
Soil Metaproteomics as a Tool for Environmental Monitoring of Minelands

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
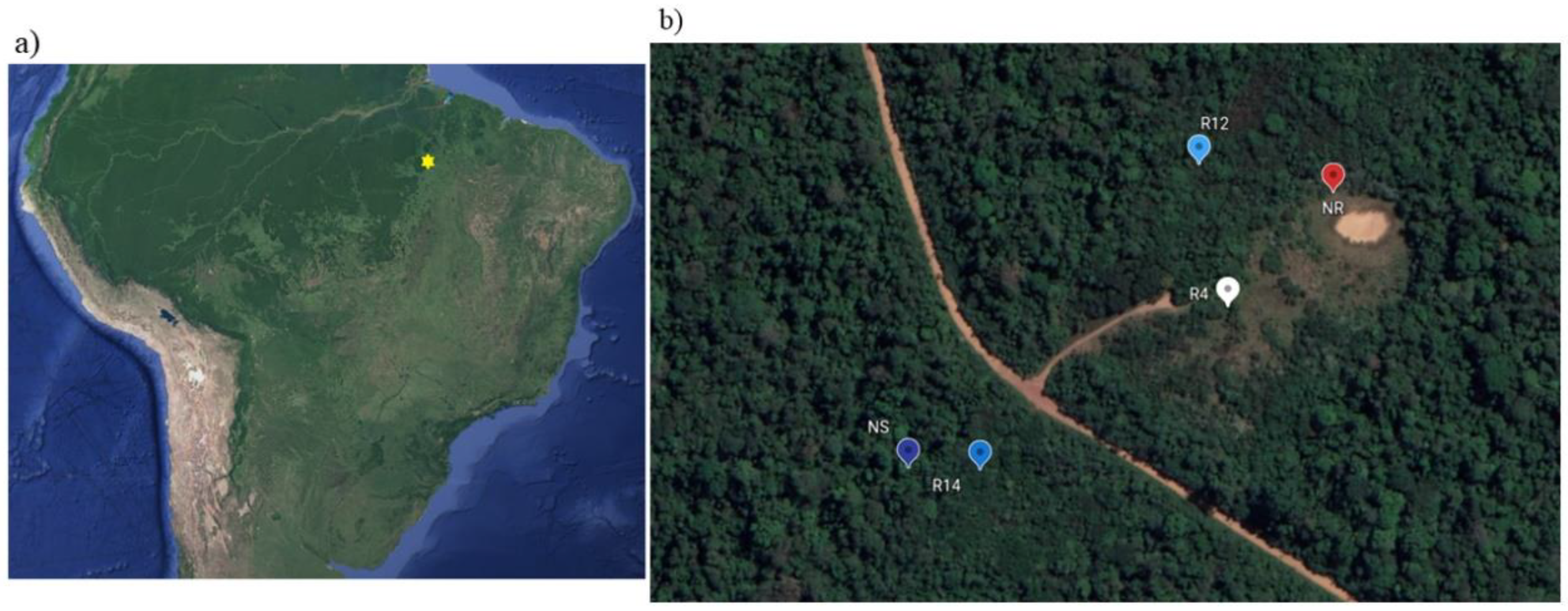
2.1. Study Area and Rehabilitation Activities
2.2. Soil Sampling
2.3. Environmental Data
2.4. Soil Protein Extraction and Identification
2.5. Data Analysis
3. Results
3.1. Environmental Variables
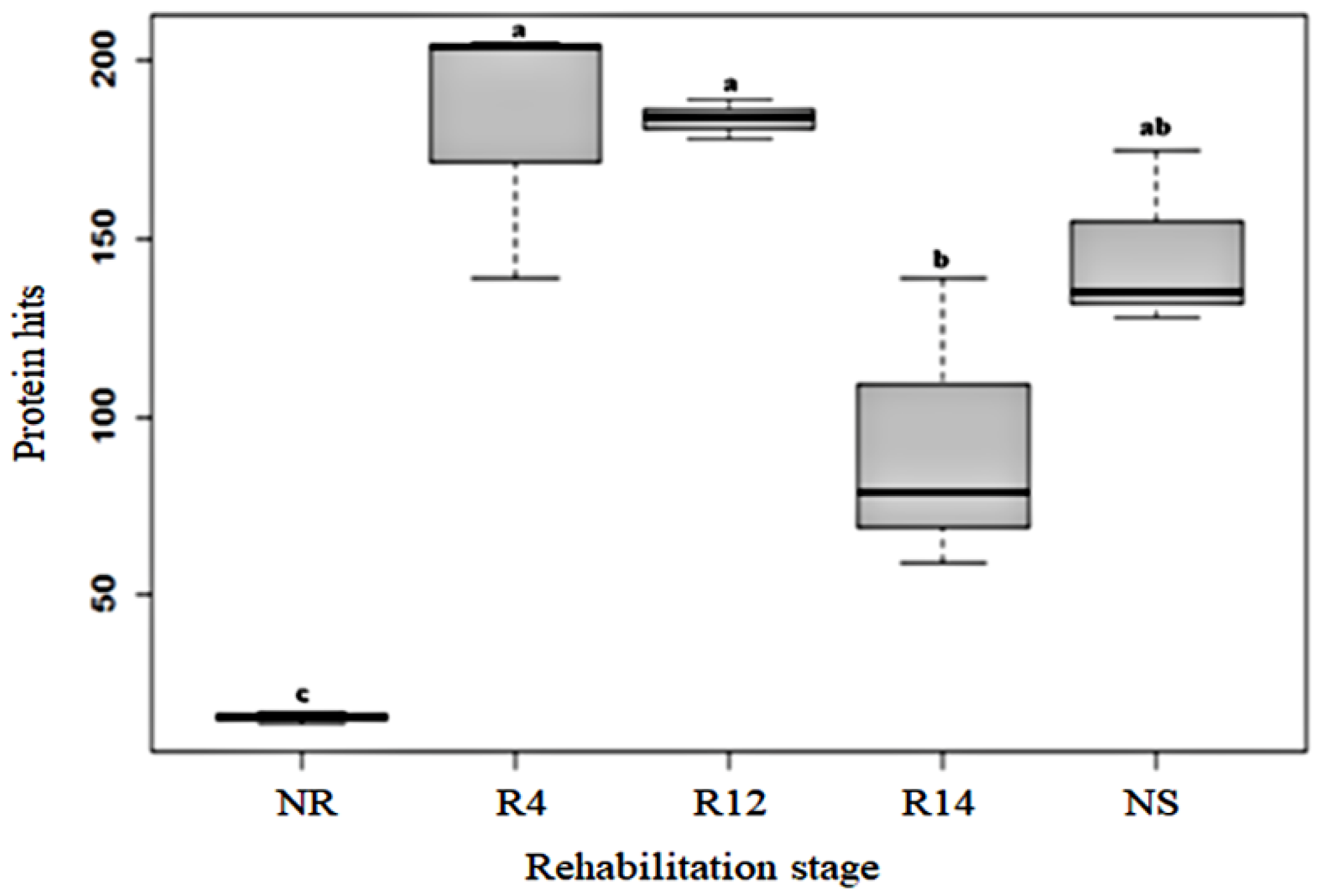
3.2. Amount of Identified Proteins
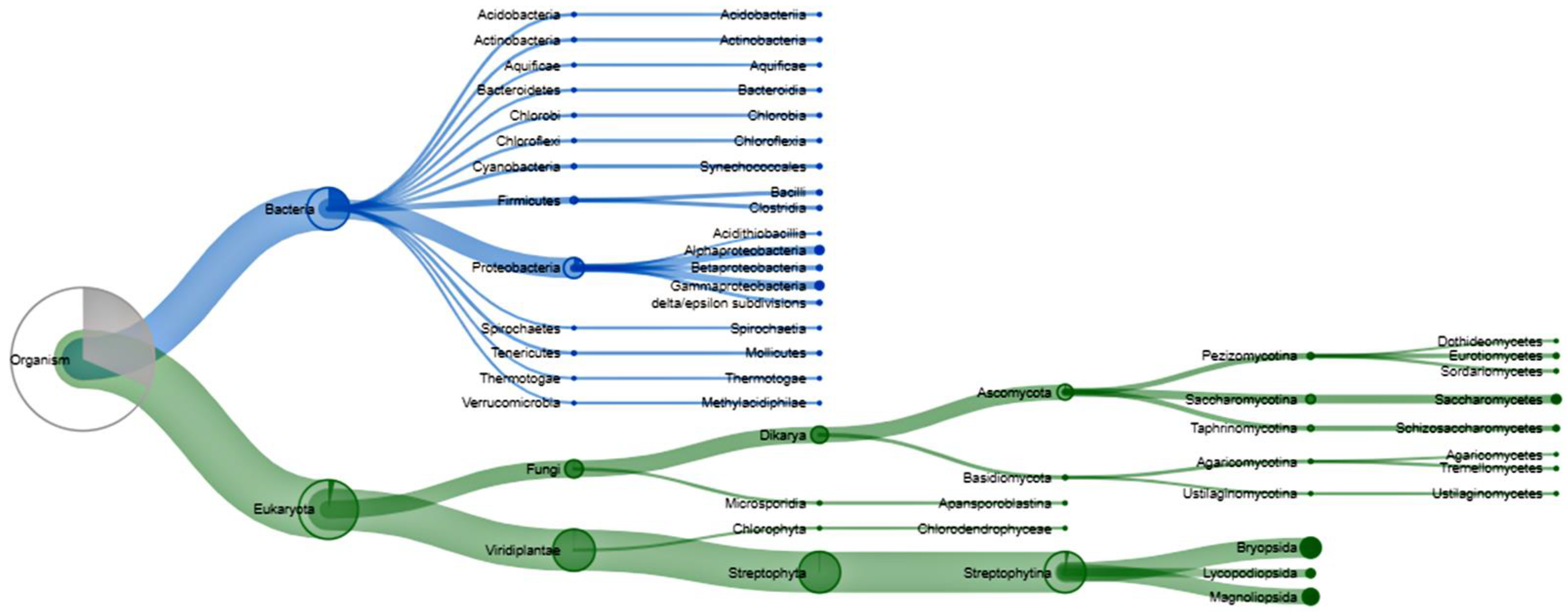
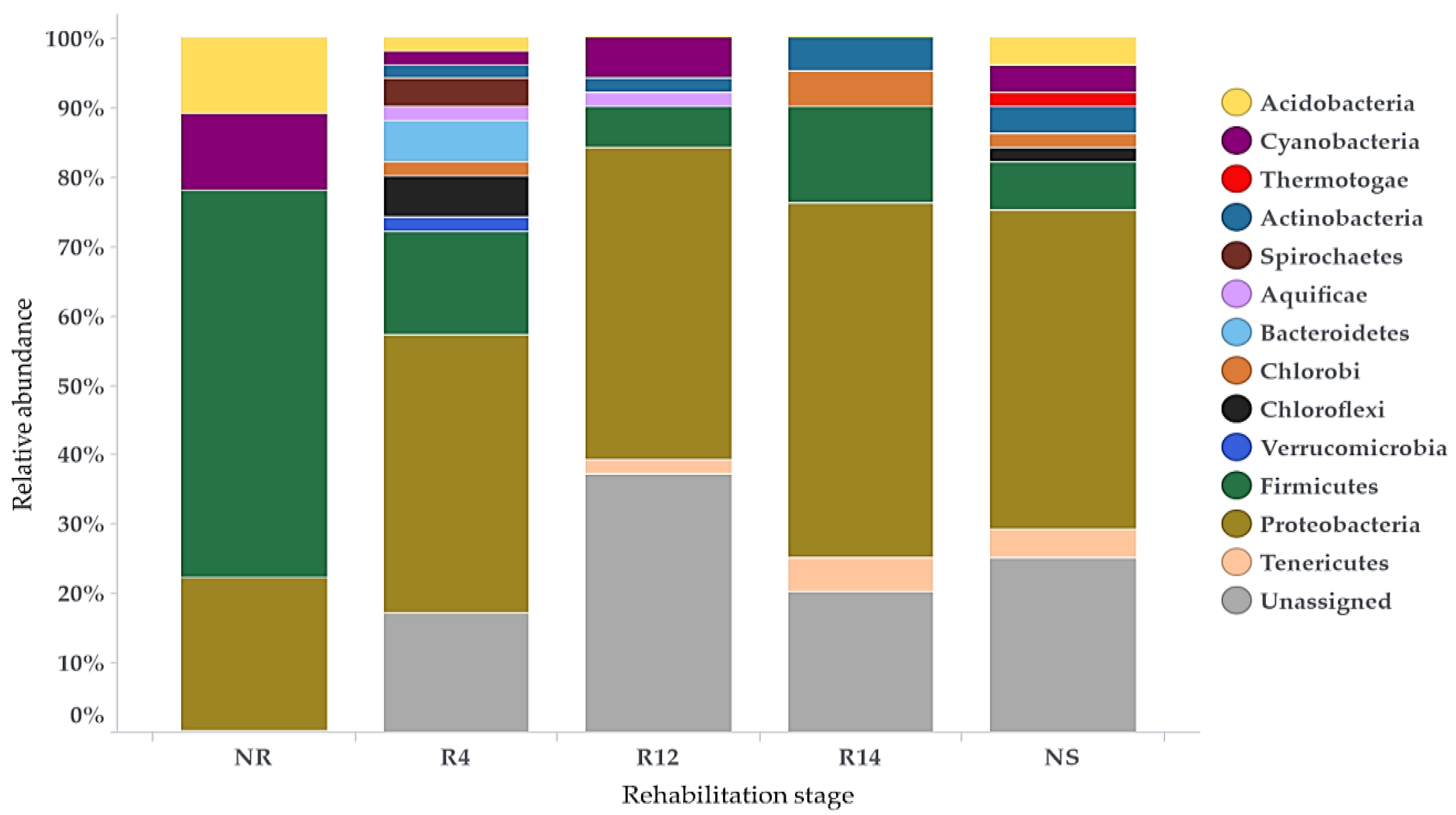
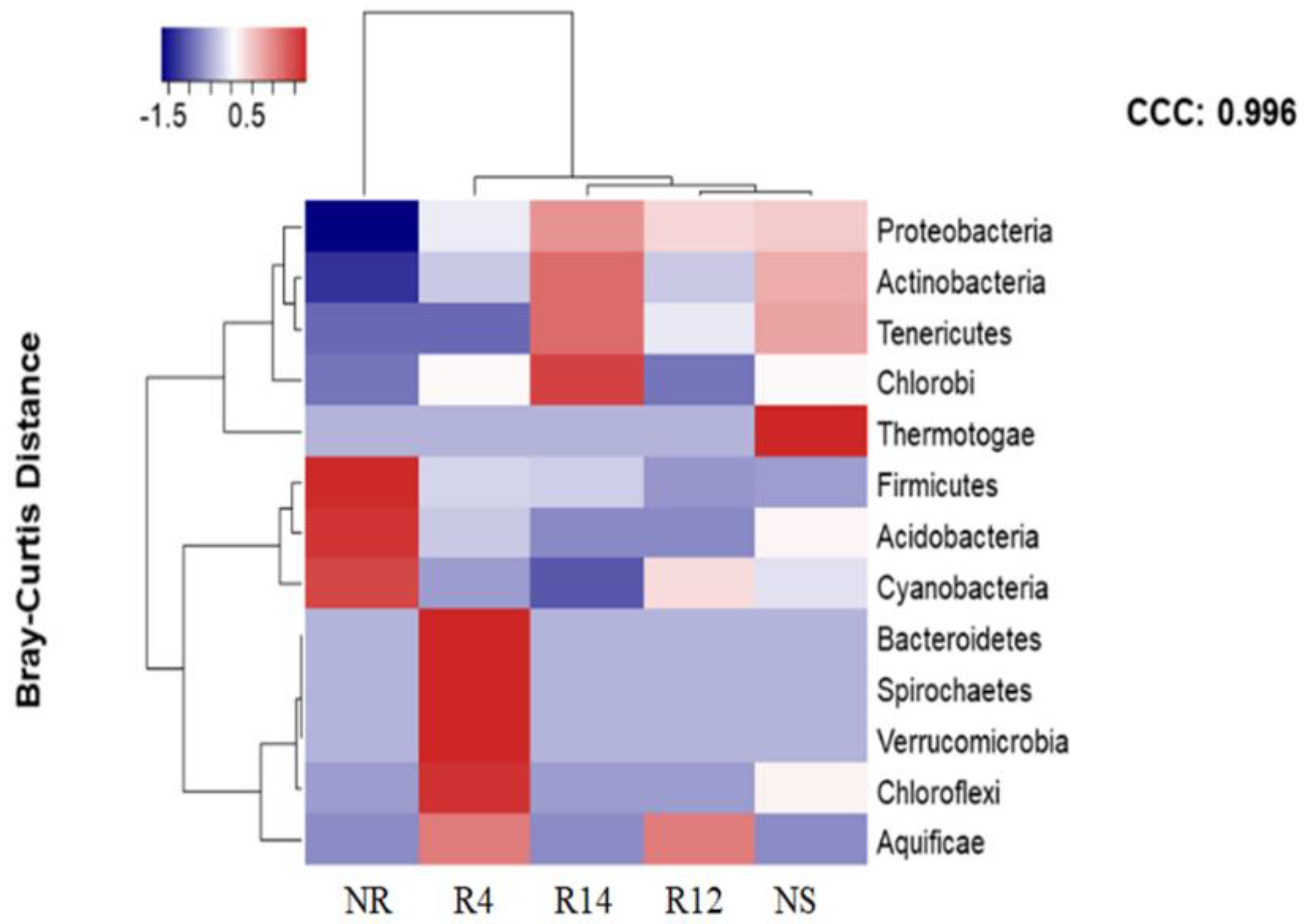
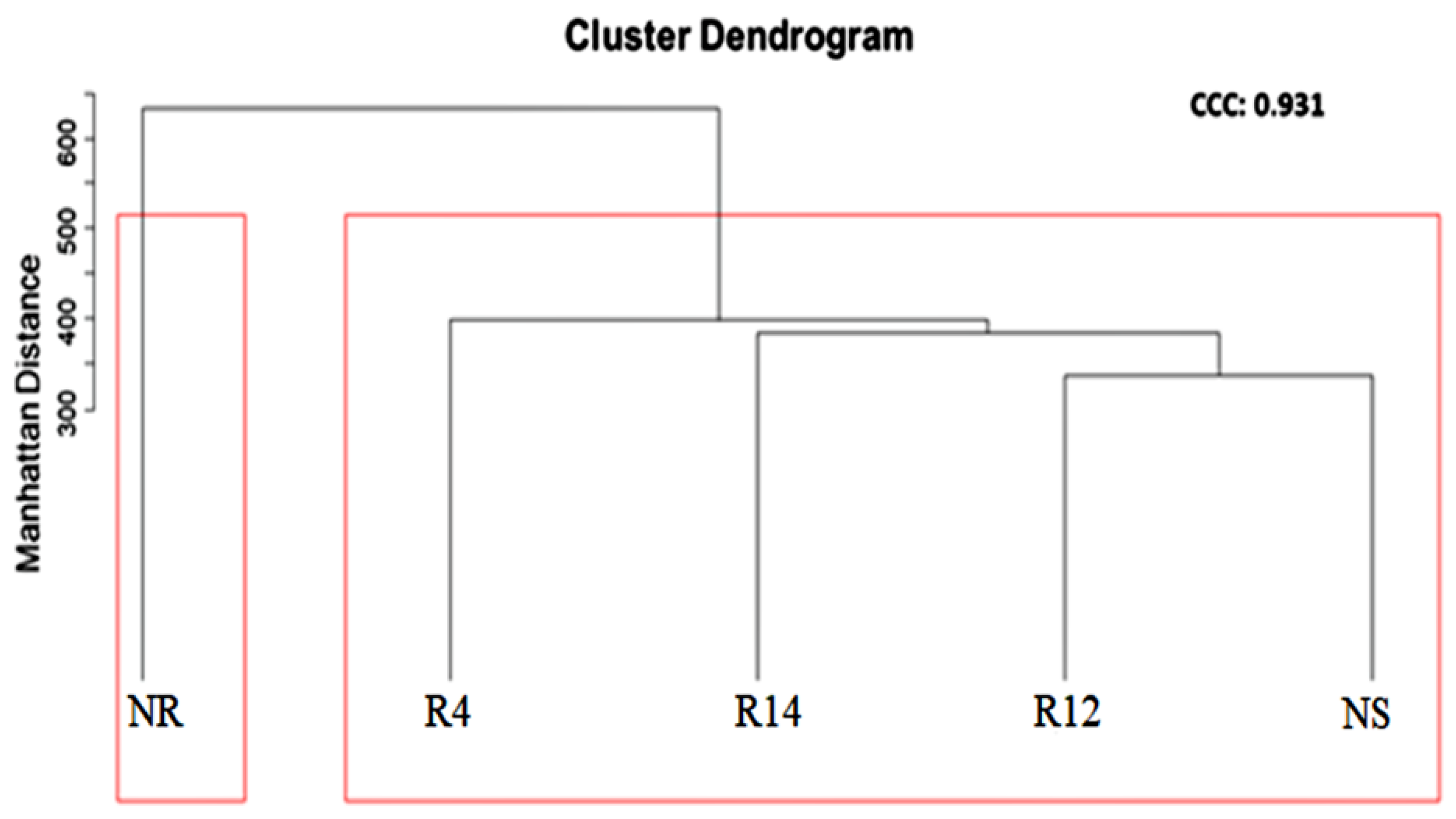
3.3. Bacterial Populations during Rehabilitation
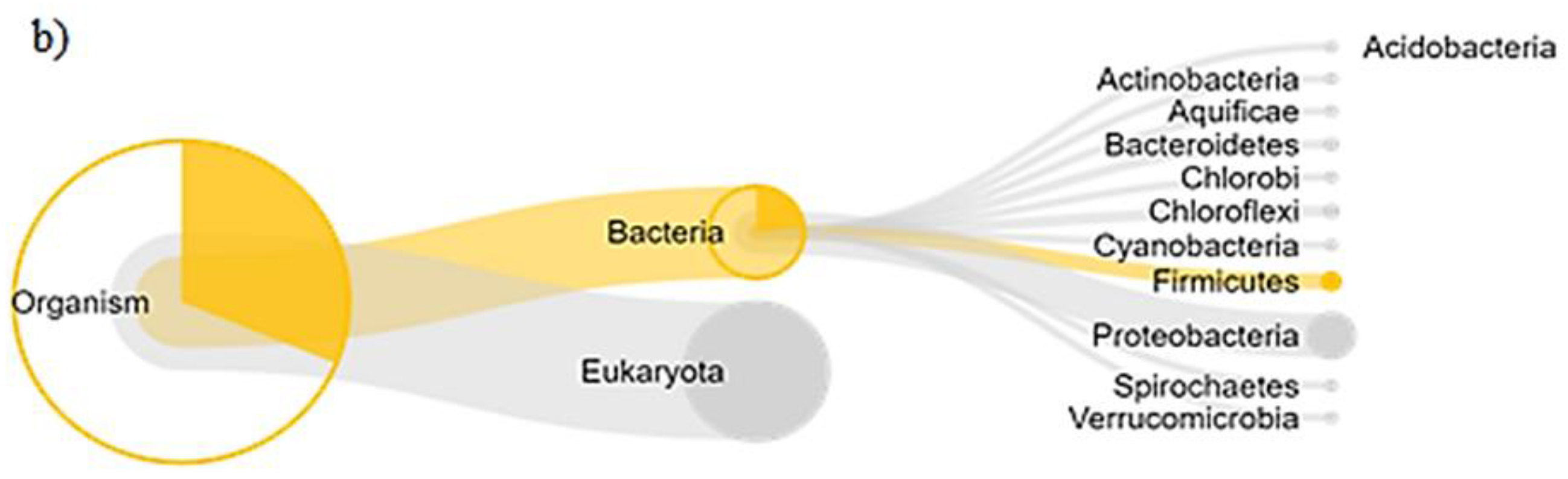
3.4. Functional Similarity of Enzymes of Soil Organisms during Rehabilitation
4. Discussion
4.1. Higher Protein Content in the Native and Rehabilitation Stages Indicates Improvement of Soil Biological Attributes during Environmental Rehabilitation
4.2. Bacterial Populations during Rehabilitation
4.3. Protein Functional Analysis in the Community of Soil Organisms
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Losfeld, G.; L’Huillier, L.; Fogliani, B.; Jaffré, T.; Grison, C. Mining in New Caledonia: Environmental stakes and restoration opportunities. Environ. Sci. Pollut. Res. 2014, 22, 5592–5607. [Google Scholar] [CrossRef]
- Gastauer, M.; Filho, P.W.M.S.; Ramos, S.J.; Caldeira, C.F.; Silva, J.R.; Siqueira, J.O.; Neto, A.E.F. Mine land rehabilitation in Brazil: Goals and techniques in the context of legal requirements. Ambio 2019, 48, 74–88. [Google Scholar] [CrossRef]
- Abiraami, T.V.; Singh, S.; Nain, L. Soil metaproteomics as a tool for monitoring functional microbial communities: Promises and challenges. Rev. Environ. Sci. Bio/Technol. 2020, 19, 73–102. [Google Scholar] [CrossRef]
- Biswas, R.; Sarkar, A. ‘Omics’ Tools in Soil Microbiology: The State of the Art. Microb. Rejuvenation Polluted Environ. 2018, 3, 35–64. [Google Scholar]
