
Diversity and Functional Potential of Prokaryotic Communities in Depth Profile of Boreo-Nemoral Minerotrophic Pine Swamp (European Russia)
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
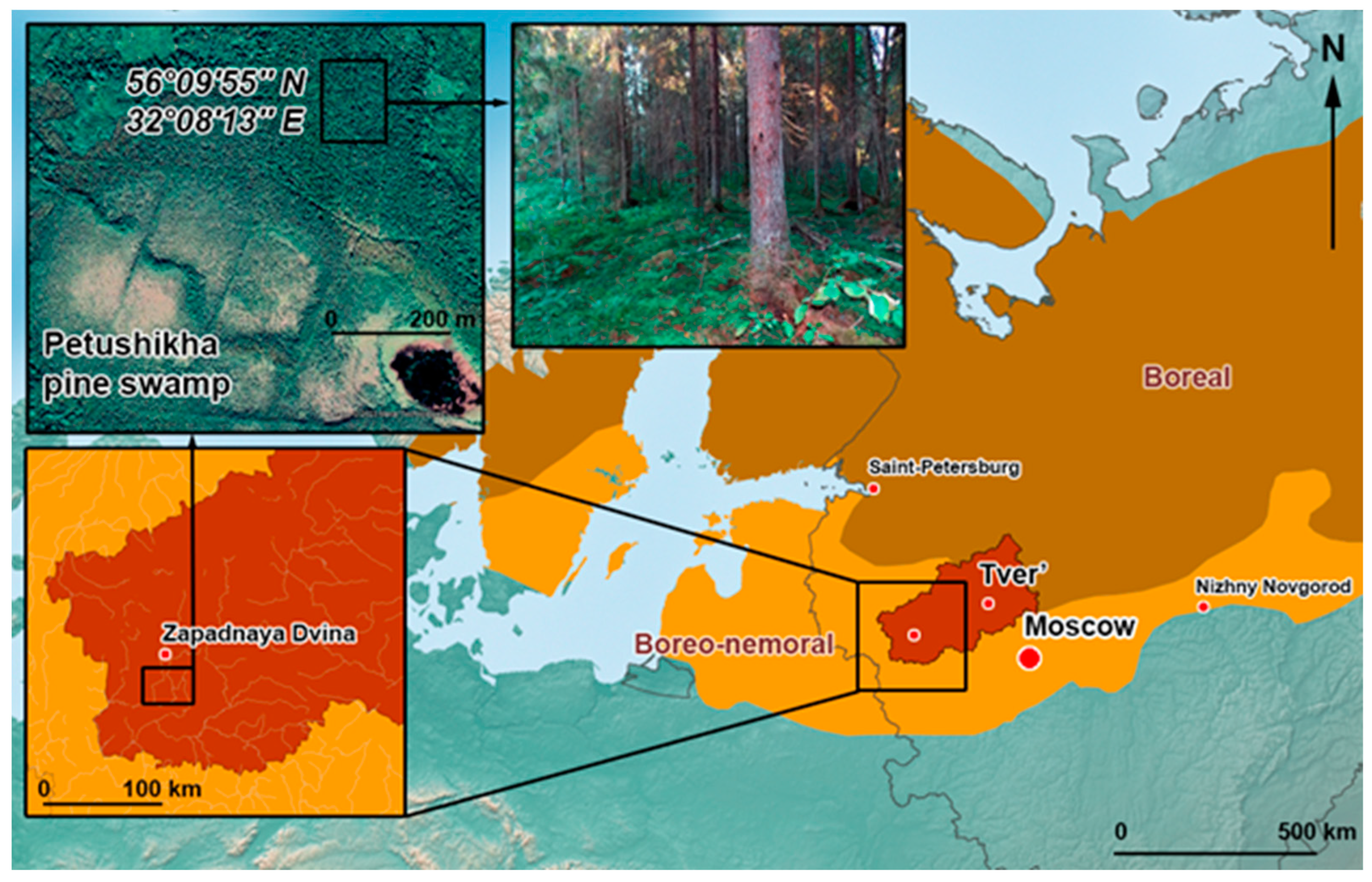
2.1. Study Location
2.2. Phytosociological Records
2.3. Soil Cover
2.4. Sampling Procedure
2.5. Sample Characterization
2.6. DNA Isolation, Amplification, and Sequencing of the 16S rRNA Gene
2.7. Bioinformatics Analysis
2.8. Statistical Analysis and Functional Characterization
2.9. Data Availability
3. Results
3.1. Characterization of Peat Samples and Isolation of Acrotelm and Catotelm
3.2. Characterization of Prokaryotic Community Diversity
3.3. Taxonomic Composition of Prokaryotic Communities at the Bacteria/Archaea Domains
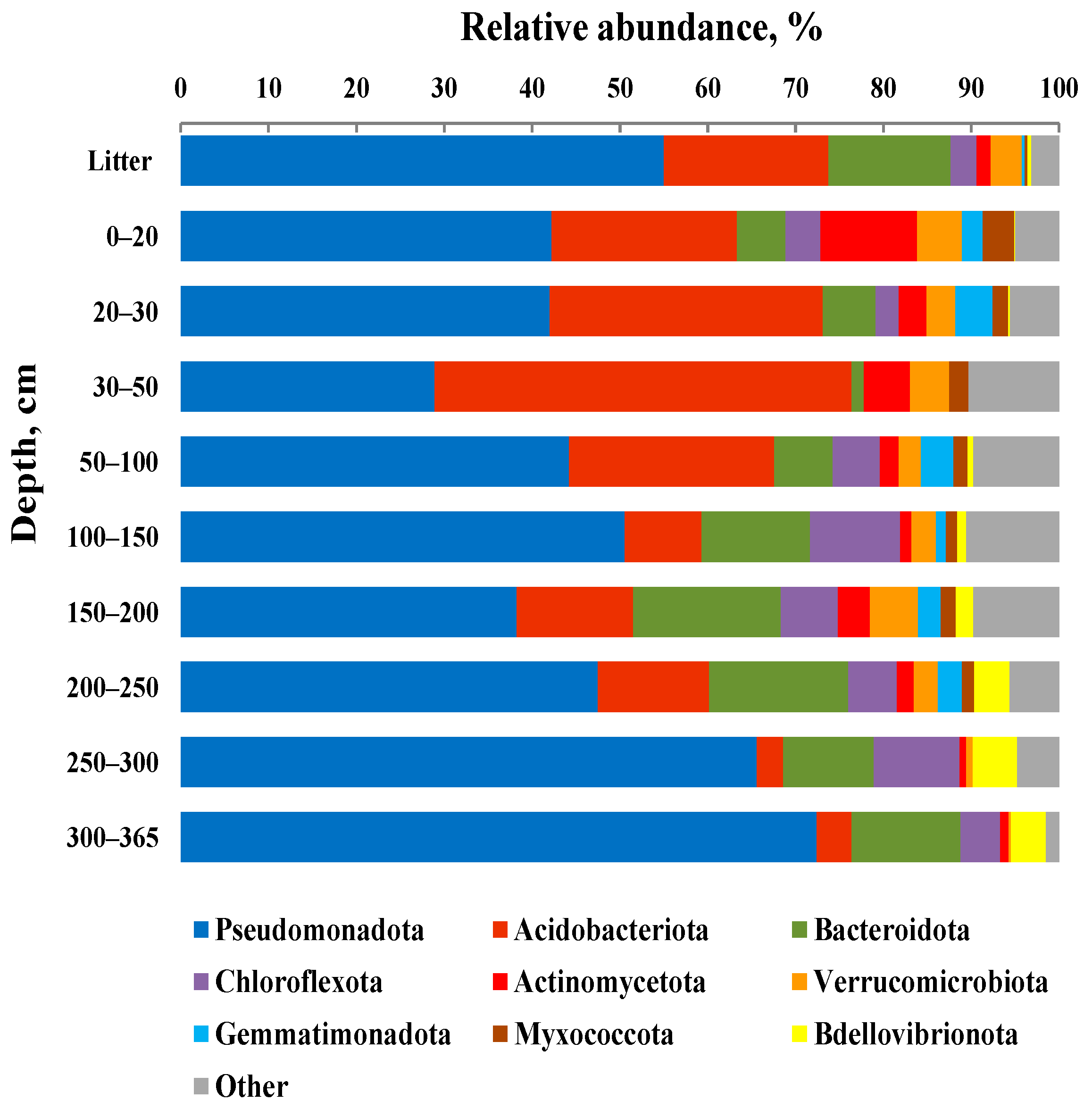
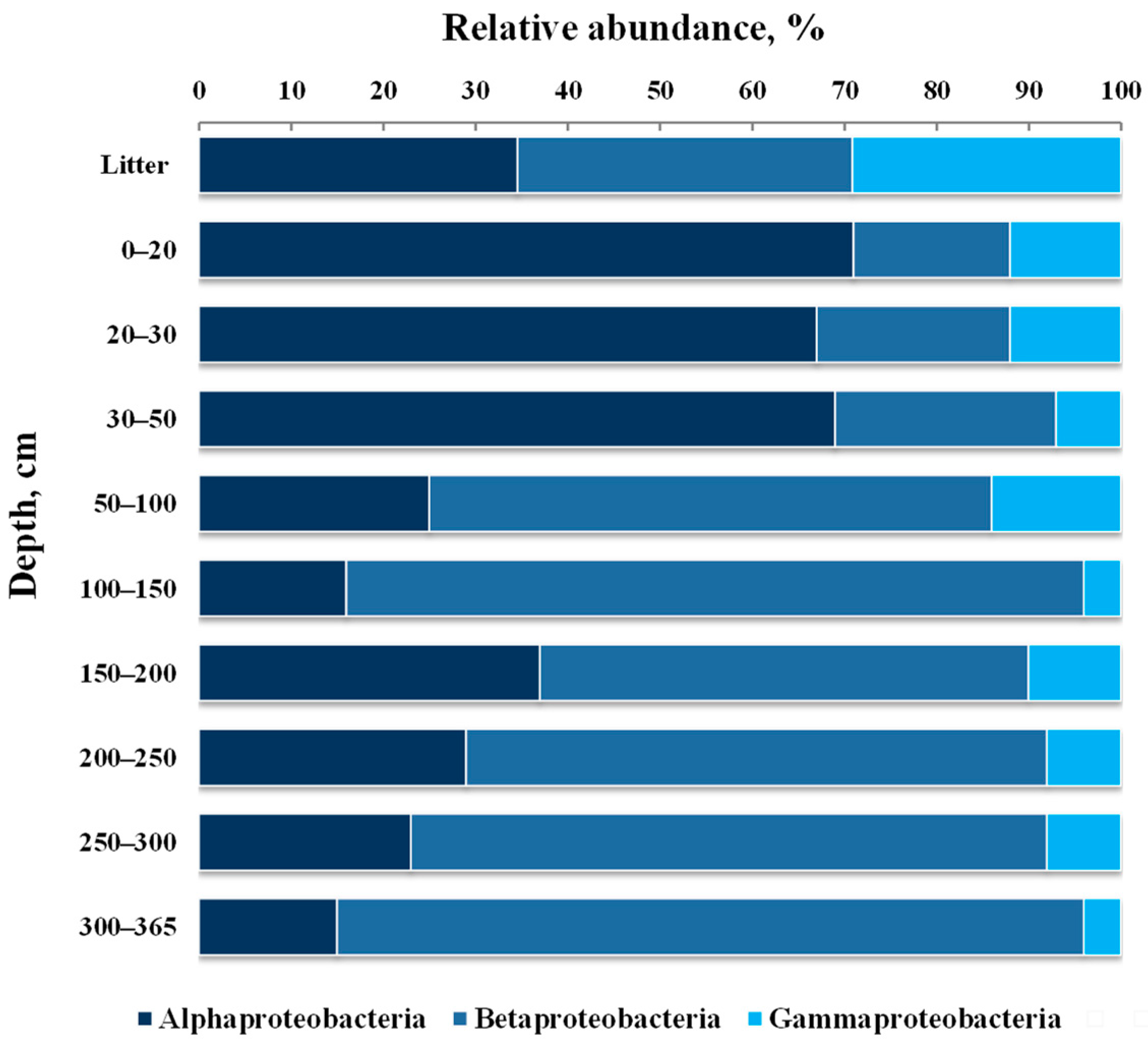
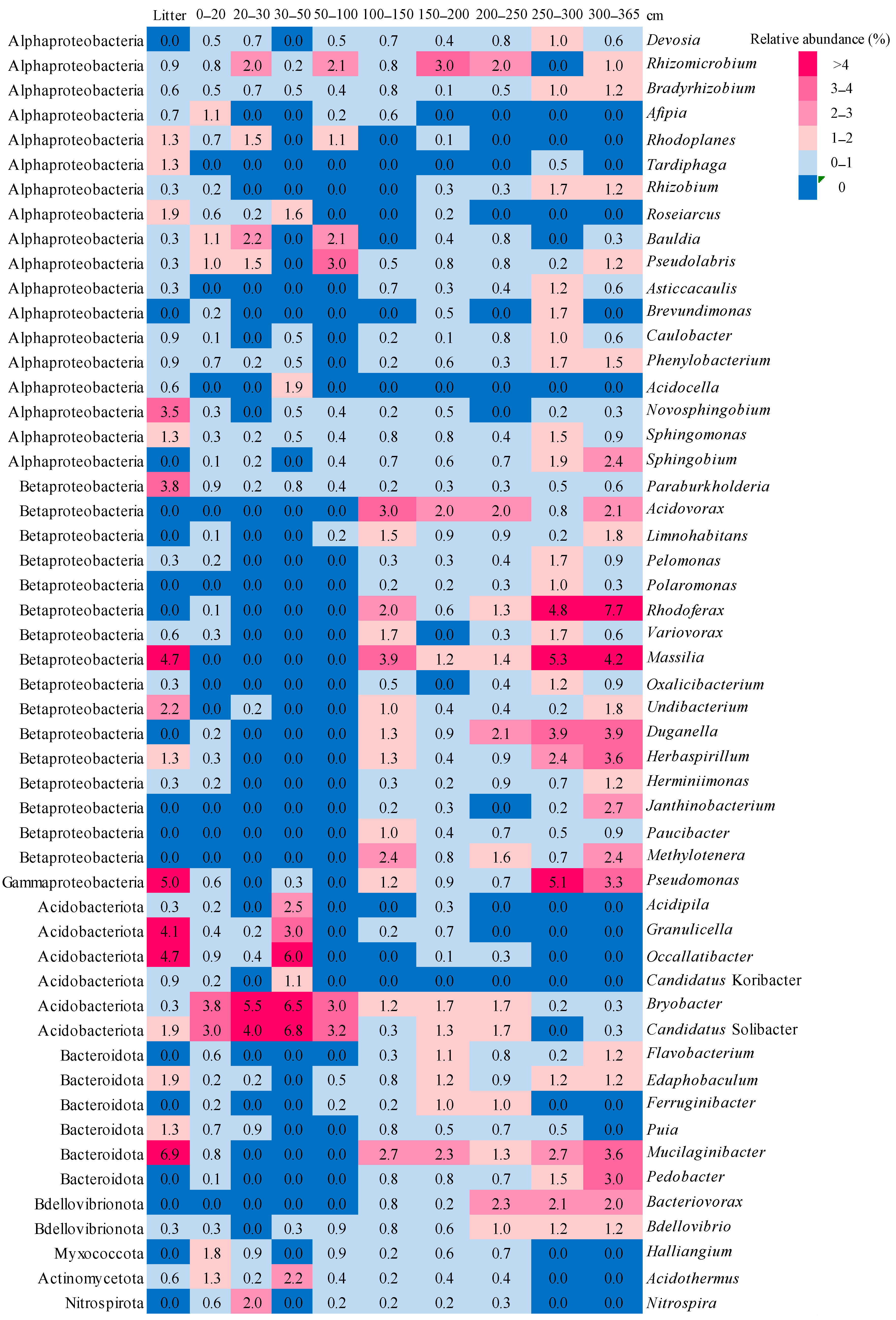
3.4. Taxonomic composition of Bacterial Communities
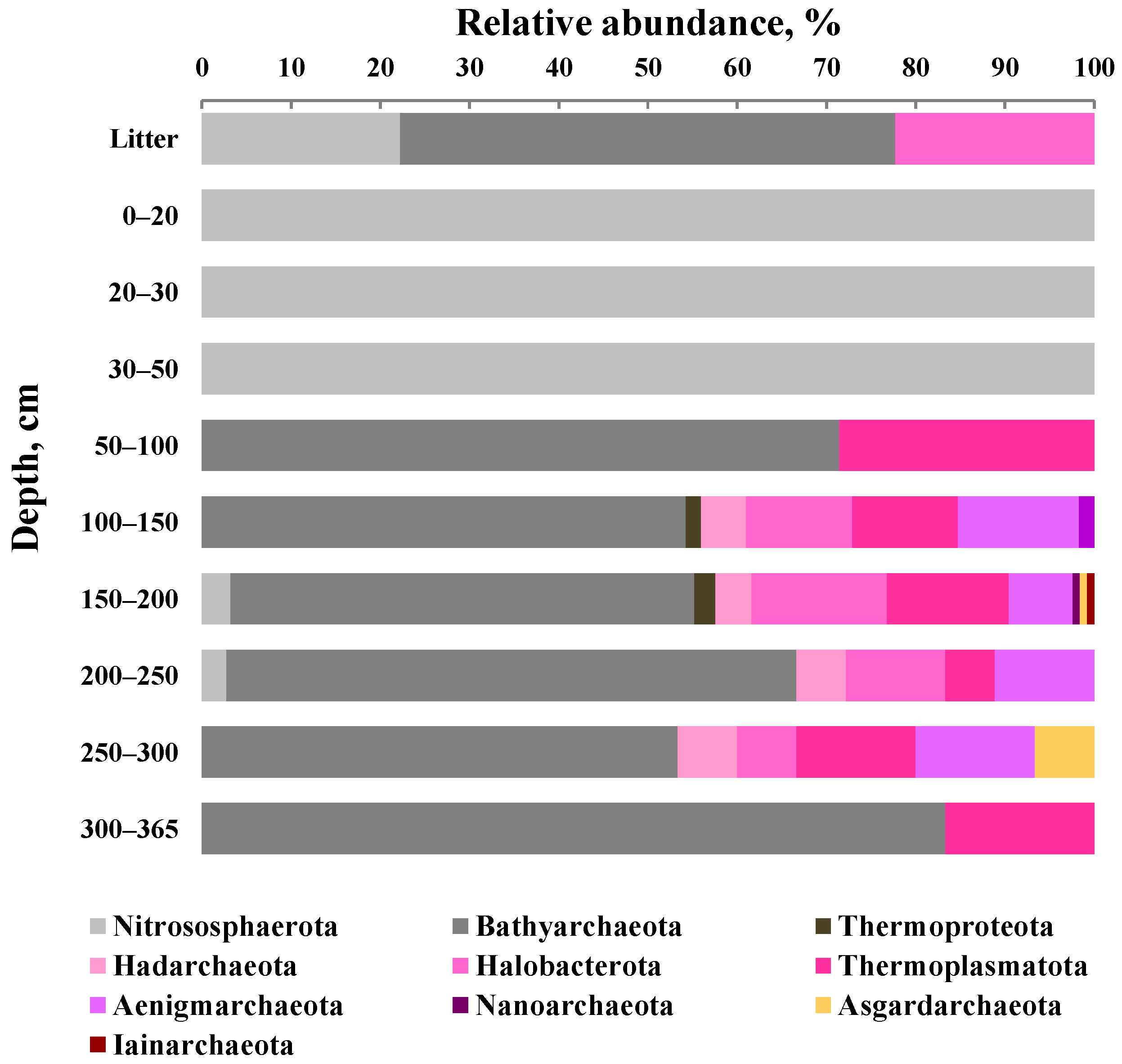
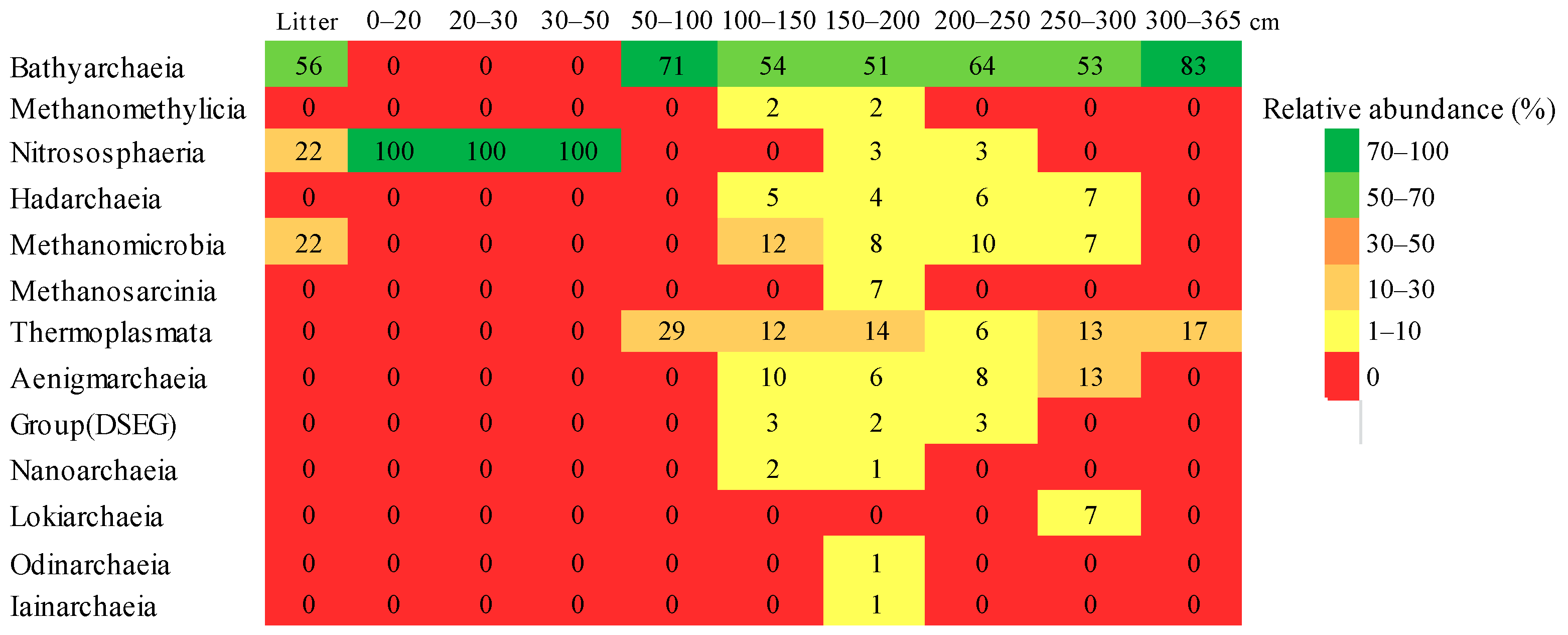
3.5. Taxonomic Composition of Archaeal Communities
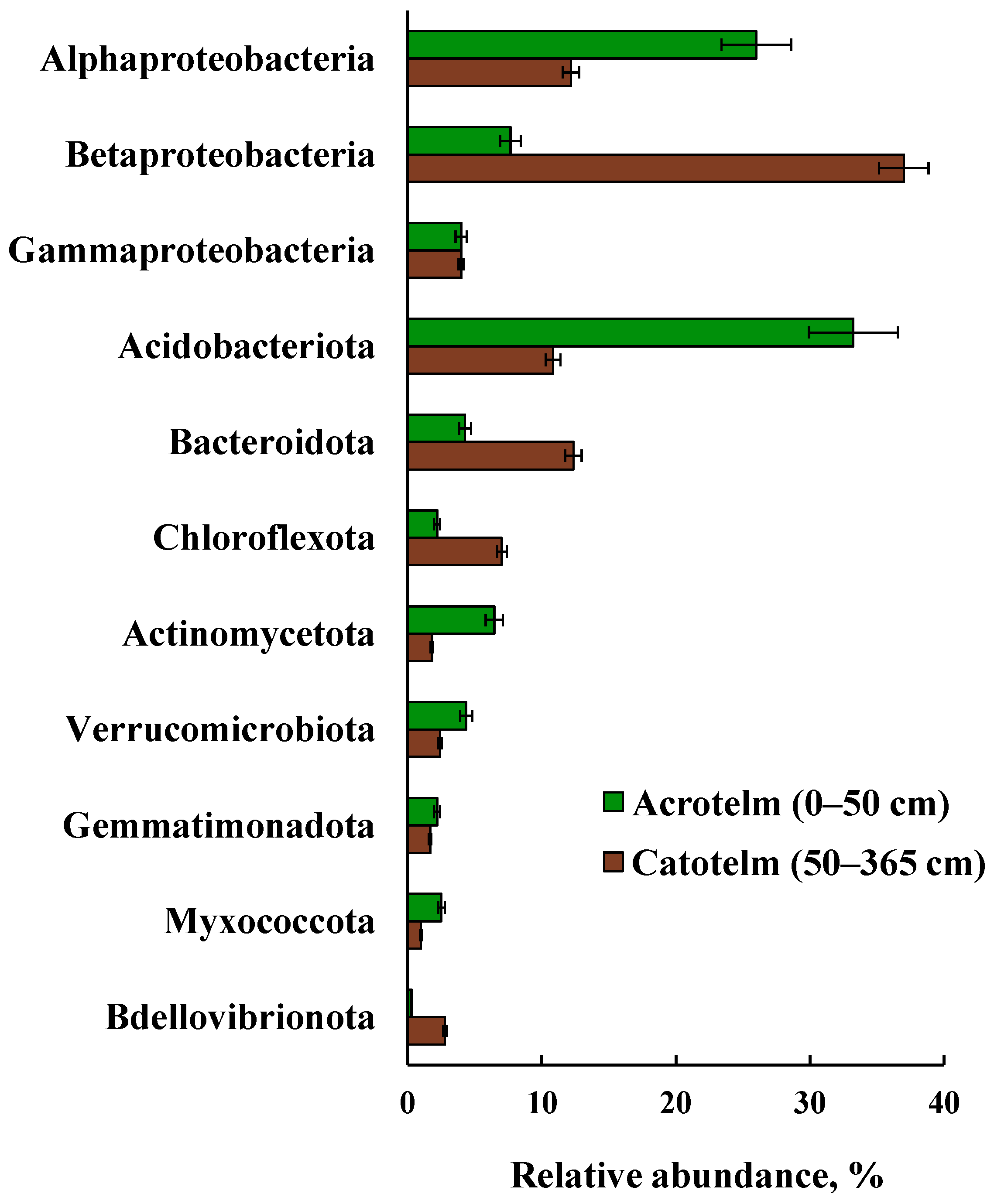
3.6. Taxonomic Composition of Prokaryotic Communities in Acrotelm and Catotelm
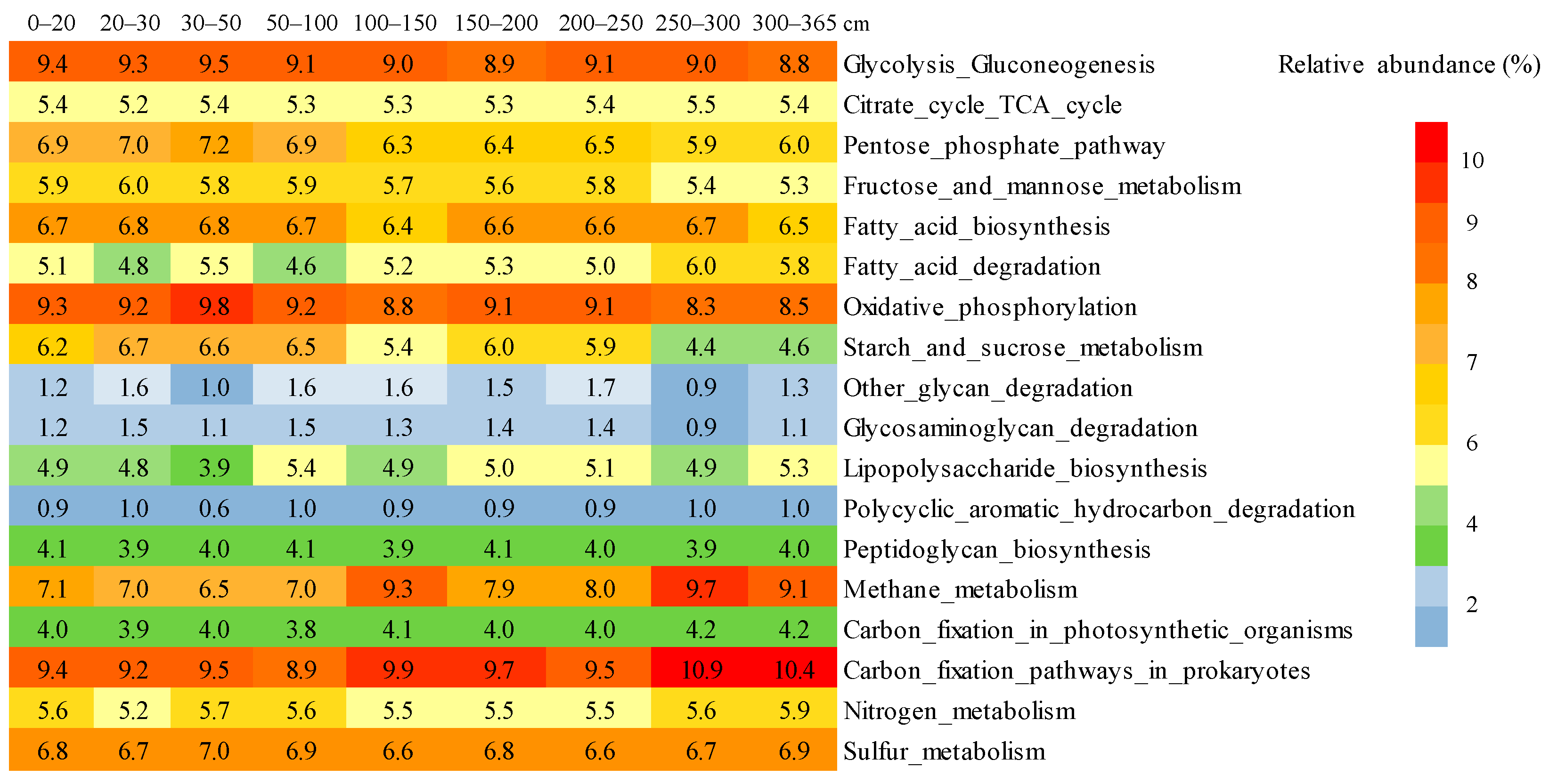
3.7. Potential Functional Characteristics of Prokaryotic Communities from fen Profile Samples
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Yu, Z.; Loisel, J.; Brosseau, D.P.; Beilman, D.W.; Hunt, S.J. Global peatland dynamics since the Last Glacial Maximum. Geophys. Res. Lett. 2010, 37, 13. [Google Scholar] [CrossRef]
- Scharlemann, J.P.; Tanner, E.V.; Hiederer, R.; Kapos, V. Global soil carbon: Understanding and managing the largest terrestrial carbon pool. Carbon Manag. 2014, 5, 81–91. [Google Scholar] [CrossRef]
- Jungblut, A.D.; Wood, S.A.; Hawes, I.; Webster-Brown, J.; Harris, C. The Pyramid Trough Wetland: Environmental and biological diversity in a newly created Antarctic protected area. FEMS Microbiol. Ecol. 2012, 82, 356–366. [Google Scholar] [CrossRef]
- Vompersky, S.E.; Sirin, A.A.; Tsyganova, O.P.; Valyaeva, N.A.; Maikov, D.A. Peatlands and paludified lands of Russia: Attempt of analyses of spatial distribution and diversity. Izv. Ross. Akad. Nauk Seriya Geogr. 2005, 5, 39–50. (In Russian) [Google Scholar]
- Vompersky, S.E.; Sirin, A.A.; Sal’nikov, A.A.; Tsyganova, O.P.; Valyaeva, N.A. Estimation of forest cover extent over peatlands and paludified shallow-peat lands in Russia. Contemp. Probl. Ecol. 2011, 4, 734–741. [Google Scholar] [CrossRef]
- Parish, F.; Sirin, A.A.; Charman, D.; Joosten, H.; Minaeva, T.Y.; Silvius, M. Assessment on Peatlands, Biodiversity and Climate Change; Wetlands International: Kuala Lumpur, Wageningen, Malaysia, 2008; p. 179. [Google Scholar]
- Dobrovol’skaya, T.G.; Golovchenko, A.V.; Pankratov, T.A.; Lysak, L.V.; Zvyagintsev, D.G. Assessment of the bacterial diversity in soils: Evolution of approaches and methods. Eurasian Soil Sci. 2009, 42, 1138–1147. [Google Scholar] [CrossRef]
- Andersen, R.; Chapman, S.J.; Artz, R.R.E. Microbial communities in natural and disturbed peatlands: A review. Soil Biol. Biochem. 2013, 57, 979–994. [Google Scholar] [CrossRef]
- Juan-Ovejeroa, R.; Brionesa, M.J.I.; Öpikb, M. Fungal diversity in peatlands and its contribution to carbon cycling. Appl. Soil Ecol. 2020, 146, 103393. [Google Scholar] [CrossRef]
