
Integrative Analysis of the Transcriptome and Metabolome Reveals the Mechanism of Chinese Fir Seed Germination

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experiment Materials and Settings
2.2. Determination of SS, SP, MDA, Pro, SOD, and POD
2.3. Total RNA Extraction and Transcriptome Sequencing
2.4. Quantitative Real-Time PCR (qRT-PCR) Analysis
2.5. Metabolite Extraction and Analysis
2.6. Statistical Analysis of Data
3. Results
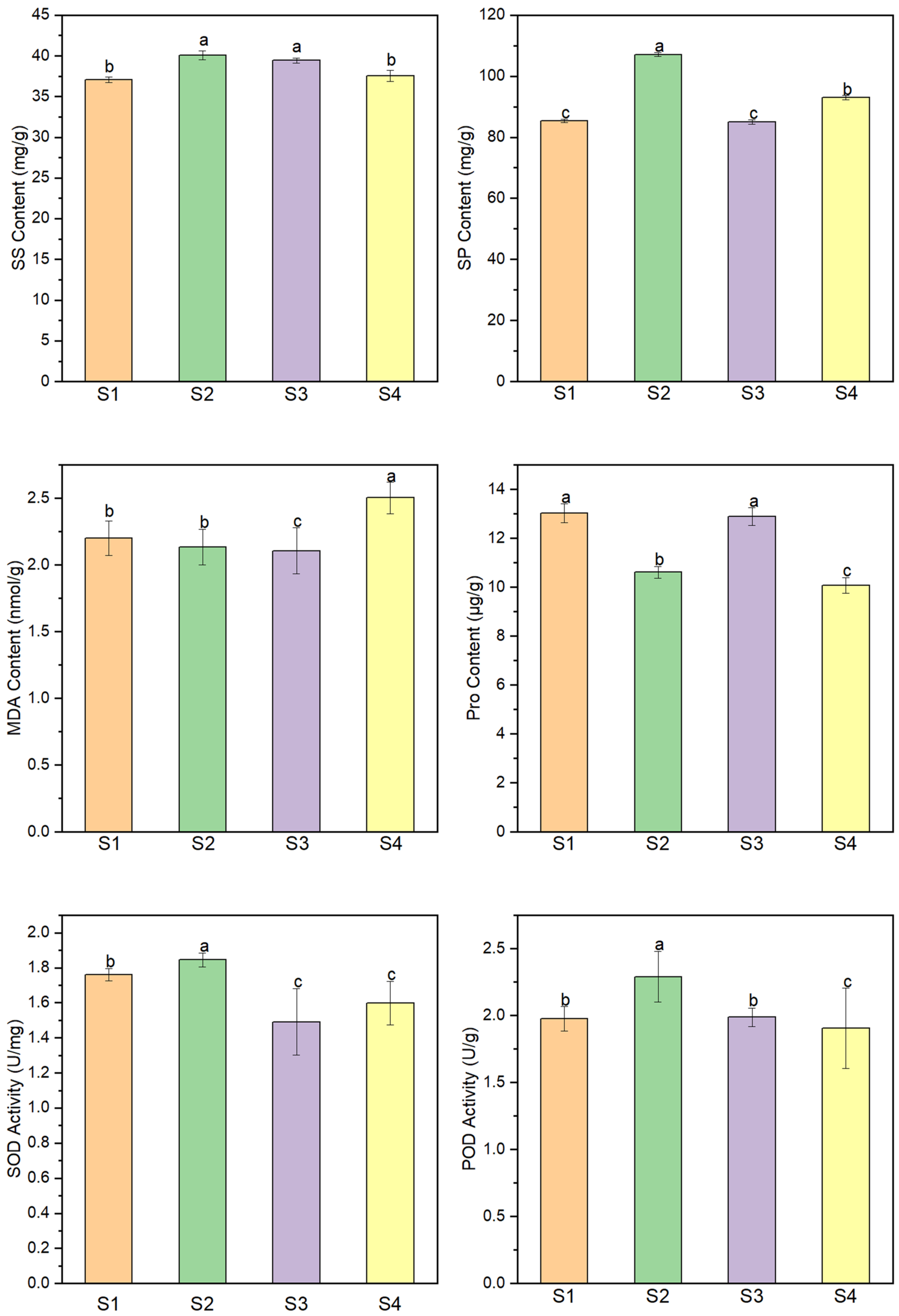
3.1. Physiological Changes of Chinese Fir Seeds
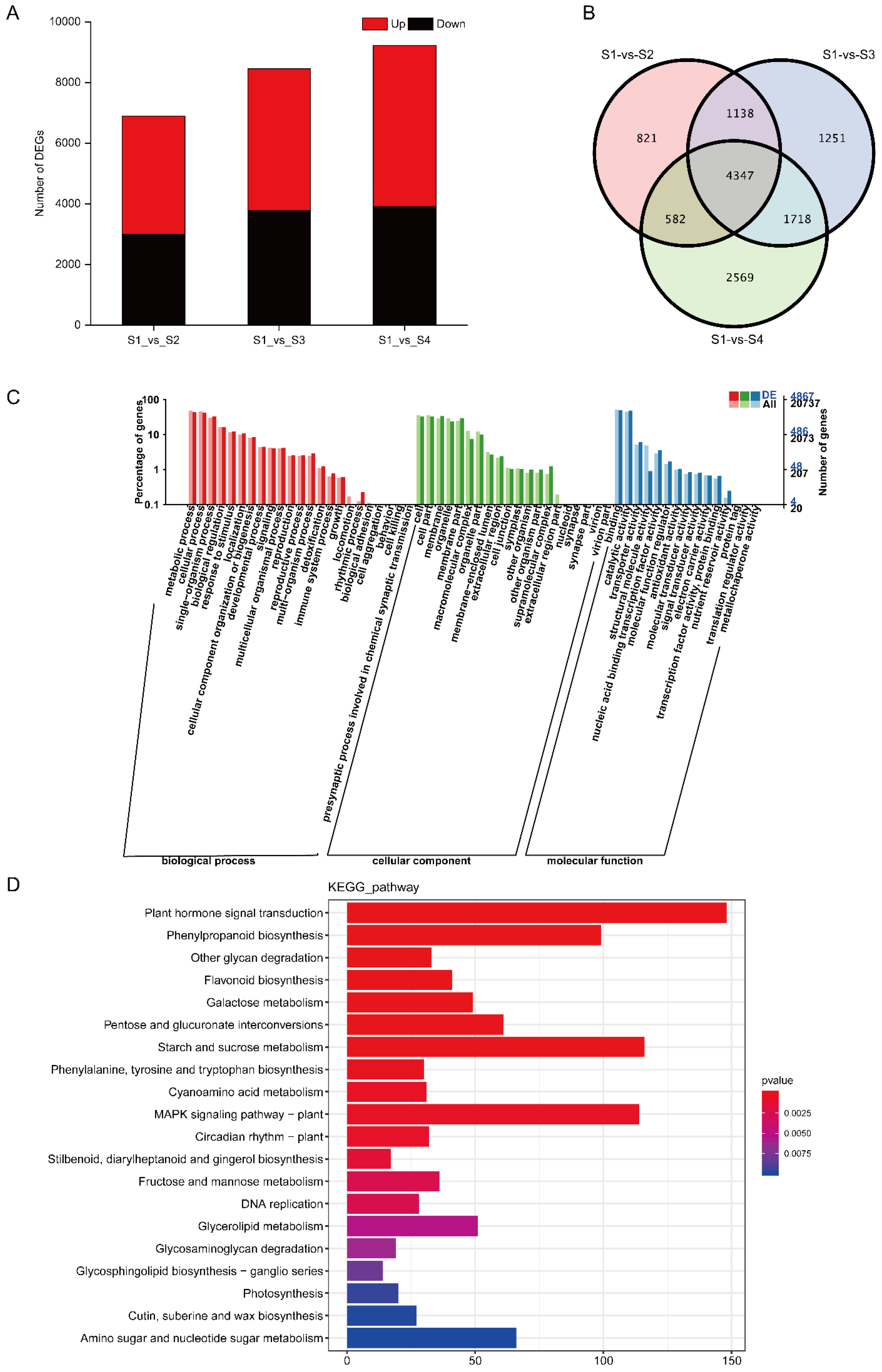
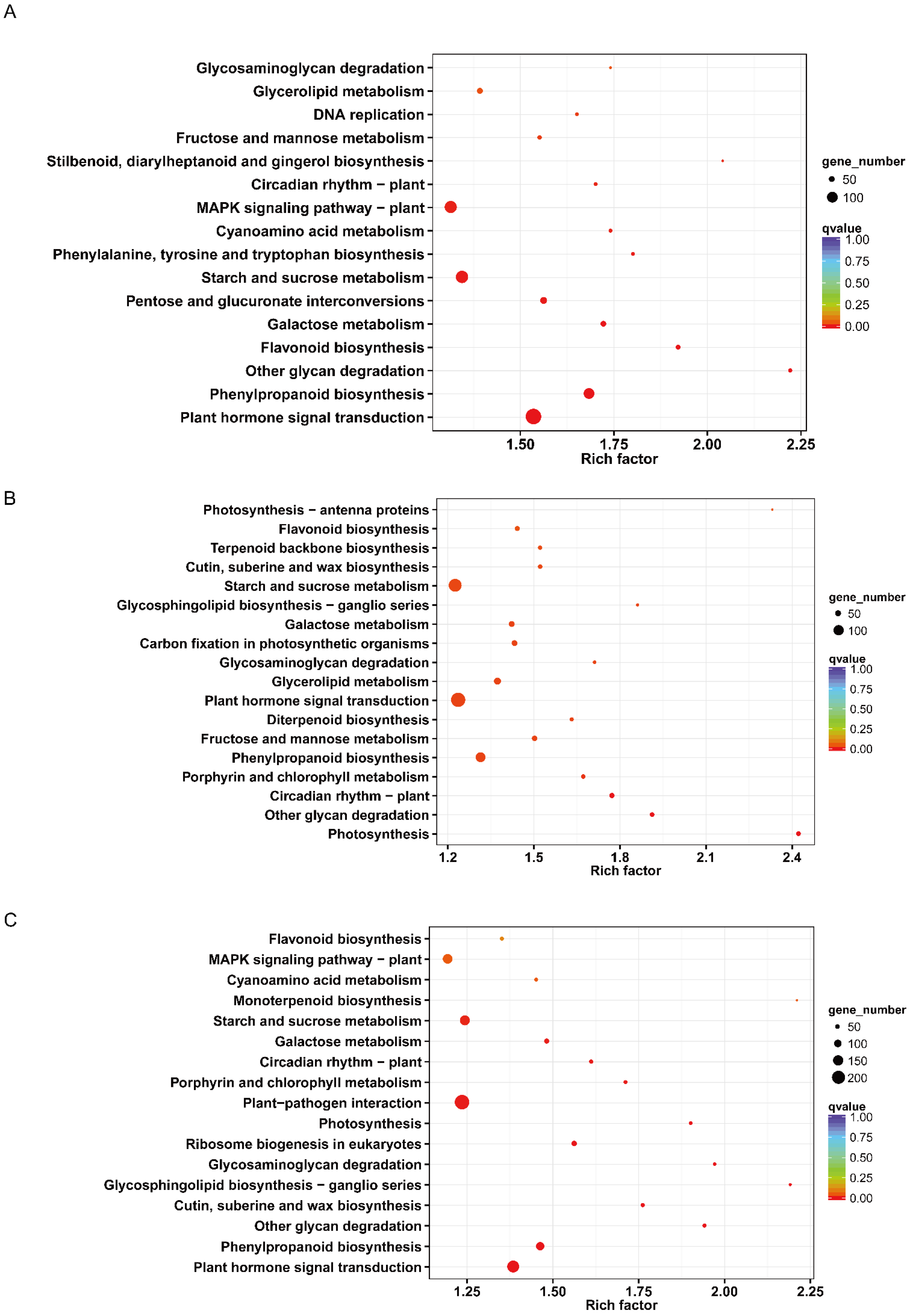
3.2. Transcription Analysis
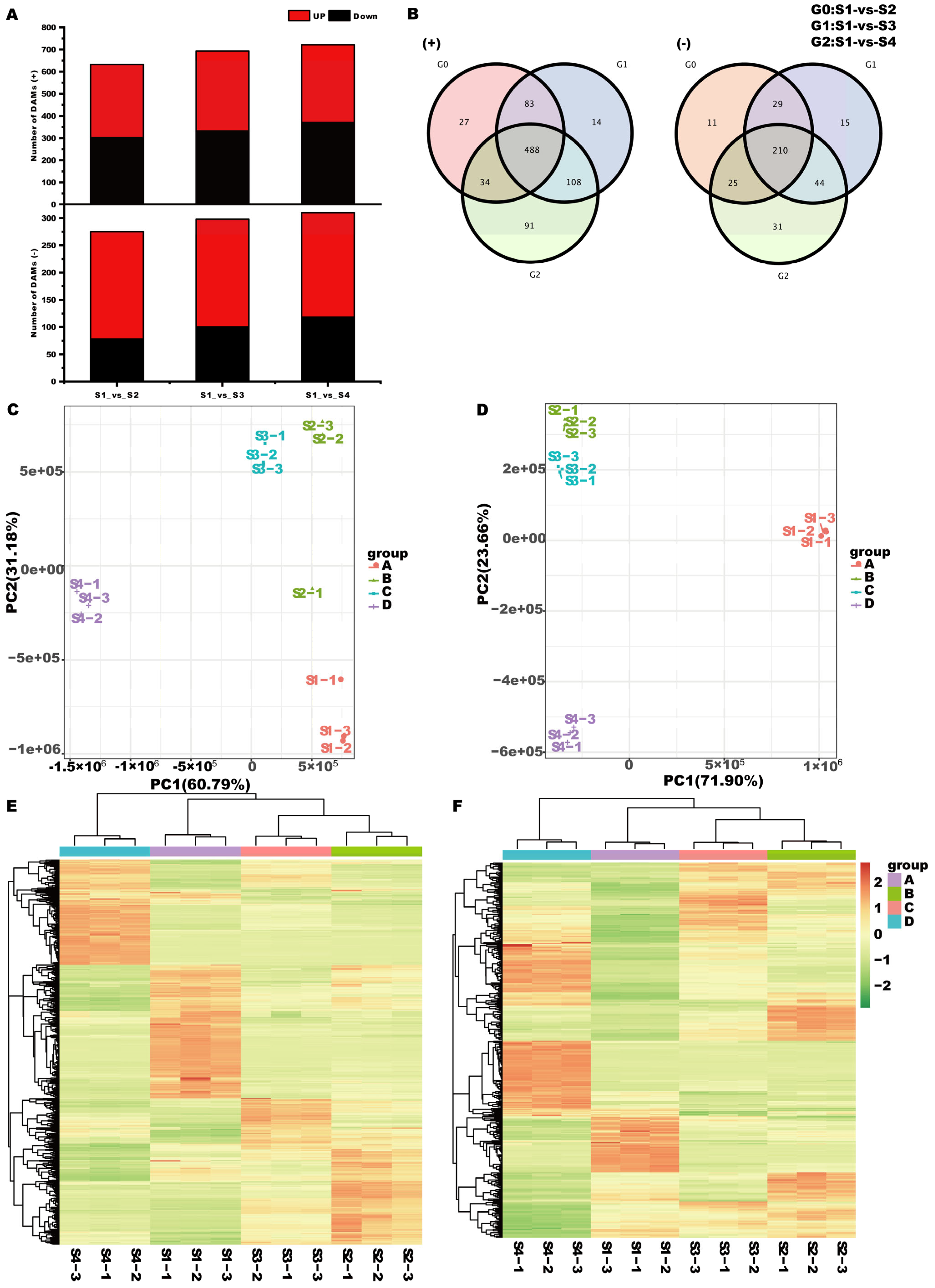
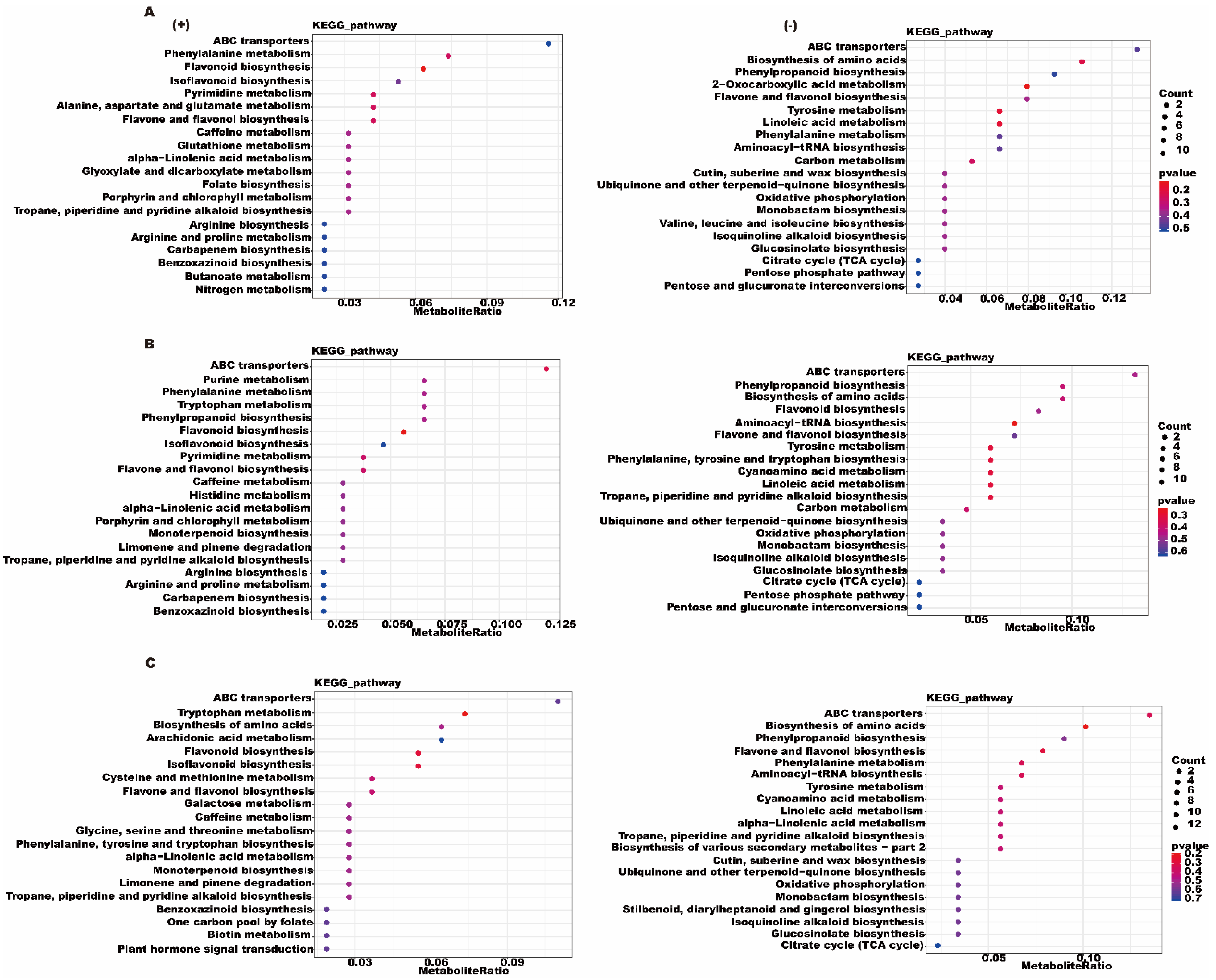
3.3. Metabolome Analysis
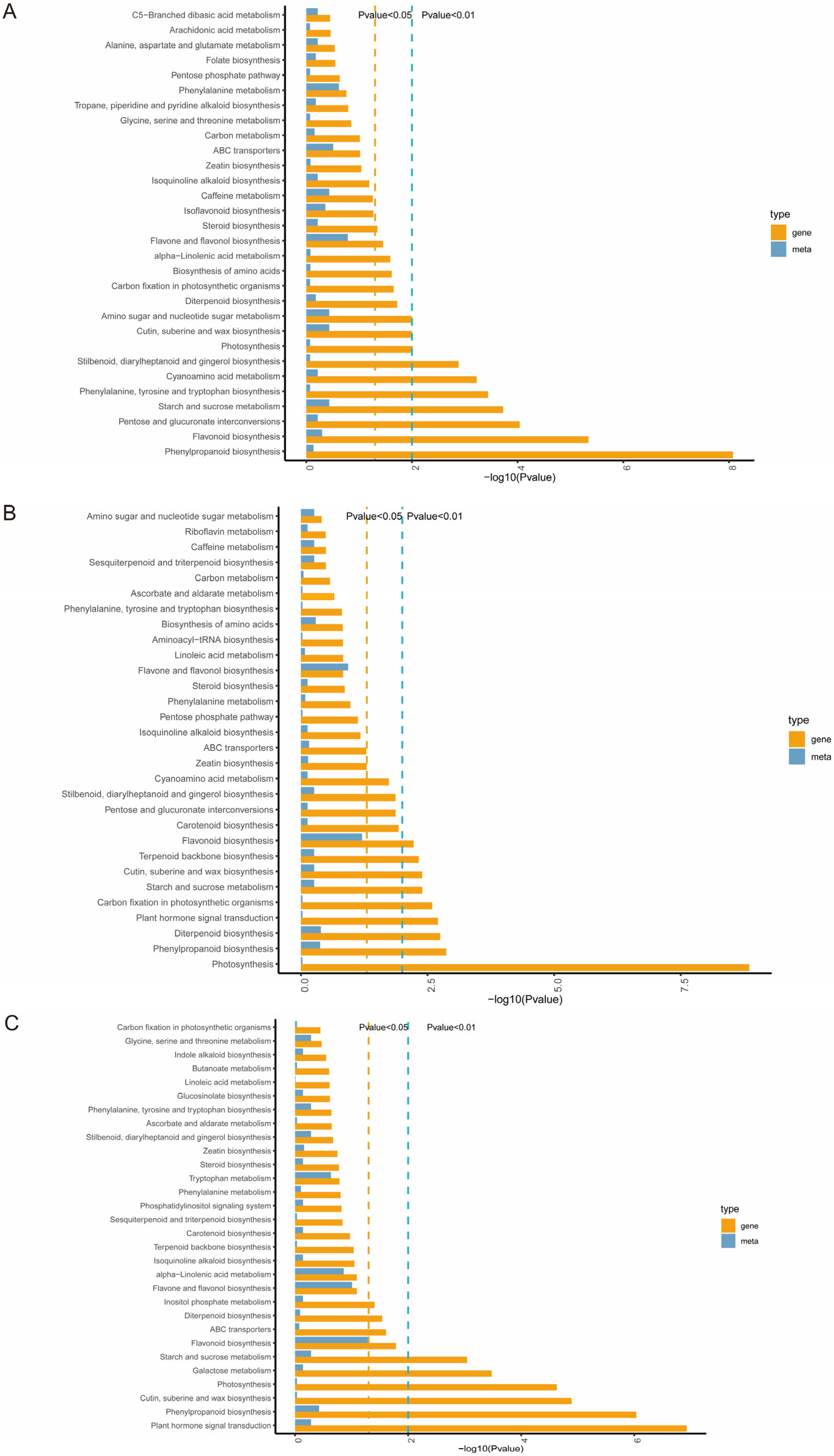
3.4. Integrated Metabolomic and Transcriptomic Analyzes
4. Discussion
4.1. Dynamic Changes of Physiological Indexes during Seed Germination
4.2. Changes of Transcriptome and Metabolome Levels during Seed Germination
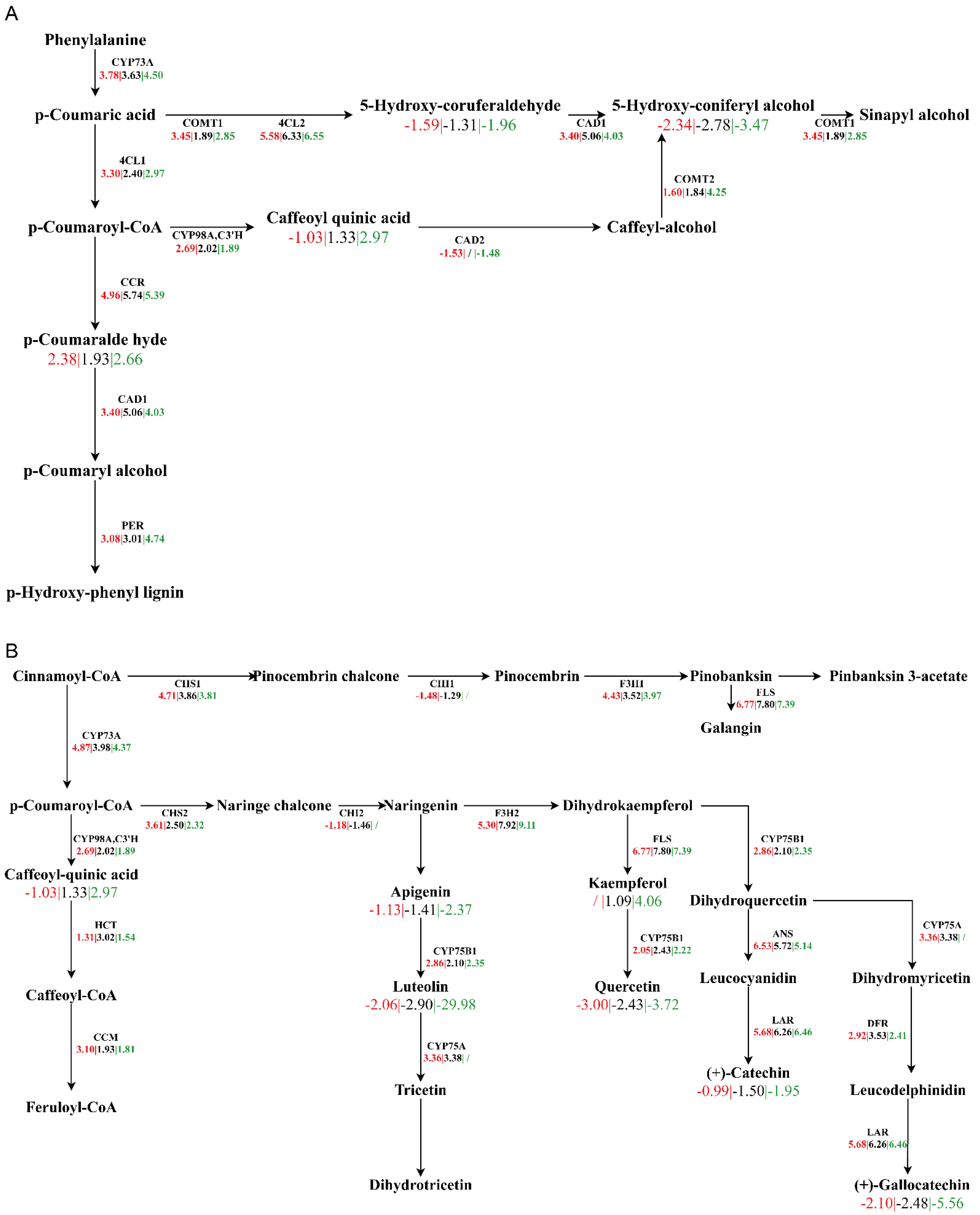
4.3. The Key Pathways during Seed Germination
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, Q.; Wang, S.; He, T.; Liu, L.; Wu, J. Response of organic carbon mineralization and microbial community to leaf litter and nutrient additions in subtropical forest soils. Soil Biol. Biochem. 2014, 71, 13–20. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, W.; Sun, T.; Chen, L.; Pang, X.; Wang, Y.; Xiao, F. N and P fertilization reduced soil autotrophic and heterotrophic respiration in a young Cunninghamia lanceolata forest. Agric. For. Meteorol. 2017, 232, 66–73. [Google Scholar] [CrossRef]
- Yao, Y.; Bingjun, L.; Zhijun, H.; Hui, Z.; Xiaojian, W.; Hassan, F.T.; Pengfei, W.; Ming, L.; Xiangqing, M. Characteristics and Driving Factors of Rhizosphere Bacterial Communities of Chinese Fir Provenances. Forests 2021, 12, 1362. [Google Scholar]
- Zhang, X.; Cao, Q.V.; Xiang, C.; Duan, A.; Zhang, J. Predicting total and component biomass of Chinese fir using a forecast combination method. Iforest Biogeosci. For. 2017, 10, 687–691. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.-K.; Wang, S.-L.; Zhong, M.-C. Ecosystem carbon storage and soil organic carbon stability in pure and mixed stands of Cunninghamia lanceolata and Michelia macclurei. Plant Soil 2013, 370, 295–304. [Google Scholar] [CrossRef]
- Ming, A.; Yang, Y.; Liu, S.; Nong, Y.; Li, H.; Tao, Y.; Sun, D.; Lei, L.; Zeng, J.; An, N. The Impact of Near Natural Forest Management on the Carbon Stock and Sequestration Potential of Pinus massoniana (Lamb.) and Cunninghamia lanceolata (Lamb.) Hook. Plantations. Forests 2019, 10, 626. [Google Scholar] [CrossRef] [Green Version]