- Trindade, F.C.; Ramos, S.J.; Gastauer, M.; Saraiva, A.M.M.; Caldeira, C.F.; Oliveira, G.; Valadares, R.B.D.S. Metaproteomes reveal increased capacity for stress tolerance of soil microbes in ferruginous tropical rocky outcrops. Pedobiologia 2020, 81, 150664. [Google Scholar] [CrossRef]
- Garris, H.W.; Baldwin, S.A.; Van Hamme, J.; Gardner, W.C.; Fraser, L.H. Genomics to assist mine reclamation: A review. Restor. Ecol. 2016, 24, 165–173. [Google Scholar] [CrossRef]
- Li, Y.; Wen, H.; Chen, L.; Yin, T. Succession of Bacterial Community Structure and Diversity in Soil along a Chronosequence of Reclamation and Re-Vegetation on Coal Mine Spoils in China. PLoS ONE 2014, 9, e115024. [Google Scholar] [CrossRef]
- Cardoso, E.J.B.N.; Vasconcellos, R.L.F.; Bini, D.; Miyauchi, M.Y.H.; Dos Santos, C.A.; Alves, P.R.L.; De Paula, A.M.; Nakatani, A.S.; Pereira, J.D.M.; Nogueira, M.A. Soil health: Looking for suitable indicators. What should be considered to assess the effects of use and management on soil health? Sci. Agric. 2013, 70, 274–289. [Google Scholar] [CrossRef] [Green Version]
- Bastida, F.; Selevsek, N.; Torres, I.F.; Hernández, T.; García, C. Soil restoration with organic amendments: Linking cellular functionality and ecosystem processes. Sci. Rep. 2015, 5, 15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastida, F.; Jehmlich, N. It’s all about functionality: How can metaproteomics help us to discuss the attributes of ecological relevance in soil? J. Proteom. 2016, 144, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, E.; Chiapello, M.; Daghino, S.; Bonfante, P.; Mello, A. Soil metaproteomics reveals an inter-kingdom stress response to the presence of black truffles. Sci. Rep. 2016, 6, 25773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tartaglia, M.; Bastida, F.; Sciarrillo, R.; Guarino, C. Soil Metaproteomics for the Study of the Relationships Between Microorganisms and Plants: A Review of Extraction Protocols and Ecological Insights. Int. J. Mol. Sci. 2020, 21, 8455. [Google Scholar] [CrossRef] [PubMed]
