
Genetic Diversity and Structure of a Diverse Population of Picea sitchensis Using Genotyping-by-Sequencing
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
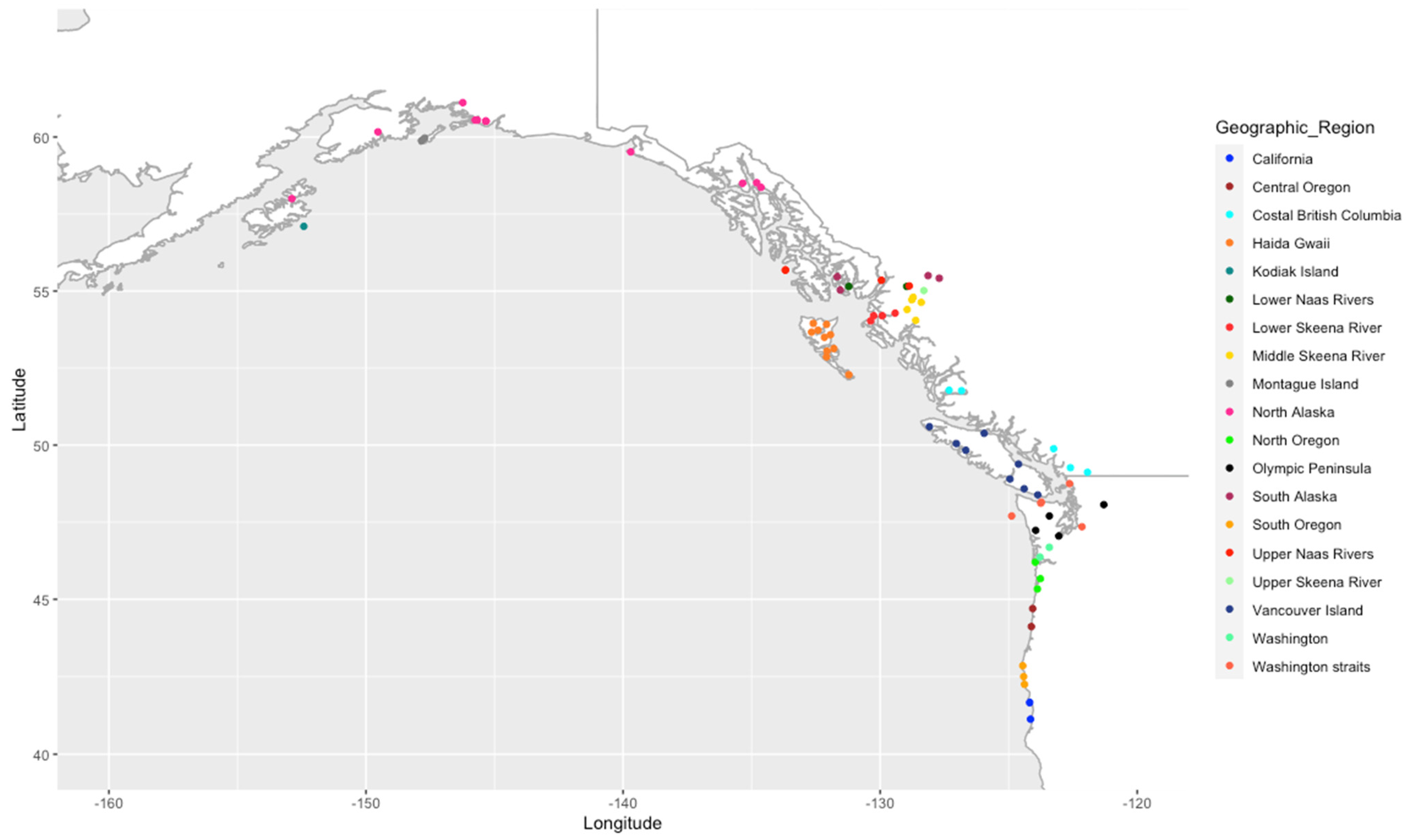
2.1. Sample Populations
2.2. DNA Extraction and Sequencing
2.3. Variant Calling
2.4. Genetic Diversity Statistics
2.5. Population Structure
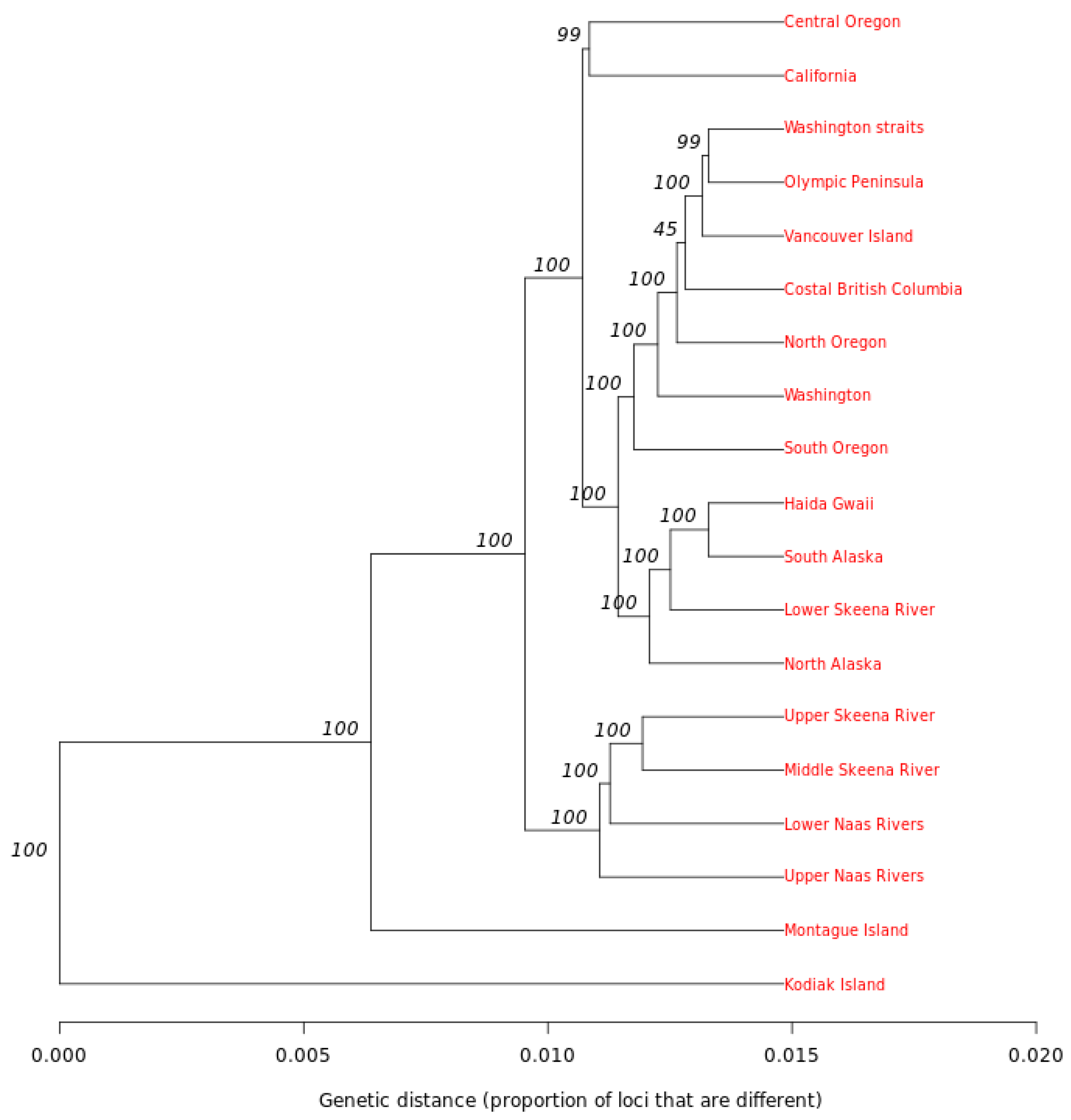
2.6. Phylogenetic Analysis
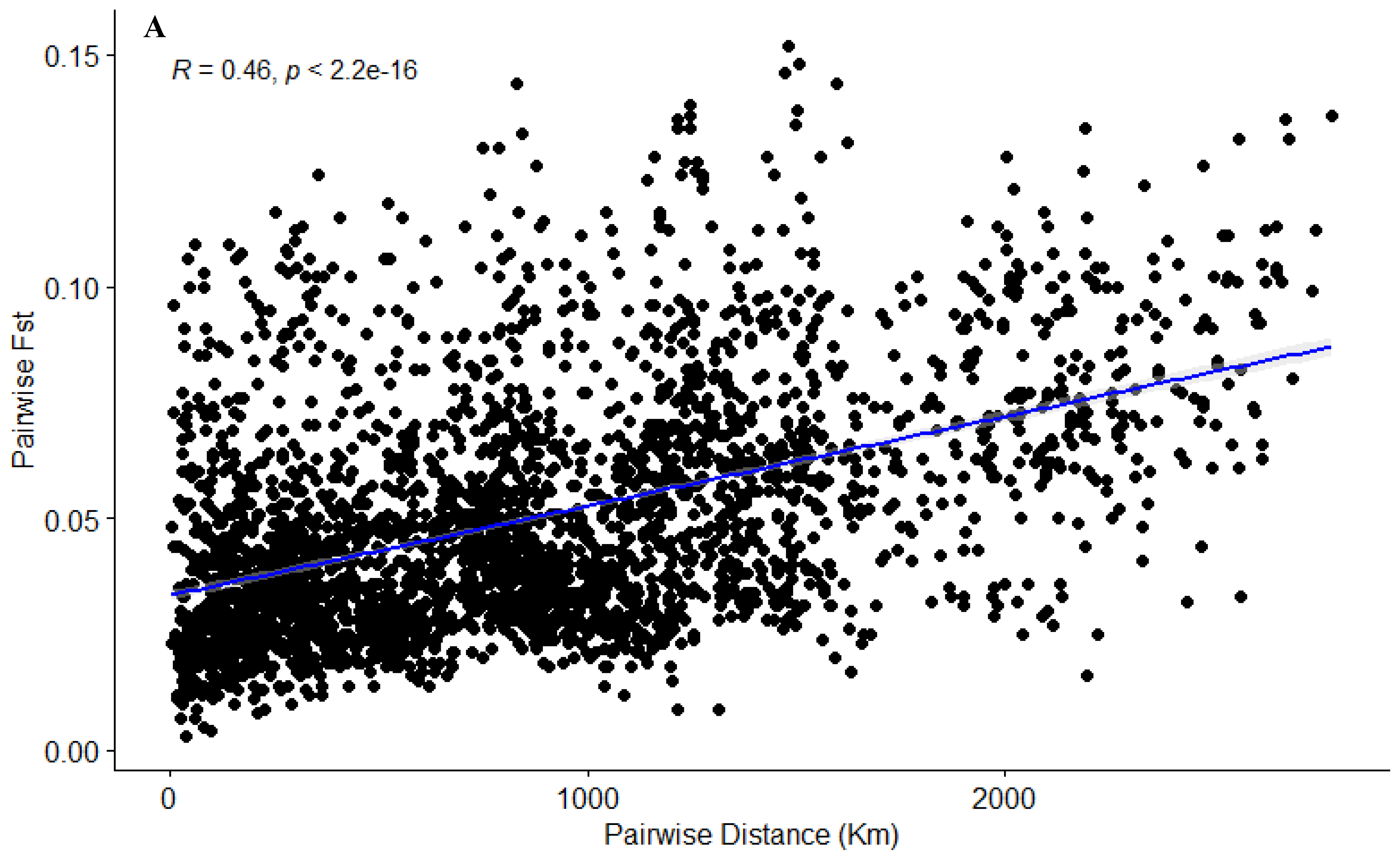
2.7. Isolation by Distance
2.8. Admixture Analysis
3. Results and Discussion
3.1. Genotyping a Large Sitka Spruce Population, Covering Its Native Geographic Range
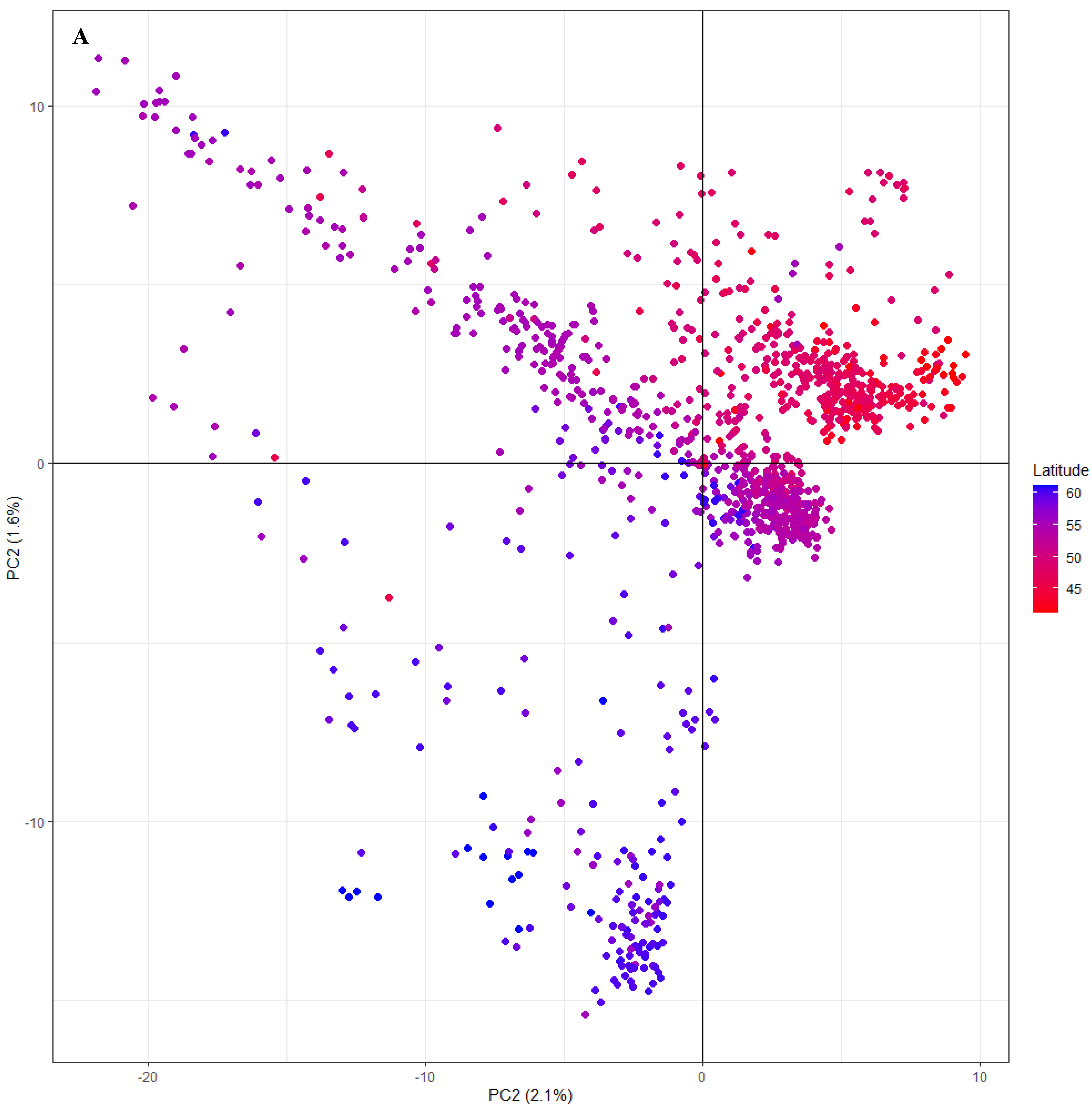
3.2. Geographic and Genetic Diversity
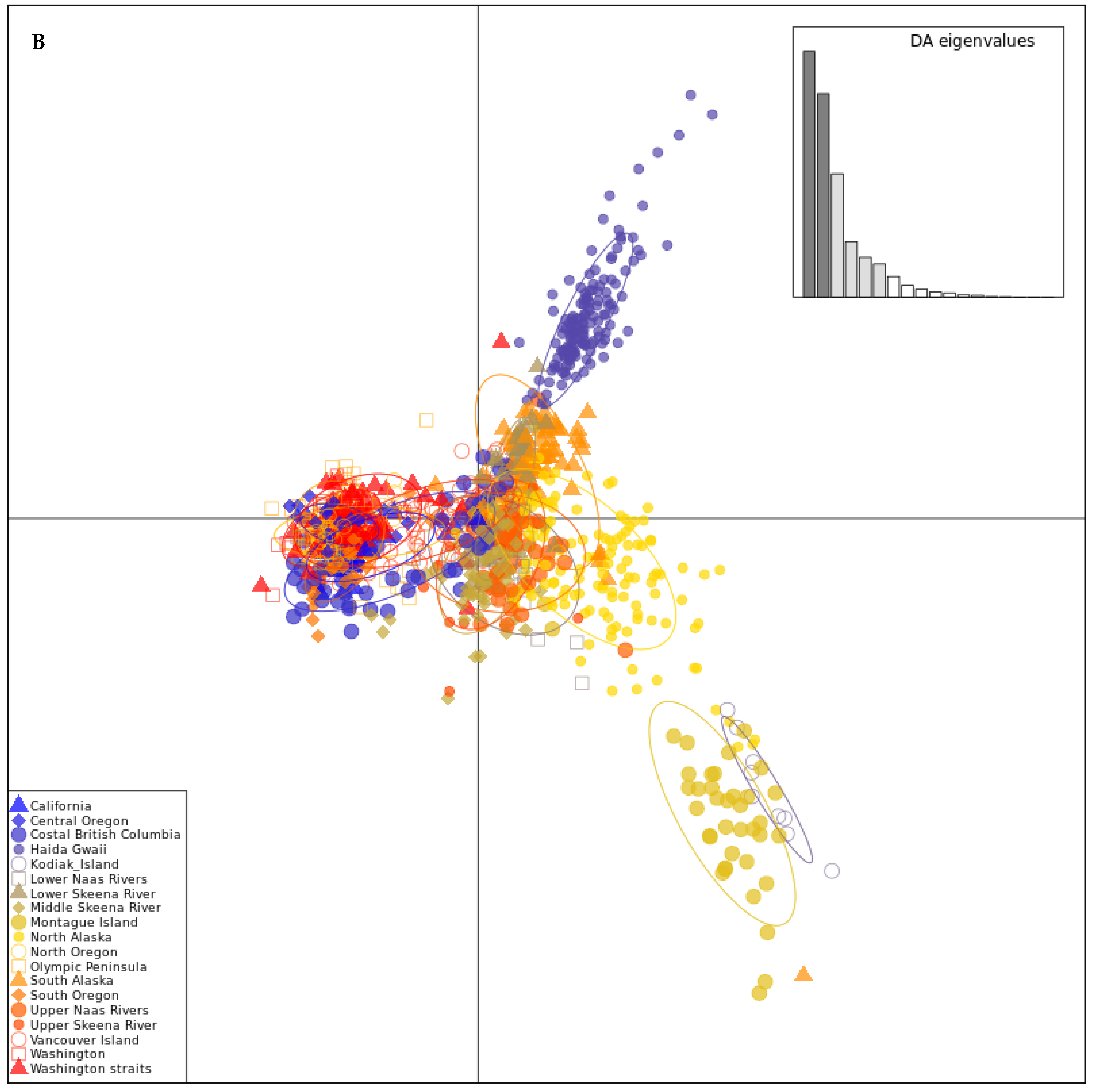
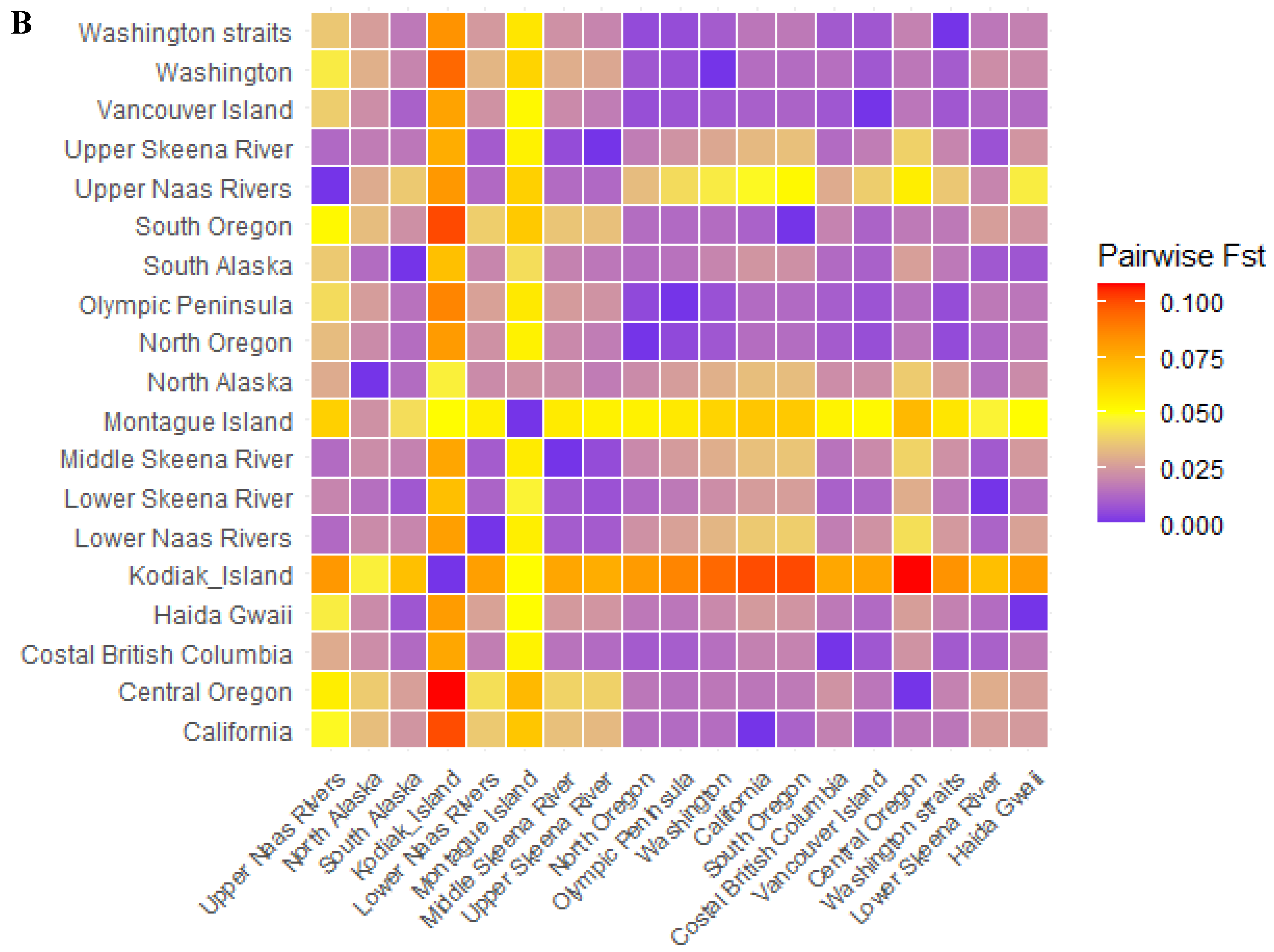