- Waksman, S.A.; Stevens, K.R. Contribition to the chemical composition of peats. V. The role of microorganisms in peat formation and decomposition. Soil Sci. 1929, 28, 315–339. [Google Scholar] [CrossRef]
- Belikova, N.M. From the results of microbiological analysis of peat (on the decomposition of fiber in peat). Torfyanoye Delo 1929, 10–11, 427–430. (In Russian) [Google Scholar]
- Waksman, S.A.; Purvis, E.R. The microbiological population of peat. Soil Sci. 1932, 34, 95–109. [Google Scholar] [CrossRef]
- Gilbert, D.; Mitchell, E.A.D. Microbial diversity in Sphagnum peatlands. Dev. Earth Surf. Process. 2006, 9, 287–318. [Google Scholar] [CrossRef]
- Aslam, Z.; Yasir, M.; Khaliq, A.; Matsui, K.; Chung, Y.R. Too much bacteria still unculturable. Crop Environ. 2010, 1, 59–60. [Google Scholar]
- Rheims, H.; Spröer, C.; Rainey, F.A.; Stackebrandt, E. Molecular biological evidence for the occurrence of uncultured members of the actinomycete line of descent in different environments and geographical locations. Microbiology 1996, 142, 2863–2870. [Google Scholar] [CrossRef] [PubMed]
- Dedysh, S.; Pankratov, T.; Belova, S.; Kulichevskaya, I.; Liesack, W. Phylogenetic Analysis and In Situ Identification of Bacteria Community Composition in an Acidic Sphagnum Peat Bog. Appl. Environ. Microbiol. 2006, 72, 2110–2117. [Google Scholar] [CrossRef]
- Pankratov, T.A.; Ivanova, A.O.; Dedysh, S.N.; Liesack, W. Bacterial populations and environmental factors controlling cellulose degradation in an acidic Sphagnum peat. Environ. Microbiol. 2011, 13, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Green, S.; Tfaily, M.M.; Prakash, O.; Konstantinidis, K.T.; Corbett, J.E.; Chanton, J.P.; Cooper, W.T.; Kostka, J.E. Microbial community structure and activity linked to contrasting biogeochemical gradients in bog and fen environments of the Glacial Lake Agassiz Peatland. Appl. Environ. Microbiol. 2012, 78, 7023–7031. [Google Scholar] [CrossRef]
- Serkebaeva, Y.M.; Kim, Y.; Liesack, W.; Dedysh, S.N. Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS ONE 2013, 21, e63994. [Google Scholar] [CrossRef]
- Tsitko, I.; Lusa, M.; Lehto, J.; Parviainen, L.; Ikonen, A.T.K.; Lahdenperä, A.M.; Bomberg, M. The Variation of Microbial Communities in a Depth Profile of an Acidic, Nutrient-Poor Boreal Bog in Southwestern Finland. Open J. Ecol. 2014, 4, 832–859. [Google Scholar] [CrossRef]
- Kim, H.M.; Jung, J.Y.; Yergeau, E.; Hwang, C.Y.; Hinzman, L.; Nam, S.; Hong, S.G.; Kim, O.S.; Chun, J.; Lee, Y.K. Bacterial community structure and soil properties of a subarctic tundra soil in Council, Alaska. FEMS Microbiol. Ecol. 2014, 89, 465–475. [Google Scholar] [CrossRef]
- Danilova, O.V.; Belova, S.E.; Gagarinova, I.V.; Dedysh, S.N. Microbial Community Composition and Methanotroph Diversity of a Subarctic Wetland in Russia. Microbiology 2016, 85, 583–591. [Google Scholar] [CrossRef]
- Birnbaum, C.; Wood, J.; Lilleskov, E.; Lamit, L.; Shannon, J.; Brewer, M.; Grover, S. Degradation Reduces Microbial Richness and Alters Microbial Functions in an Australian Peatland. Microb. Ecol. 2022, 85, 875–891. [Google Scholar] [CrossRef]