- Bastida, F.; Hernandez, T.; García, C. Metaproteomics of soils from semiarid environment: Functional and phylogenetic information obtained with different protein extraction methods. J. Proteom. 2014, 101, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Bastida, F.; García, C.; von Bergen, M.; Moreno, J.L.; Richnow, H.; Jehmlich, N. Deforestation fosters bacterial diversity and the cyanobacterial community responsible for carbon fixation processes under semiarid climate: A metaproteomics study. Appl. Soil Ecol. 2015, 93, 65–67. [Google Scholar] [CrossRef]
- Gastauer, M.; Caldeira, C.F.; Ramos, S.J.; Silva, D.F.; Siqueira, J. Active rehabilitation of Amazonian sand mines converges soils, plant communities and environmental status to their predisturbance levels. Land Degrad. Dev. 2019, 31, 607–618. [Google Scholar] [CrossRef]
- Gastauer, M.; Caldeira, C.F.; Ramos, S.J.; Trevelin, L.C.; Jaffé, R.; Oliveira, G.; Vera, M.P.O.; Pires, E.; Santiago, F.L.D.A.; Carneiro, M.A.C.; et al. Integrating environmental variables by multivariate ordination enables the reliable estimation of mineland rehabilitation status. J. Environ. Manag. 2020, 256, 109894. [Google Scholar] [CrossRef]
- Viana, P.L.; Mota, N.F.D.O.; Gil, A.D.S.B.; Salino, A.; Zappi, D.; Harley, R.M.; Ilkiu-Borges, A.L.; Secco, R.; Almeida, T.E.; Watanabe, M.; et al. Flora das cangas da Serra dos Carajás, Pará, Brasil: História, área de estudos e metodologia. Rodriguésia 2016, 67, 1107–1124. [Google Scholar] [CrossRef]
- Instituto Brasileiro de Geografia e Estatística—IBGE. Manual Técnico da Vegetação Brasileira. Manuais Téc. Geociênc. 2012, 2, 275. [Google Scholar]