3.3. Genetic Structure
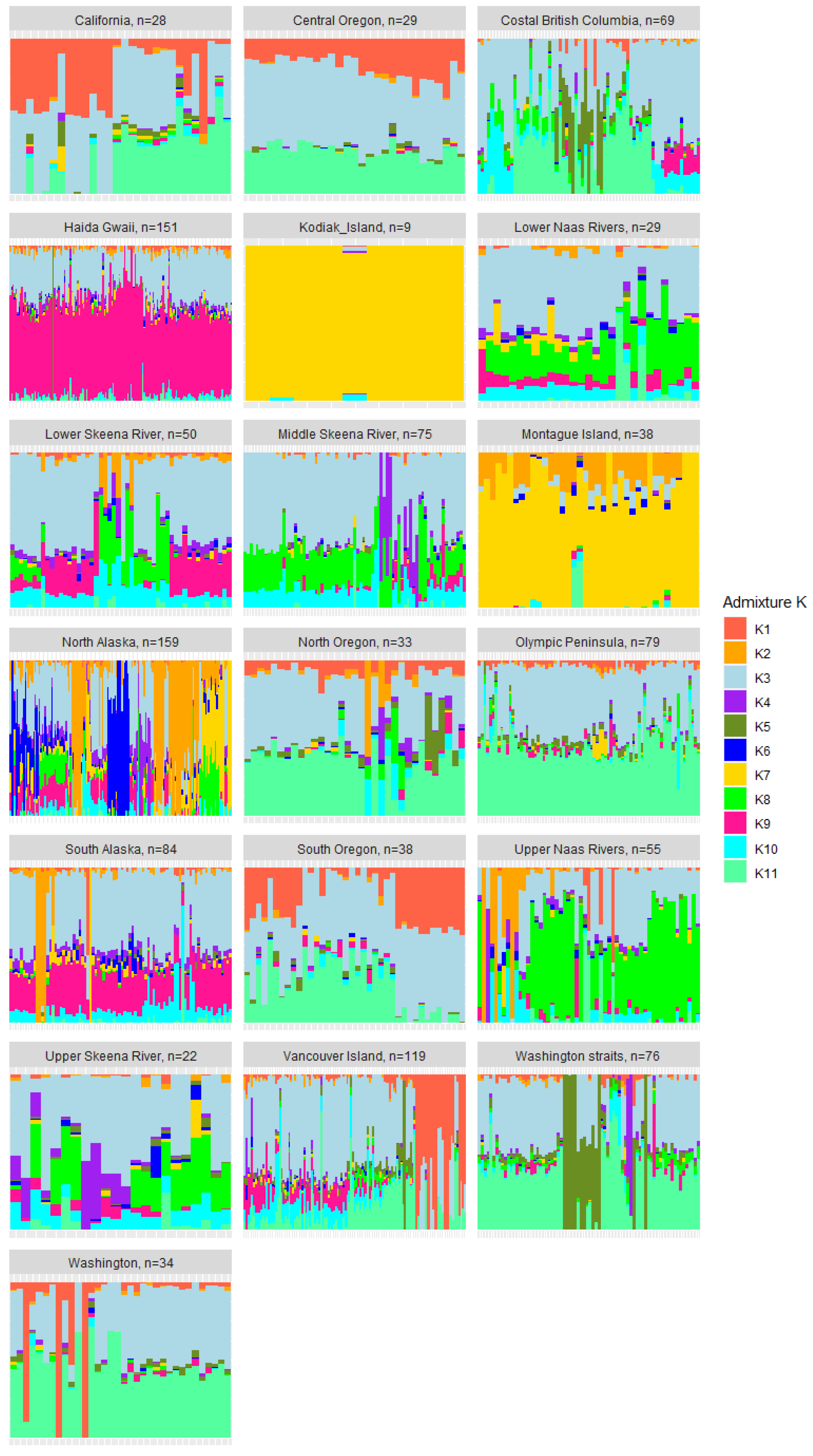
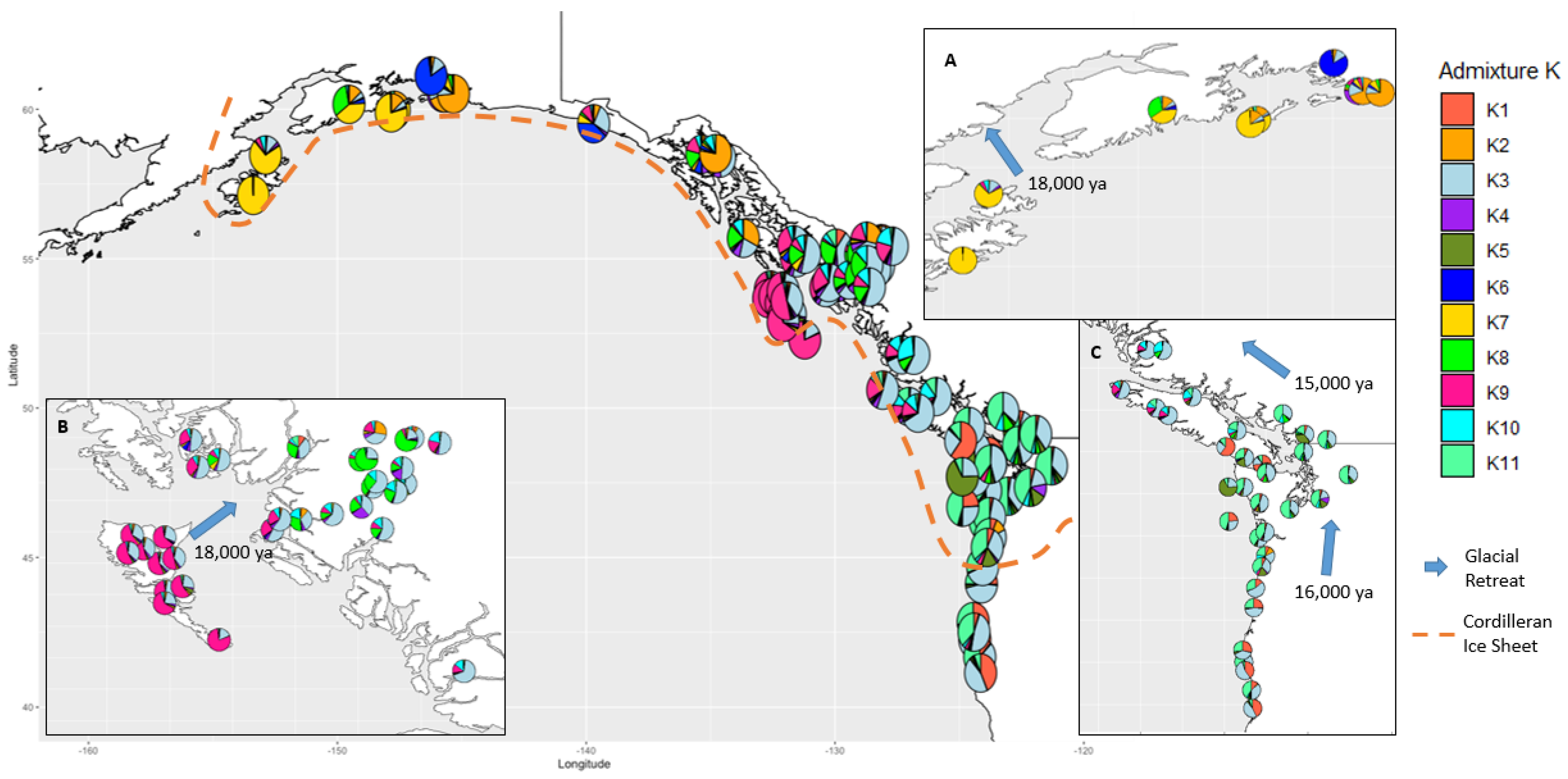
3.4. Genetic Admixture and Ancestry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Griffith, R. Picea sitchensis. In Fire Effects Information System; Forest Services: Washington, DC, USA; Available online: https://www.fs.fed.us/database/feis/plants/tree/picsit/all.html (accessed on 14 April 2022).
- Hamilton, J.A.; Aitken, S.N. Genetic and Morphological Structure of a Spruce Hybrid (Picea Sitchensis x P. Glauca) zone along a Climatic Gradient. Am. J. Bot. 2013, 100, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Degner, J. Spruce hybridization in British Columbia. For. Genet. Counc. BC 2015, 1–2. [Google Scholar]