- Dobrovolskaya, T.G.; Golovchenko, A.V.; Zvyagintsev, D.G. Analysis of ecological factors limiting the destruction of high-moor peat. Eurasian Soil Sci. 2014, 47, 182–193. [Google Scholar] [CrossRef]
- Golovchenko, A.V.; Tikhonova, E.Y.; Zvyagintsev, D.G. Abundance, biomass, structure, and activity of the microbial complexes of minerotrophic and ombrotrophic peatlands. Microbiology 2007, 76, 630–637. [Google Scholar] [CrossRef]
- Dobrovolskaya, T.G.; Golovchenko, A.V.; Kukharenko, O.S. The structure of the microbial communities in low-moor and high-moor peat bogs of Tomsk oblast. Eurasian Soil Sci. 2012, 45, 273–281. [Google Scholar] [CrossRef]
- Golovchenko, A.V.; Dmitrienko, Y.D.; Morozov, A.A.; Pozdnyakov, L.A.; Glukhova, T.V.; Inisheva, L.I. Microbial Biomass in Eutrophic Peatlands: Stock, Structure and Activity. Eurasian Soil Sci. 2021, 54, 1068–1077. [Google Scholar] [CrossRef]
- Bogdanova, O.Y.; Golovchenko, A.V.; Lysak, L.V.; Glukhova, T.V.; Zvyagintsev, D.G. Viability of bacteria in peatlands. Eurasian Soil Sci. 2014, 47, 297–303. [Google Scholar] [CrossRef]
- Concheri, G.; Stevanato, P.; Zaccone, C.; Shotyk, W.; D’Orazio, V.; Miano, T.; Piffanelli, P.; Rizzi, V.; Ferrandi, C.; Squartini, A. Rapid peat accumulation favours the occurrence of both fen and bog microbial communities within a Mediterranean, free-floating peat island. Sci. Rep. 2017, 7, 8511. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Beletsky, A.V.; Rakitin, A.L.; Kadnikov, V.V.; Philippov, D.A.; Mardanov, A.V.; Ravin, N.V.; Dedysh, S.N. Closely Located but Totally Distinct: Highly Contrasting Prokaryotic Diversity Patterns in Raised Bogs and Eutrophic Fens. Microorganisms 2020, 8, 484. [Google Scholar] [CrossRef]
- Seward, J.; Carson, M.A.; Lamit, L.J. Peatland Microbial Community Composition Is Driven by a Natural Climate Gradient. Microb. Ecol. 2020, 80, 593–602. [Google Scholar] [CrossRef]
- Lin, X.; Tfaily, M.M.; Green, S.J.; Steinweg, J.M.; Chanton, P.; Imvittaya, A.; Chanton, J.P.; Cooper, W.; Schadt, C.; Kostka, J.E. Microbial metabolic potential for carbon degradation and nutrient (nitrogen and phosphorus) acquisition in an ombrotrophic peatland. Appl. Environ. Microbiol. 2014, 80, 3531–3540. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Z.; Tian, L. Microbial communities in peatlands along a chronosequence on the Sanjiang Plain, China. Sci. Rep. 2017, 7, 9567. [Google Scholar] [CrossRef]
- Bakhnov, V.K. Soil Formation (A Look into the Past and Present); SO RAN: Novosibirsk, Russia, 2002; p. 117. (In Russian) [Google Scholar]
- Hytteborn, H.; Maslov, A.A.; Nazimova, D.I.; Rysin, L.P. Boreal forests of Eurasia. In Ecosystems of the World; Elsevier: Amsterdam, The Netherlands, 2005; pp. 23–99. [Google Scholar]
- Vompersky, S.E.; Solovyov, S.A.; Vakurov, A.D. Biogeocenological Study of Swamp Forests in Connection with Experimental Hydromelioration; Nauka: Moscow, Russia, 1982; p. 208. (In Russian) [Google Scholar]
- Vompersky, S.E.; Sirin, A.A.; Glukhov, A.I. Formation and Regime of Runoff during Hydroforestry; Nauka: Moscow, Russia, 1988; p. 168. (In Russian) [Google Scholar]
- Semensky, E.P. Technical Analysis of Peat; Nedra: Moscow, Russia, 1966; p. 232. (In Russian) [Google Scholar]
- Arinushkina, E.V. Manual on Chemical Analysis of Soils; MSU Publishing House: Moscow, Russia, 1970; p. 487. (In Russian) [Google Scholar]