- Donagema, G.K.; Campos, D.V.B.; Calderano, S.B.; Teixeira, W.G.; Viana, J.H.M. Manual de Métodos de Análise do Solo, 2nd ed.; Centro Nacional de Pesquisa de Solos: Rio de Janeiro, Brazil, 2011. [Google Scholar]
- Wang, W.; Vignani, R.; Scali, M.; Cresti, M. A universal and rapid protocol for protein extraction from recalcitrant plant tissues for proteomic analysis. Electrophoresis 2006, 27, 2782–2786. [Google Scholar] [CrossRef]
- Nascimento, S.V.D.; Magalhães, M.M.; Cunha, R.L.; Costa, P.H.D.O.; Alves, R.C.D.O.; De Oliveira, G.C.; Valadares, R.B.D.S. Differential accumulation of proteins in oil palms affected by fatal yellowing disease. PLoS ONE 2018, 13, e0195538. [Google Scholar] [CrossRef] [Green Version]
- Searle, B.C. Scaffold: A bioinformatic tool for validating MS/MS-based proteomic studies. Proteomics 2010, 10, 1265–1269. [Google Scholar] [CrossRef]
- Nesvizhskii, A.I.; Keller, A.; Kolker, E.; Aebersold, R. A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry abilities that proteins are present in a sample on the basis. Anal. Chem. 2003, 75, 4646–4658. [Google Scholar] [CrossRef] [PubMed]
- Mesuere, B.; Debyser, G.; Aerts, M.; Devreese, B.; Vandamme, P.; Dawyndt, P. The Unipept metaproteomics analysis pipeline. Proteomics 2015, 15, 1437–1442. [Google Scholar] [CrossRef] [PubMed]
- Verschaffelt, P.; Bossche, T.V.D.; Martens, L.; Dawyndt, P.; Mesuere, B. Unipept Desktop: A Faster, More Powerful Metaproteomics Results Analysis Tool. J. Proteome Res. 2021, 20, 2005–2009. [Google Scholar] [CrossRef] [PubMed]
- Miralles, I.; Domingo, F.; Cantón, Y.; Trasar-Cepeda, C.; Leirós, M.C.; Gil-Sotres, F. Hydrolase enzyme activities in a successional gradient of biological soil crusts in arid and semi-arid zones. Soil Biol. Biochem. 2012, 53, 124–132. [Google Scholar] [CrossRef]
- Bautista-Cruz, A.; Ortiz-Hernández, Y.D. Hydrolytic soil enzymes and their response to fertilization: A short review. Comun. Sci. 2015, 6, 255. [Google Scholar] [CrossRef]