- Gapare, W.J.; Aitken, S.N.; Ritland, C.E. Genetic diversity of core and peripheral Sitka spruce (Picea sitchensis (Bong.) Carr) populations: Implications for conservation of widespread species. Biol. Conserv. 2005, 123, 113–123. [Google Scholar] [CrossRef]
- Leitch, A.R.; Leitch, I.J. Ecological and genetic factors linked to contrasting genome dynamics in seed plants. New Phytol. 2012, 194, 629–646. [Google Scholar] [CrossRef] [PubMed]
- Florin, R. The distribution of conifer and taxad genera in time and space. Ann. De Geogr. 1964, 73, 712–713. [Google Scholar]
- Buschiazzo, E.; Ritland, C.; Bohlmann, J.K.R. Slow but not low: Genomic comparisons reveal slower evolutionary rate and higher dN/dS in conifers compared to angiosperms. BMC Evol. Biol. 2012, 12. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Birol, I.; Bousquet, J.; Ingvarsson, P.K.; Jansson, S.; Jones, S.J.; Keeling, C.I.; MacKay, J.; Nilsson, O.; Ritland, K.; et al. Insights into conifer giga-genomes. Plant Physiol. 2014, 166, 1724–1732. [Google Scholar] [CrossRef]
- Prunier, J.; Verta, J.P.; MacKay, J.J. Conifer genomics and adaptation: At the crossroads of genetic diversity and genome function. New Phytol. 2016, 209, 44–62. [Google Scholar] [CrossRef]
- Wright, J. Species crossability in spruce in relation to distribution and taxonomy. For. Sci 1955, 30. [Google Scholar]
- Lockwood, J.D.; Aleksic, J.M.; Zou, J.; Wang, J.; Liu, J.; Renner, S.S. A new phylogeny for the genus Picea from plastid, mitochondrial, and nuclear sequences. Mol. Phylogenet. Evol. 2013, 69, 717–727. [Google Scholar] [CrossRef]