- Takahashi, S.; Tomita, J.; Nishioka, K.; Hisada, T.; Nishijima, M. Development of a prokaryotic universal primer for simultaneousanalysis of Bacteria and Archaea using next-generation sequencing. PLoS ONE 2014, 9, e105592. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N. QIIME allows analysis of high-throughput community sequencing sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 21, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Kindt, R.; Legendre, P.; O’Hara, B.; Stevens, M.H.H.; Oksanen, M.J.; Suggests, M.A.S.S. The vegan package. Community Ecol. Package 2007, 10, 631–637. [Google Scholar]
- Nagpal, S.; Haque, M.M.; Singh, R.; Mande, S.S. iVikodak—A Platform and standard workflow for inferring, analyzing, comparing, and visualizing the functional potential of microbial communities. Front. Microbiol. 2019, 9, 3336. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Hunger, S.; Gößner, A.S.; Drake, H.L. Anaerobic trophic interactions of contrasting methane-emitting mire soils: Processes versus taxa. FEMS Microbiol. Ecol. 2015, 91, fiv045. [Google Scholar] [CrossRef]
- Bergkessel, M.; Delavaine, L. Diversity in starvation survival strategies and outcomes among heterotrophic Proteobacteria. Microb. Physiol. 2021, 31, 146–162. [Google Scholar] [CrossRef]
- Ueki, A.; Kodama, Y.; Kaku, N.; Shiromura, T.; Satoh, A.; Watanabe, K.; Ueki, K. Rhizomicrobium palustre Gen. Nov., Sp. Nov., a Facultatively Anaerobic, Fermentative Stalked Bacterium in the Class Alphaproteobacteria Isolated from Rice Plant Roots. J. Gen. Appl. Microbiol. 2010, 56, 193–203. [Google Scholar] [CrossRef]
- Yee, B.; Oertli, G.E.; Fuerst, J.A.; Staley, J.T. Reclassification of the polyphyletic genus Prosthecomicrobium to form two novel genera, Vasilyevaea gen. nov. and Bauldia gen. nov. with four new combinations: Vasilyevaea enhydra comb. nov., Vasilyevaea mishustinii comb. nov., Bauldia consociata comb. nov. and Bauldia litoralis comb. nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 2960–2966. [Google Scholar] [CrossRef]
- Kämpfer, P.; Young, C.; Arun, A.B.; Shen, F.T.; Jäckel, U.; Rosselló-Mora, R.; Lai, W.A.; Rekha, P.D. Pseudolabrys taiwanensis gen. nov., sp. nov., an alphaproteobacterium isolated from soil. Int. J. Syst. Evol. Microb. 2006, 56, 2469–2472. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, M.; Hamana, K.; Hiraishi, A. Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 2001, 51, 1405–1417. [Google Scholar] [CrossRef] [PubMed]
- Kämpfer, P.; Young, C.C.; Busse, H.J. Novosphingobium soli sp. nov., isolated from soil. Int. J. Syst. Evol. Microbiol. 2011, 61, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Wolińska, A.; Kruczyńska, A.; Grządziel, J.; Gałązka, A.; Marzec-Grządziel, A.; Szałaj, K.; Kuźniar, A. Functional and Seasonal Changes in the Structure of Microbiome Inhabiting Bottom Sediments of a Pond Intended for Ecological King Carp Farming. Biology 2022, 11, 913. [Google Scholar] [CrossRef]
- Satola, B.; Wübbeler, J.H.; Steinbüchel, A. Metabolic Characteristics of the Species Variovorax paradoxus. Appl. Microbiol. Biotechnol. 2013, 97, 541–560. [Google Scholar] [CrossRef]