- Zago, L.D.M.S.; Oliveira, R.N.; Bombonatto, A.K.G.; Moreira, L.M.D.O.; Melo, E.N.D.P.; Caramori, S.S. Enzimas Extracelulares de Solos de Cerrado como Bioindicadores de Qualidade em Áreas Agricultáveis em Goiás, Brasil. Front. J. Soc. Technol. Environ. Sci. 2016, 5, 104–127. [Google Scholar] [CrossRef]
- Starke, R.; Jehmlich, N.; Bastida, F. Using proteins to study how microbes contribute to soil ecosystem services: The current state and future perspectives of soil metaproteomics. J. Proteom. 2019, 198, 50–58. [Google Scholar] [CrossRef]
- Lekevičius, E. Vacant niches in nature, ecology, and evolutionary theory: A mini-review. Ekologija 2009, 55, 165–174. [Google Scholar] [CrossRef]
- Yang, Y.; Weiner, J.; Wang, G.; Ren, Z. Convergence of community composition during secondary succession on Zokor rodent mounds on the Tibetan Plateau. J. Plant Ecol. 2017, 11, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Gastauer, M.; Silva, J.R.; Junior, C.F.C.; Ramos, S.J.; Souza-Filho, P.W.M.; Neto, A.E.F.; Siqueira, J.O. Mine land rehabilitation: Modern ecological approaches for more sustainable mining. J. Clean. Prod. 2018, 172, 1409–1422. [Google Scholar] [CrossRef]
- Banning, N.C.; Gleeson, D.; Grigg, A.H.; Grant, C.D.; Andersen, G.; Brodie, E.L.; Murphy, D.V. Soil Microbial Community Successional Patterns during Forest Ecosystem Restoration. Appl. Environ. Microbiol. 2011, 77, 6158–6164. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Zhou, Y.; Tan, Y.; Wu, Z.; Lu, P.; Zhang, G.; Yu, F. Restoration with pioneer plants changes soil properties and remodels the diversity and structure of bacterial communities in rhizosphere and bulk soil of copper mine tailings in Jiangxi Province, China. Environ. Sci. Pollut. Res. 2018, 25, 22106–22119. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.; Jackson, R.B. Toward an Ecological Classification of Soil Bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Madigan, M.T.; Schaaf, N.A.V.; Sattley, W.M. The Chlorobiaceae, Chloroflexaceae, and Heliobacteriaceae. In Modern Topics in the Phototrophic Prokaryotes; Springer Science and Business Media LLC: Berlin, Germany, 2017; pp. 139–161. [Google Scholar]