- Su, L.; Wan, S.; Zhou, J.; Shao, Q.S.; Xing, B. Transcriptional regulation of plant seed development. Physiol. Plant. 2021, 173, 2013–2025. [Google Scholar] [CrossRef]
- Xia, K.; Zhu, Z.Q. Characterization of physiological traits during development of the recalcitrant seeds of Quercus serrata. Plant Biol. 2021, 23, 1000–1005. [Google Scholar] [CrossRef]
- Steven, P.; Yi, L.; Gilday, A.D.; Stuart, G.; Graham, I.A. Arabidopsis ABA INSENSITIVE4 regulates lipid mobilization in the embryo and reveals repression of seed germination by the endosperm. Plant Cell 2006, 18, 1887–1899. [Google Scholar]
- Verma, V.; Ravindran, P.; Kumar, P.P. Plant hormone-mediated regulation of stress responses. BMC Plant Biol. 2016, 16, 86. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Lu, J.; Xing, J.; Du, M.; Wang, M.; Zhang, L.; Li, Y.; Zhang, C.; Wu, Y. Transcriptome and metabolome analyses revealing the potential mechanism of seed germination in Polygonatum cyrtonema. Sci. Rep. 2021, 11, 12161. [Google Scholar] [CrossRef]
- Fu, F.-f.; Peng, Y.-s.; Wang, G.-b.; El-Kassaby, Y.A.; Cao, F.-l. Integrative analysis of the metabolome and transcriptome reveals seed germination mechanism in Punica granatum L. J. Integr. Agric. 2021, 20, 132–146. [Google Scholar] [CrossRef]