- OECD. Section 5—Sitka Spruce (PICEA SITCHENSIS (BONG.) CARR.). OECD Publishing: Paris, France, 2006. [Google Scholar]
- Critchfield, W.B. Impact of the Pleistocene on the genetic structure of North American conifers. In Proceedings of the Proceedings of the 8th North American Forest Biology Workshop, Utah State University, Logan, UT, USA, 30 July–1 August 1984; pp. 70–118. [Google Scholar]
- Galeano, E.; Bousquet, J.; Thomas, B.R. SNP-based analysis reveals unexpected features of genetic diversity, parental contributions and pollen contamination in a white spruce breeding program. Sci. Rep. 2021, 11, 4990. [Google Scholar] [CrossRef]
- Korecky, J.; Cepl, J.; Stejskal, J.; Faltinova, Z.; Dvorak, J.; Lstiburek, M.; El-Kassaby, Y.A. Genetic diversity of Norway spruce ecotypes assessed by GBS-derived SNPs. Sci. Rep. 2021, 11. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Hao, Y.; Xia, X.; Khan, A.; Xu, Y.; Varshney, R.K.; He, Z. Crop breeding chips and genotyping platforms: Progress, challenges, and perspectives. Mol Plant 2017, 10, 1047–1064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira-Dias, L.; Vilanova, S.; Fita, A.; Prohens, J.; Rodriguez-Burruezo, A. Genetic diversity, population structure, and relationships in a collection of pepper (Capsicum spp.) landraces from the Spanish centre of diversity revealed by genotyping-by-sequencing (GBS). Hortic. Res. 2019, 6, 54. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, J. Sitka Spruce International Ten Provenance Experiment. Available online: https://www.fao.org/3/l1807e/L1807E06.htm (accessed on 14 April 2022).