- Eisenlord, S.; Zak, D. Simulated Atmospheric Nitrogen Deposition Alters Actinobacterial Community Composition in Forest Soils. Soil Biol. Biochem. 2009, 74, 1157–1166. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.M.D.; Junior, N.P. Produção, propriedades e aplicação de celulases na hidrólise de resíduos agroindustriais. Quim. Nova 2010, 33, 181–188. [Google Scholar] [CrossRef]
- Makoi, J.H.; Ndakidemi, P.A. Selected soil enzymes: Examples of their potential roles in the ecosystem. Afr. J. Biotechnol. 2008, 7, 3. [Google Scholar]
- Shallom, D.; Shoham, Y. Microbial hemicellulases. Curr. Opin. Microbiol. 2003, 6, 219–228. [Google Scholar] [CrossRef]
- Yao, Q.; Li, Z.; Song, Y.; Wright, S.J.; Guo, X.; Tringe, S.G.; Tfaily, M.M.; Paša-Tolić, L.; Hazen, T.C.; Turner, B.L.; et al. Community proteogenomics reveals the systemic impact of phosphorus availability on microbial functions in tropical soil. Nat. Ecol. Evol. 2018, 2, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Bach, H.-J.; Munch, J.C. Identification of bacterial sources of soil peptidases. Biol. Fertil. Soils 2000, 31, 219–224. [Google Scholar] [CrossRef]
- Vranova, V.; Rejsek, K.; Formánek, P. Proteolytic activity in soil: A review. Appl. Soil Ecol. 2013, 70, 23–32. [Google Scholar] [CrossRef]
- Adetunji, A.T.; Lewu, F.; Mulidzi, R.; Ncube, B. The biological activities of β-glucosidase, phosphatase and urease as soil quality indicators: A review. J. Soil Sci. Plant Nutr. 2017, 17, 794–807. [Google Scholar] [CrossRef] [Green Version]
- Carter, D.O.; Yellowlees, D.; Tibbett, M. Autoclaving kills soil microbes yet soil enzymes remain active. Pedobiologia 2007, 51, 295–299. [Google Scholar] [CrossRef]