- Kampfer, P.; Lodders, N.; Martin, K.; Falsen, E. Revision of the genus Massilia La Scola et al. 2000, with an emended description of the genus and inclusion of all species of the genus Naxibacter as new combinations, and proposal of Massilia consociata sp. nov. Int. J. Syst. Evol. Microbiol. 2011, 61, 1528–1533. [Google Scholar] [CrossRef]
- Lincoln, S.P.; Fermor, T.R.; Tindall, B.J. Janthinobacterium agaricidamnosum sp. nov., a soft rot pathogen of Agaricus bisporus. Int. J. Syst. Evol. Microbiol. 2009, 49, 1577–1589. [Google Scholar] [CrossRef]
- Zhang, J.; Kim, Y.J.; Hoang, V.A.; Nguyen, N.L. Duganella ginsengisoli sp. nov., isolated from ginseng soil. Int. J. Syst. Evol. Microbiol. 2016, 66, 56–61. [Google Scholar] [CrossRef]
- Kämpfer, P.; Rosselló-Mora, R.; Hermansson, M.; Persson, F.; Huber, B.; Falsen, E.; Busse, H.J. Undibacterium pigrum gen. nov., sp. nov., isolated from drinking water. Int. J. Syst. Evol. Microbiol. 2007, 57, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Willems, A.; Falsen, E.; Pot, B.; Jantzen, E.; Hoste, B.; Vandamme, P.; Gillis, M.; Kersters, K.; De Ley, J. Acidovorax, a New Genus for Pseudomonas facilis, Pseudomonas delafieldii, E. Falsen (EF) Group 13, EF Group 16, and Several Clinical Isolates, with the Species Acidovorax facilis comb. nov., Acidovorax delaJieldii comb. nov., and Acidovorax temperans sp. nov. Int. J. Syst. Bacteriol. 1990, 40, 384–398. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Rainey, F.A.; Nobre, M.F.; Pinhal, I.; Folhas, F.; Da Costa, M.S. Herminiimonas fonticola gen. nov., sp. nov., a Betaproteobacterium isolated from a source of bottled mineral water. Syst. Appl. Microbiol. 2005, 28, 596–603. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W.; Kasalický, V.; Jezbera, J.; Brandt, U.; Jezberová, J.; Šimek, K. Limnohabitans curvus gen. nov., sp. nov., a planktonic bacterium isolated from a freshwater lake. Int. J. Syst. Bacteriol. 2010, 60, 1358–1365. [Google Scholar] [CrossRef]
- Tamer, A.U.; Aragno, M.; Sahin, N. Isolation and characterization of a new type of aerobic, oxalic acid utilizing bacteria, and proposal of Oxalicibacterium flavum gen. nov., sp. nov. Syst. Appl. Microbiol. 2002, 25, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Irgens, R.L.; Gosink, J.J.; Staley, J.T. Polaromonas vacuolata gen. nov., sp. nov., a psychrophilic, marine, gas vacuolate bacterium from Antarctica. Int. J. Syst. Bacteriol. 1996, 46, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.H.; Yokota, A. Reclassification of Alcaligenes latus strains IAM 12599T and IAM 12664 and Pseudomonas saccharophila as Azohydromonas lata gen. nov., comb. Nov., Azohydromonas australica sp. nov. and Pelomonas saccharophila gen. nov., comb. nov., respectively. Int. J. Syst. Evol. Microbiol. 2005, 55, 2419–2425. [Google Scholar] [CrossRef]
- Baldani, J.I.; Baldani, V.D.L.; Dobereiner, J. Characterization of Herbaspirillum seropedicae gen. nov., sp. nov., a Root-Associated Nitrogen-Fixing Bacterium. Int. J. Syst. Evol. Microbiol. 1986, 36, 86–93. [Google Scholar] [CrossRef]
- Kalyuzhnaya, M.G.; Bowerman, S.; Lara, J.S.; Lidstrom, M.E.; Chistoserdova, L. Methylotenera mobilis gen. nov., sp. nov., an obligately methylamine-utilizing bacterium within the family Methylophilaceae. Int. J. Syst. Evol. Microbiol. 2006, 56, 2819–2823. [Google Scholar] [CrossRef]