- Yijia, J.; YanLi, M.; Ping, Z.; Gui-Guang, C.; Jiexin, Z.; Shengbao, C. Effects of different oligochitosans on isoflavone metabolites, antioxidant activity and isoflavone biosynthetic genes in soybean (Glycine max) seeds during germination. J. Agric. Food Chem. 2019, 67, 4652–4661. [Google Scholar]
- Fei, X.; Qi, Y.; Lei, Y.; Wang, S.; Hu, H.; Wei, A. Transcriptome and metabolite analysis reveals key genes for melanin synthesis during the development of Zanthoxylum bungeanum seeds. Ind. Crops Prod. 2021, 165, 113419. [Google Scholar] [CrossRef]
- Wang, Y.; Nie, L.; Ma, J.; Zhou, B.; Han, X.; Cheng, J.; Lu, X.; Fan, Z.; Li, Y.; Cao, Y. Transcriptomic Variations and Network Hubs Controlling Seed Size and Weight During Maize Seed Development. Front. Plant Sci 2022, 13, 828923. [Google Scholar] [CrossRef]
- Pang, X.; Suo, J.; Liu, S.; Xu, J.; Yang, T.; Xiang, N.; Wu, Y.; Lu, B.; Qin, R.; Liu, H.; et al. Combined transcriptomic and metabolomic analysis reveals the potential mechanism of seed germination and young seedling growth in Tamarix hispida. BMC Genom. 2022, 23, 109. [Google Scholar] [CrossRef]
- Yemm, E.W.; Willis, A.J. The estimation of carbohydrates in plant extracts by anthrone. Biochem. J. 1954, 57, 508–514. [Google Scholar] [CrossRef] [Green Version]
- Howarth, R.E.; McArthur, J.M.; Goplen, B.P. Bloat Investigations: Determination of Soluble Protein Concentration in Alfalfa1. Crop Sci. 1973, 13, 677–680. [Google Scholar] [CrossRef]