- Bastida, F.; Crowther, T.W.; Prieto, I.; Routh, D.; García, C.; Jehmlich, N. Climate shapes the protein abundance of dominant soil bacteria. Sci. Total. Environ. 2018, 640, 18–21. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.A.; Puissant, J.; Buckeridge, K.; Goodall, T.; Jehmlich, N.; Chowdhury, S.; Gweon, H.S.; Peyton, J.M.; Mason, K.E.; Van Agtmaal, M.; et al. Land use driven change in soil pH affects microbial carbon cycling processes. Nat. Commun. 2018, 9, 3591. [Google Scholar] [CrossRef]
- Schneider, T.; Keiblinger, K.M.; Schmid, E.; Sterflinger-Gleixner, K.; Ellersdorfer, G.; Roschitzki, B.; Richter, A.; Eberl, L.; Zechmeister-Boltenstern, S.; Riedel, K. Who is who in litter decomposition? Metaproteomics reveals major microbial players and their biogeochemical functions. ISME J. 2012, 6, 1749–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rehabilitation Techniques | Description |
|---|---|
| Liming | Study sites were limed, 1.5 t of dolomitic lime per ha, 45 days before planting |
| Planting | Seeds and seedlings from 15 to 20 native species, comprising both pioneer and nonpioneer species, were planted at a planting density of 1700 seedlings/ha |
| Fertilizing | 300 g of NPK fertilizer (4-14-8) enriched with fritted glass material (FTE BR 12 containing the micronutrients S, B, Cu, Mn, and Zn) and 1 kg of dried manure were applied |
| Other | Until canopy closure, the herbaceous layer surrounding the seedlings was mowed every three to four months |
| Study Site | Rehabilitation Stage | Onset of Rehabilitation Activities | Latitude | Longitude |
|---|---|---|---|---|
| NR | Nonrehabilitated | - | −6.09634 | −50.2279 |
| R4 | Early rehabilitation (4 years) | 2014 | −6.09711 | −50.2286 |
| R12 | Intermediate rehabilitation (12 years) | 2006 | −6.09613 | −50.2288 |
| R14 | Advanced rehabilitation (14 years) | 2004 | −6.09821 | −50.2303 |