- O’Driscoll, J. Sitka Spruce, its distribution and genetic variation. Ir. For. 1977, 2, 11. [Google Scholar]
- O’Driscoll, J. Working Plan for International Ten Provenance Experiment; Forest and Wildlife Service: Dublin, Ireland, 1972. [Google Scholar]
- Van de Sype, H.; Roman-Amat, B. Genetic Variability of Sitka Spruce of the IUFRO Collection; IUFRO: Montreal, QC, Canada, 1990; p. 1. [Google Scholar]
- King, J.; Alfaro, R.; Cartwright, C. Genetic resistance of Sitka spruce (Picea sitchensis) populations to the white pine weevil (Pissodes strobi): Distribution of resistance. Forestry 2004, 77, 7. [Google Scholar] [CrossRef]
- Parra-Salazar, A.; Gomez, J.; Lozano-Arce, D.; Reyes-Herrera, P.H.; Duitama, J. Robust and efficient software for reference-free genomic diversity analysis of genotyping-by-sequencing data on diploid and polyploid species. Mol. Ecol. Resour. 2022, 22, 439–454. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Gagalova, K.K.; Warren, R.L.; Coombe, L.; Wong, J.; Nip, K.M.; Yuen, M.M.S.; Whitehill, J.G.A.; Celedon, J.M.; Ritland, C.; Taylor, G.A.; et al. Spruce giga-genomes: Structurally similar yet distinctive with differentially expanding gene families and rapidly evolving genes. Plant J. 2022, 111, 1469–1485. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [PubMed]
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Meeus, T.; Goudet, J. A step-by-step tutorial to use HierFstat to analyse populations hierarchically structured at multiple levels. Infect Genet Evol 2007, 7, 731–735. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grunwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.M.; Cullingham, C.I.; Peery, R.M. The influence of a priori grouping on inference of genetic clusters: Simulation study and literature review of the DAPC method. Hered. (Edinb) 2020, 125, 269–280. [Google Scholar] [CrossRef]
- Highton, R. The relationship between the number of loci and the statistical support for the topology of UPGMA trees obtained from genetic distance data. Mol. Phylogenet. Evol. 1993, 2, 337–343. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-Statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinform. 2011, 12, 246. [Google Scholar] [CrossRef]
- Yu, G. Scatterpie: Scatter Pie Plot. Available online: https://CRAN.R-project.org/package=scatterpie (accessed on 14 April 2022).
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- South, S. Rnaturalearth: World Map Data from Natural Earth. Available online: https://github.com/ropensci/rnaturalearth (accessed on 14 April 2022).
- He, J.; Zhao, X.; Laroche, A.; Lu, Z.X.; Liu, H.; Li, Z. Genotyping-by-sequencing (GBS), an ultimate marker-assisted selection (MAS) tool to accelerate plant breeding. Front. Plant Sci. 2014, 5, 484. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Yuan, Y.; Wang, H.; Yu, D.; Liu, Y.; Zhang, A.; Gowda, M.; Nair, S.K.; Hao, Z.; Lu, Y.; et al. Applications of genotyping-by-sequencing (GBS) in maize genetics and breeding. Sci. Rep. 2020, 10, 16308. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, S.J.; Puritz, J.B.; Willis, S.C.; Hollenbeck, C.M.; Portnoy, D.S. These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists (vol 27, pg 3193, 2018). Mol. Ecol. 2019, 28, 3459. [Google Scholar] [CrossRef]
- Veeckman, E.; Van Glabeke, S.; Haegeman, A.; Muylle, H.; van Parijs, F.R.D.; Byrne, S.L.; Asp, T.; Studer, B.; Rohde, A.; Roldan-Ruiz, I.; et al. Overcoming challenges in variant calling: Exploring sequence diversity in candidate genes for plant development in perennial ryegrass (Lolium perenne). DNA Res. 2019, 26, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Pavy, N.; Namroud, M.C.; Gagnon, F.; Isabel, N.; Bousquet, J. The heterogeneous levels of linkage disequilibrium in white spruce genes and comparative analysis with other conifers. Heredity 2012, 108, 273–284. [Google Scholar] [CrossRef]
- Qu, J.; Kachman, S.D.; Garrick, D.; Fernando, R.L.; Cheng, H. Exact distribution of linkage disequilibrium in the presence of mutation, selection, or minor allele frequency filtering. Front. Genet. 2020, 11, 362. [Google Scholar] [CrossRef]
- Chen, B.W.; Cole, J.W.; Grond-Ginsbach, C. Departure from Hardy Weinberg Equilibrium and Genotyping Error. Front. Genet. 2017, 8, 167. [Google Scholar] [CrossRef]
- Pavan, S.; Delvento, C.; Ricciardi, L.; Lotti, C.; Ciani, E.; D’Agostino, N. Recommendations for Choosing the Genotyping Method and Best Practices for Quality Control in Crop Genome-Wide Association Studies. Front. Genet. 2020, 11, 447. [Google Scholar] [CrossRef]
- Blanco-Pastor, J.L.; Barre, P.; Keep, T.; Ledauphin, T.; Escobar-Gutierrez, A.; Roschanski, A.M.; Willner, E.; Dehmer, K.J.; Hegarty, M.; Muylle, H. Canonical correlations reveal adaptive loci and phenotypic responses to climate in perennial ryegrass. Mol. Ecol. Resour. 2021, 21, 849–870. [Google Scholar] [CrossRef]