- Velikova, V.; Yordanov, I.; Edreva, A. Oxidative stress and some antioxidant systems in acid rain-treated bean plants. Plant Sci. 2000, 151, 59–66. [Google Scholar] [CrossRef]
- Bates, L.S.; Waldren, R.P.; Teare, I.D. Rapid determination of free proline for water-stress studies. Plant Soil 1973, 39, 205–207. [Google Scholar] [CrossRef]
- Giannopolitis, C.N.; Ries, S.K. Superoxide Dismutases: I. Occurrence in Higher Plants 1 2. Plant Physiol. 1977, 59, 309–314. [Google Scholar] [CrossRef]
- Hernández, J.A.; Jiménez, A.; Mullineaux, P.; Sevilia, F. Tolerance of pea (Pisum sativum L.) to long-term salt stress is associated with induction of antioxidant defences. Plant Cell Environ. 2001, 23, 853–862. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar]
- Chen, X.; Xizeng, M.; Jiaju, H.; Yang, D.; Jianmin, W.; Shan, D.; Lei, K.; Ge, G.; Chuan-Yun, L.; Liping, W. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar]
- Wishart, D.S.; Djoumbou, F.Y.; Ana, M.; Chi, G.A.; Kevin, L.; Rosa, V.-F.; Tanvir, S.; Daniel, J.; Carin, L.; Naama, K.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Eoin, F.; Manish, S.; Dawn, C.; Shankar, S. LIPID MAPS online tools for lipid research. Nucleic Acids Res. 2007, 35, W606–W612. [Google Scholar]
- Guangchuang, Y.; Li-Gen, W.; Yanyan, H.; Qing-Yu, H. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar]