- Hiraishi, A.; Hoshino, Y.; Satoh, T. Rhodoferax fermentans gen. nov., sp. nov., a phototrophic purple nonsulfur bacterium previously referred to as the Rhodocyclus gelatinosus-like group. Arch. Microbiol. 1991, 155, 330–336. [Google Scholar] [CrossRef]
- Pheng, S.; Lee, J.J.; Eom, M.K. Paucibacter oligotrophus sp. nov., isolated from fresh water, and emended description of the genus Paucibacter. Int. J. Syst. Evol. Microbiol. 2017, 67, 2231–2235. [Google Scholar] [CrossRef]
- Kalam, S.; Basu, A.; Ahmad, I.; Sayyed, R.Z.; El-Enshasy, H.A.; Dailin, D.J.; Suriani, N.L. Recent understanding of soil Acidobacteria and their ecological significance: A critical review. Front. Microbiol. 2020, 30, 580024. [Google Scholar] [CrossRef]
- Dedysh, S.N.; Ivanova, A.A.; Begmatov, S.H.; Beletsky, A.V.; Rakitin, A.L.; Mardanov, A.V.; Philippov, D.A.; Ravin, N.V. Acidobacteria in fens: Phylogenetic diversity and genome analysis of the key representatives. Microbiology 2022, 91, 685–694. [Google Scholar] [CrossRef]
- Chaves, M.G.; Silva, G.G.Z.; Rossetto, R.; Edwards, R.A.; Tsai, S.M.; Navarette, A.A. Acidobacteria subgroups and their metabolic potential for carbon degradation in sugarcane soil amended with vinasse and nitrogen fertilizers. Front. Microbiol. 2019, 10, 1680. [Google Scholar] [CrossRef] [PubMed]
- Männistö, M.K.; Kurhela, E.; Tiirola, M.; Häggblom, M.M. Acidobacteria dominate the active bacterial communities of Arctic tundra with widely divergent winter-time snow accumulation and soil temperatures. FEMS Microbiol. Ecol. 2013, 84, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Kulichevskaya, I.S.; Suzina, N.E.; Liesack, W.; Dedysh, S.N. Bryobacter aggregatus gen. nov., sp. nov., a peat-inhabiting, aerobic chemo-organotroph from subdivision 3 of the Acidobacteria. Int. J. Syst. Evol. Microbiol. 2010, 60, 301–306. [Google Scholar] [CrossRef]
- Ward, N.L.; Challacombe, J.F.; Janssen, P.H.; Henrissat, B. Three genomes from the phylum Acidobacteria provide insight into their lifestyles in soils. Appl. Environ. Microbiol. 2009, 75, 2046–2056. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Kawai, A.; Yamada, T.; Hiraishi, A. Acidipila rosea gen. nov., sp. nov., an acidophilic chemoorganotrophic bacterium belonging to the phylum Acidobacteria. FEMS Microbiol. Lett. 2011, 317, 138–142. [Google Scholar] [CrossRef]
- Foesel, B.U.; Mayer, S.; Luckner, M.; Wanner, G.; Rohde, M.; Overmann, J. Occallatibacter riparius gen. nov., sp. nov. and Occallatibacter avannae sp. nov., acidobacteria isolated from Namibian soils and emended description of the family Acidobacteriaceae. Int. J. Syst. Evol. Microbiol. 2016, 66, 219–229. [Google Scholar] [CrossRef]
- Dobrovolskaya, T.G.; Golovchenko, A.V.; Lysak, L.V.; Yurchenko, E.N. Taxonomic Structure of Bacterial Communities of Rhizospheric Soil under Bogs’ Plants. Mosc. Univ. Soil Sci. Bull. 2020, 75, 93–100. [Google Scholar] [CrossRef]
- Pankratov, T.; Dedysh, S.N. Granulicella paludicola gen. nov., sp. nov., G. pectinivorans sp. nov., G. aggregans sp. nov. and G. rosea sp. nov., novel acidophilic, polymer-degrading Acidobacteria from Sphagnum peat bogs. Int. J. Syst. Evol. Microbiol. 2010, 60, 2951–2959. [Google Scholar] [CrossRef] [PubMed]