- De La Torre, A.R.; Wilhite, B.; Neale, D.B. Environmental Genome-Wide Association Reveals Climate Adaptation Is Shaped by Subtle to Moderate Allele Frequency Shifts in Loblolly Pine. Genome Biol. Evol. 2019, 11, 2976–2989. [Google Scholar] [CrossRef]
- A’Hara, S.W.; Cottrell, J.E. A set of microsatellite markers for use in Sitka spruce (Picea sitchensis) developed from Picea glauca ESTs. Mol. Ecol. Notes 2004, 4, 4. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Lexer, C.; Aitken, S.N. Differential introgression reveals candidate genes for selection across a spruce (Picea sitchensis x P. glauca) hybrid zone. New Phytol. 2013, 197, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Holliday, J.A.; Suren, H.; Aitken, S.N. Divergent selection and heterogeneous migration rates across the range of Sitka spruce (Picea sitchensis). Proc. Biol. Sci. 2012, 279, 1675–1683. [Google Scholar] [CrossRef] [PubMed]
- Holliday, J.A.; Ritland, K.; Aitken, S.N. Widespread, ecologically relevant genetic markers developed from association mapping of climate-related traits in Sitka spruce (Picea sitchensis). New Phytol. 2010, 188, 501–514. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Sun, X.; Dong, L.; Zhang, S. Mating patterns and pollen dispersal in a Japanese larch (Larix kaempferi) clonal seed orchard: A case study. Sci. China Life Sci. 2018, 61, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, A.; Lawson, S.S.; Frank, G.S.; Coggeshall, M.V.; Woeste, K.E.; McKenna, J.R. Pollen flow and paternity in an isolated and non-isolated black walnut (Juglans nigra L.) timber seed orchard. PLoS ONE 2018, 13, e0207861. [Google Scholar] [CrossRef]
- O’Connell, L.M.; Mosseler, A.; Rajora, O.P. Extensive long-distance pollen dispersal in a fragmented landscape maintains genetic diversity in white spruce. J. Hered. 2007, 98, 640–645. [Google Scholar] [CrossRef]
- Farris, D.W.H.; Haeussler, P.J. Selected geologic maps of the Kodiak batholith and other Paleocene intrusive rocks, Kodiak Island, Alaska. US Geol. Surv. Sci. Investig. Map 2020, 3441, 10. [Google Scholar] [CrossRef]
- Menounos, B.; Goehring, B.M.; Osborn, G.; Margold, M.; Ward, B.; Bond, J.; Clarke, G.K.C.; Clague, J.J.; Lakeman, T.; Koch, J.; et al. Cordilleran Ice Sheet mass loss preceded climate reversals near the Pleistocene Termination. Science 2017, 358, 781–784. [Google Scholar] [CrossRef] [Green Version]
- Haro, H. Animating the Temporal Progression of Cordilleran Deglaciation and Vegetation Succession in the Pacific Northwest during the late Quaternary Period. In Proceedings of the Western Cedar, Western Washington University, Belligham, DC, USA, 17 May 2017. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Definition | Overall | |
|---|---|---|
| Ho | Observed Heterozygosity | 0.21 |
| Hs | Within Population Gene Diversity | 0.198 |
| Ht | Overall Gene Diversity | 0.204 |
| Dst | Gene Diversity among samples | 0.006 |
| Fst | Fixation index | 0.029 |
| Fis | Inbreeding Coefficient | 0 |
| Dest * | Population Differentiation | 0.0078 |
| Gst ** | Population Differentiation | 0.0284 |
| Geographic Regions | Ho | He | Fst | Allelic Richness |
|---|---|---|---|---|
| California | 0.18 | 0.18 | 0.117 | 1.180 |
| Central Oregon | 0.17 | 0.17 | 0.162 | 1.171 |
| Coastal British Columbia | 0.23 | 0.21 | −0.021 | 1.209 |
| Haida Gwaii | 0.19 | 0.19 | 0.091 | 1.186 |
| Kodiak Island | 0.24 | 0.18 | 0.086 | NA |
| Lower Naas Rivers | 0.22 | 0.21 | −0.028 | 1.21 |
| Lower Skeena River | 0.23 | 0.21 | −0.032 | 1.211 |
| Middle Skeena River | 0.23 | 0.22 | −0.055 | 1.216 |
| Montague Island | 0.19 | 0.18 | 0.108 | 1.182 |
| North Alaska | 0.22 | 0.21 | −0.025 | 1.209 |
| North Oregon | 0.23 | 0.2 | 0.003 | 1.204 |
| Olympic Peninsula | 0.2 | 0.19 | 0.072 | 1.19 |
| South Alaska | 0.2 | 0.19 | 0.050 | 1.194 |
| South Oregon | 0.18 | 0.18 | 0.128 | 1.178 |
| Upper Naas Rivers | 0.23 | 0.23 | −0.117 | 1.228 |
| Upper Skeena River | 0.24 | 0.22 | −0.079 | 1.22 |
| Vancouver Island | 0.21 | 0.2 | 0.028 | 1.199 |
| Washington | 0.2 | 0.19 | 0.074 | 1.189 |
| Washington Straits | 0.22 | 0.2 | 0.010 | 1.202 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Byrne, T.; Farrelly, N.; Kelleher, C.; Hodkinson, T.R.; Byrne, S.L.; Barth, S. Genetic Diversity and Structure of a Diverse Population of Picea sitchensis Using Genotyping-by-Sequencing. Forests 2022, 13, 1511. https://doi.org/10.3390/f13091511
Byrne T, Farrelly N, Kelleher C, Hodkinson TR, Byrne SL, Barth S. Genetic Diversity and Structure of a Diverse Population of Picea sitchensis Using Genotyping-by-Sequencing. Forests. 2022; 13(9):1511. https://doi.org/10.3390/f13091511
Chicago/Turabian StyleByrne, Tomás, Niall Farrelly, Colin Kelleher, Trevor R. Hodkinson, Stephen L. Byrne, and Susanne Barth. 2022. "Genetic Diversity and Structure of a Diverse Population of Picea sitchensis Using Genotyping-by-Sequencing" Forests 13, no. 9: 1511. https://doi.org/10.3390/f13091511