleic acid detection/Large in size, less application | |
| Virus | Genome | CRISPR-Cas for Genetic Targeting | Dependent or Positive Regulators | Restriction Factors |
|---|---|---|---|---|
| HIV | +ssRNA | CRISPR-SpCas9, CRISPRa CRISPR-SaCas9 (Cpf1, FnCas9, C2c1/3, C2c2/Cas13) | CCR5, CXCR4, LEDGF/p75, TNPO3 (PSIP1, Nup358, CPSF6, ALCAM) | APOBEC3G, APOBEC3B, IFNγ (TRIM5α, BST-2/Tetherin, SAMHD-1, Mx2, Serinc3/5) |
| HBV | relaxed circular (RC) DNA | CRISPR-SpCas9 (CRISPR-SaCas9, Cpf1, FnCas9, C2c1/3, C2c2/Cas13) | (NTCP, Hsc70, Hsp90, TDP2, γ2-adaptin) | IFNγ (BST-2/tetherin) |
| HPV | dsDNA | CRISPR-SpCas9 (CRISPR-SaCas9, Cpf1, FnCas9, C2c1/3, C2c2/Cas13) | (SIRT1, Brd4, CXCR4, KLF4/13, ORC2) | (Sp100, miR-145, p56, IFI16, C/EBPβ, p53, EVERs, APOBEC, FANCD2) |
| EBV | dsDNA | CRISPR-SpCas9 (CRISPR-SaCas9, Cpf1, FnCas9, C2c1/3, C2c2/Cas13) | IKKβ, HOIP, p52, RBP-Jκ, WDR48, MDM2/4, CTBP1, BCL6,SYK,BTK,BLNK,PTEN, CD19/81, TNFRSF1A, BATF, IRF4, IRF2 | (c-CBL) |
| HCV | +ssRNA | FnCas9 (C2c2/Cas13) | DGCR8, CLDN1, OCLN, CD81 | (NLRX1, SMYD3, VAP-C) |
| ZIKV | +ssRNA | C2c2/Cas13 (FnCas9) | (OST, AXL, RAB5C, RABGEF, NDST1, EXT1, EMC) | - |
| DENV | +ssRNA | C2c2/Cas13 (FnCas9) | (OST, RAB5C, RABGEF, NDST1, EXT1, EMC) | - |
| WNV | +ssRNA | (FnCas9, C2c2/Cas13) | (SPCS, EMC2, EMC3, SEL1L, DERL2, UBE2G2, UBE2J1, HRD1) | (HSP70) |
| IAV | −ssRNA | (FnCas9, C2c2/Cas13) | (IFITM, B4GALNT2, α2,3 linked sialic acid receptor) | (DcR3) |
| EBOV | +ssRNA | (FnCas9, C2c2/Cas13) | (NPC1, Tim-1) | - |
| SARS-CoV | +ssRNA | (FnCas9, C2c2/Cas13) | (ACE2, IFITM) | - |
| MERS-CoV | +ssRNA | (FnCas9, C2c2/Cas13) | (CD26/DDP4, CD9) | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, S.; Yu, X.; Guo, D. CRISPR-Cas Targeting of Host Genes as an Antiviral Strategy. Viruses 2018, 10, 40. https://doi.org/10.3390/v10010040
Chen S, Yu X, Guo D. CRISPR-Cas Targeting of Host Genes as an Antiviral Strategy. Viruses. 2018; 10(1):40. https://doi.org/10.3390/v10010040
Chicago/Turabian StyleChen, Shuliang, Xiao Yu, and Deyin Guo. 2018. "CRISPR-Cas Targeting of Host Genes as an Antiviral Strategy" Viruses 10, no. 1: 40. https://doi.org/10.3390/v10010040