- Geoffrey, L.; Ying-Chih, C.; Yi, W.; Jing-Ke, W. Mechanistic basis for the evolution of chalcone synthase catalytic cysteine reactivity in land plants. J. Biol. Chem. 2018, 293, 18601–18612. [Google Scholar]
- Dixon, R.A.; Achnine, L.; Kota, P.; Liu, C.J.; Reddy, M.S.; Wang, L. The phenylpropanoid pathway and plant defence-a genomics perspective. Mol. Plant Pathol. 2002, 3, 371–390. [Google Scholar] [CrossRef]
- Li, J.; Chen, X.; Zhan, R.; He, R. Transcriptome profiling reveals metabolic alteration in Andrographis paniculata in response to continuous cropping. Ind. Crops Prod. 2019, 137, 585–596. [Google Scholar] [CrossRef]
- Pan, J.; Li, Z.; Dai, S.; Ding, H.; Wang, Q.; Li, X.; Ding, G.; Wang, P.; Guan, Y.; Liu, W. Integrative analyses of transcriptomics and metabolomics upon seed germination of foxtail millet in response to salinity. Sci. Rep. 2020, 10, 13660. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, L.; Zhao, Y.; Zhao, X.; Yuan, Z. Systematic Analysis and Expression Profiles of the 4-Coumarate: CoA Ligase (4CL) Gene Family in Pomegranate (Punica granatum L.). Int. J. Mol. Sci. 2022, 23, 3509. [Google Scholar] [CrossRef]
- Chen, X.; Su, W.; Zhang, H.; Zhan, Y.; Zeng, F. Fraxinus mandshurica 4-coumarate-CoA ligase 2 enhances drought and osmotic stress tolerance of tobacco by increasing coniferyl alcohol content. Plant Physiol. Biochem. 2020, 155, 697–708. [Google Scholar] [CrossRef]