- François, T.; Hehemann, J.-H.; Rebuffet, E.; Czjzek, M.; Michel, G. Environmental and Gut Bacteroidetes: The Food Connection. Front. Microbiol. 2011, 2, 93. [Google Scholar]
- Kuramae, E.E. Soil characteristics more strongly influence soil bacterial communities than land-use type. FEMS Microbiol Ecol. 2012, 79, 12–24. [Google Scholar] [CrossRef]
- Peralta, R.M.; Ahn, C.; Gillevet, P.M. Characterization of soil bacterial community structure and physicochemical properties in created and natural wetlands. Sci. Total Environ. 2012, 443, 725–732. [Google Scholar] [CrossRef]
- Gavande, P.V. Functional characterization of thermotolerant microbial consortium for lignocellulolytic enzymes with central role of Firmicutes in rice straw depolymerization. Sci. Rep. 2021, 11, 3032. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, X.Y.; Zhu, L. Structure and function of the microbial consortia of activated sludge in typical municipal wastewater treatment plants in winter. Sci. Rep. 2017, 7, 17930. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Castelle, C.J.; Wrighton, K.C.; Thomas, B.C.; Sharon, I.; Frischkorn, K.R.; Banfield, J.F. Community Genomic Analyses Constrain the Distribution of Metabolic Traits across the Chloroflexi Phylum and Indicate Roles in Sediment Carbon Cycling. Microbiome 2013, 1, 1–17. [Google Scholar] [CrossRef]
- Nguyen, L.N. Genome sequencing as a new window into the microbial community of membrane bioreactors-A critical review. Sci. Total Environ. 2020, 704, 135279. [Google Scholar] [CrossRef]
- Pankratov, T.A.; Tindall, B.J.; Liesack, W.; Dedysh, S.N. Mucilaginibacter paludis gen. nov., sp. nov. and Mucilaginibacter gracilis sp. nov., pectin-, xylan- and laminarin-degrading members of the family Sphingobacteriaceae from acidic Sphagnum peat bog. Int. J. Syst. Evol. Microbiol. 2007, 57, 2349–2354. [Google Scholar] [CrossRef]
- Dahal, R.H.; Kim, J. Pedobacter humicola sp. nov., a member of the genus Pedobacter isolated from soil. Int. J. Syst. Evol. Microbiol. 2016, 66, 2205–2211. [Google Scholar] [CrossRef]
- Stams, A.J.; Plugge, C.M. Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 2009, 7, 568–577. [Google Scholar] [CrossRef]
- Walker, C.B.; de la Torre, J.R.; Klotz, M.G.; Urakawa, H.; Pinel, N.; Arp, D.J.; Stahl, D. Nitrosopumilus maritimus genome reveals unique mechanisms for nitrification and autotrophy in globally distributed marine crenarchaea. Proc. Natl. Acad. Sci. USA 2010, 107, 8818–8823. [Google Scholar] [CrossRef]
- Hawkins, A.N.; Johnson, K.W.; Bräuer, S.L. Southern Appalachian peatlands support high archaeal diversity. Microb. Ecol. 2014, 67, 587–602. [Google Scholar] [CrossRef] [PubMed]
- Xiang, X.; Wang, R.; Wang, H.; Gong, L.; Man, B.; Xu, Y. Distribution of Bathyarchaeota communities across different terrestrial settings and their potential ecological functions. Sci. Rep. 2017, 7, 45028. [Google Scholar] [CrossRef]
- Hou, J.; Wang, Y.; Zhu, P.; Yang, N.; Liang, L.; Yu, T.; Niu, M.; Konhauser, K.; Woodcroft, B.; Wang, F. Taxonomic and carbon metabolic diversification of Bathyarchaeia during its coevolution history with early Earth surface environment. Sci. Adv. 2023, 9, eadf5069. [Google Scholar] [CrossRef] [PubMed]
- Bräuer, S.L.; Basiliko, N.; Siljanen, H.; Zinder, S. Methanogenic archaea in peatlands. FEMS Microbiol. Lett. 2020, 367, fnaa172. [Google Scholar] [CrossRef]
- Clymo, R.S.; Bryant, C.L. Diffusion and mass flow of dissolved carbon dioxide, methane, and dissolved organic carbon in a 7-m deep raised peat bog. Geochim. Cosmochim. Acta 2008, 72, 2048–2066. [Google Scholar] [CrossRef]
- Tfaily, M.M.; Cooper, W.T.; Kostka, J.; Chanton, P.R.; Schadt, C.W.; Hanson, P.J.; Iversen, C.M.; Chanton, J.P. Organic matter transformation in the peat column at Marcell Experimental Forest: Humification and vertical stratification. J. Geophys. Res. Biogeosci. 2014, 119, 661–675. [Google Scholar] [CrossRef]
- Morris, P.J.; Waddington, J.M.; Benscoter, B.W.; Turetsky, M.R. Conceptual frameworks in peatland ecohydrology: Looking beyond the two-layered (acrotelm–catotelm) model. Ecohydrology 2011, 4, 1–11. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Depth of Sampling (cm) | Botanical Composition of Peat | Degree of Peat Decomposition (%) | pH | Total Carbon (%) | Total Nitrogen (%) |
|---|---|---|---|---|---|
| 0–20 | Woody–sedge | 35 | 5.8 | 48 | 3 |
| 20–30 | Woody–sedge | 37 | 5.8 | 49 | 2 |
| 30–50 | Woody | 45 | 5.6 | 50 | 3 |
| 50–100 | Woody | 45 | 5.6 | 49 | 2 |
| 100–150 | Woody | 45 | 5.7 | 50 | 3 |
| 150–200 | Woody | 47 | 5.7 | 49 | 3 |
| 200–250 | Woody | 52 | 5.6 | 49 | 3 |
| 250–300 | Woody–sedge | 42 | 5.7 | 49 | 3 |
| 300–365 | Sedge–sphagnum | 30 | 6.0 | 51 | 2 |
| Library | Number of OTUs | Chao1 Index | Shannon Index |
|---|---|---|---|
| L1 (Litter) | 210 | 493 | 5.2 |
| L2 (0–20 cm) | 934 | 3231 | 6.7 |
| L3 (20–30 cm) | 372 | 2050 | 5.8 |
| L4 (30–50 cm) | 243 | 549 | 5.3 |
| L5 (50–100 cm) | 459 | 1810 | 6.0 |
| L6 (100–150 cm) | 444 | 1394 | 6.0 |
| L7 (150–200 cm) | 1461 | 4063 | 7.1 |
| L8 (200–250 cm) | 582 | 1647 | 6.3 |
| L9 (250–300 cm) | 286 | 877 | 5.5 |
| L10 (300–365 cm) | 251 | 1087 | 5.4 |
| Depth (cm) | Litter | 0–10 | 10–30 | 30–50 | 50–100 | 100–150 | 150–200 | 200–250 | 250–300 | 300–365 |
|---|---|---|---|---|---|---|---|---|---|---|
| Bacteria | 97.2 | 99.8 | 99.6 | 98.1 | 98.9 | 90.1 | 93.9 | 95.3 | 96.4 | 98.2 |
| Archaea | 2.8 | 0.2 | 0.4 | 1.9 | 1.1 | 9.9 | 6.1 | 4.7 | 3.6 | 1.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Golovchenko, A.V.; Dobrovolskaya, T.G.; Sokolova, D.S.; Gracheva, T.A.; Dorchenkova, Y.A.; Glukhova, T.V.; Manucharova, N.A. Diversity and Functional Potential of Prokaryotic Communities in Depth Profile of Boreo-Nemoral Minerotrophic Pine Swamp (European Russia). Forests 2023, 14, 2313. https://doi.org/10.3390/f14122313
Golovchenko AV, Dobrovolskaya TG, Sokolova DS, Gracheva TA, Dorchenkova YA, Glukhova TV, Manucharova NA. Diversity and Functional Potential of Prokaryotic Communities in Depth Profile of Boreo-Nemoral Minerotrophic Pine Swamp (European Russia). Forests. 2023; 14(12):2313. https://doi.org/10.3390/f14122313
Chicago/Turabian StyleGolovchenko, Alla V., Tatiana G. Dobrovolskaya, Diyana Sh. Sokolova, Tatiana A. Gracheva, Yuliya A. Dorchenkova, Tamara V. Glukhova, and Natalia A. Manucharova. 2023. "Diversity and Functional Potential of Prokaryotic Communities in Depth Profile of Boreo-Nemoral Minerotrophic Pine Swamp (European Russia)" Forests 14, no. 12: 2313. https://doi.org/10.3390/f14122313