- Qu, C.; Zuo, Z.; Cao, L.; Huang, J.; Sun, X.; Zhang, P.; Yang, C.; Li, L.; Xu, Z.; Liu, G. Comprehensive dissection of transcript and metabolite shifts during seed germination and post-germination stages in poplar. BMC Plant Biol. 2019, 19, 279. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Mavraganis, I.; Shen, W.; Yang, H.; Cram, D.; Xiang, D.; Patterson, N.; Zou, J. Transcriptome dissection of candidate genes associated with lentil seed quality traits. Plant Biol. 2022, 24, 815–826. [Google Scholar] [CrossRef]
- Yang, K.; Han, H.; Li, Y.; Ye, J.; Xu, F. Significance of miRNA in enhancement of flavonoid biosynthesis. Plant Biol. 2022, 24, 217–226. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Kong, Z.; Huan, X.; Li, L.; Zhang, P.; Wang, Q.; Guo, Y.; Zhu, W.; Qin, P. Transcriptomics and metabolomics analyses of the mechanism of flavonoid synthesis in seeds of differently colored quinoa strains. Genomics 2022, 114, 138–148. [Google Scholar] [CrossRef]
- Xu, N.; Liu, S.; Lu, Z.; Pang, S.; Wang, L.; Wang, L.; Li, W. Gene Expression Profiles and Flavonoid Accumulation during Salt Stress in Ginkgo biloba Seedlings. Plants 2020, 9, 1162. [Google Scholar] [CrossRef]
- Ferrer, J.-L.; Austin, M.B.; Stewart, C.; Noel, J.P. Structure and function of enzymes involved in the biosynthesis of phenylpropanoids. Plant Physiol. Biochem. 2007, 46, 356–370. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, X.; Chen, H.; Lu, J.; Chen, D.; Luo, C.; Cheng, X.; Jia, Y.; Huang, C. Transcriptome and Metabolome Analyses of the Flowers and Leaves of Chrysanthemum dichrum. Front. Genet. 2021, 12, 716163. [Google Scholar] [CrossRef]
- Xu, C.; Abbas, H.M.K.; Zhan, C.; Huang, Y.; Huang, S.; Yang, H.; Wang, Y.; Yuan, H.; Luo, J.; Zeng, X. Integrative metabolomic and transcriptomic analyses reveal the mechanisms of Tibetan hulless barley grain coloration. Front. Plant Sci. 2022, 13, 1038625. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zhao, G.; Li, Y.; Wei, S.; Dong, Y.; Jiao, R. Integrative Analysis of the Transcriptome and Metabolome Reveals the Mechanism of Chinese Fir Seed Germination. Forests 2023, 14, 676. https://doi.org/10.3390/f14040676
Chen X, Zhao G, Li Y, Wei S, Dong Y, Jiao R. Integrative Analysis of the Transcriptome and Metabolome Reveals the Mechanism of Chinese Fir Seed Germination. Forests. 2023; 14(4):676. https://doi.org/10.3390/f14040676
Chicago/Turabian StyleChen, Xiangteng, Guangyu Zhao, Yanglong Li, Shumeng Wei, Yuhong Dong, and Ruzhen Jiao. 2023. "Integrative Analysis of the Transcriptome and Metabolome Reveals the Mechanism of Chinese Fir Seed Germination" Forests 14, no. 4: 676. https://doi.org/10.3390/f14040676