| NS | Native soil (reference) | - | −6.09821 | −50.2308 |
| Rehabilitation Stage | SOM (%) | LAI | Soil Respiration (µmol CO2 m−2 s−1) |
|---|---|---|---|
| NR | 0.833 ± 0.272 b | 0.274 ± 0.007 d | 1.121 ± 0.354 b |
| R4 | 1.097 ± 0.197 b | 0.579 ± 0.469 d | 4.896 ± 1.985 a |
| R12 | 2.343 ± 0.511 a | 3.105 ± 0.387 c | 2.887 ± 0.710 a,b |
| R14 | 1.917 ± 0.211 a | 4.650 ± 0.217 b | 4.654 ± 1.334 a |
| NS | 2.453 ± 0.133 a | 5.513 ± 0.292 a | 3.690 ± 0.431 a,b |
| Name (EC Number) | Function | Ecological Significance | Rehabilitation Stage |
|---|---|---|---|
| Photosystem I (1.97.1.12) | Carbon fixation | Carbon fixation by cyanobacteria in the poorest SOM content area | NR |
| D-lactate dehydrogenase (1.1.1.28) | Transformation of organic matter | Transformation of organic matter through microbial metabolism | NR |
| (1.1.2.4) | R4 | ||
| Peptidases (3.4.-.-) | Hydrolysis of peptides to amino acids | Mineralization/Nitrogen cycling | NR, R4, R12, R14, NS |
| Cysteine endopeptidases (3.4.22.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | R4, R12 |
| Aminopeptidases (3.4.11.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | R4, R14, NS |
| Serine endopeptidases (3.4.21.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | NR, R4, R12, R14, NS |
| Metallocarboxypeptidases (3.4.17.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | R12, R14 |
| Dipeptidyl-peptidase (3.4.14.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | R12 |
| Metalloendopeptidases (3.4.24.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | R14, NS |
| Omega peptidase (3.4.19.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | NS |
| Aspartic endopeptidase (3.4.23.-) | Hydrolysis of peptides to amino acids | Nitrogen cycling | NS |
| Nitrogenase (1.18.6.1) | Converts N2 to NH3 | Nitrogen fixation in the early rehabilitation area, with low SOM content. Nitrogen cycling. | R4 |
| Beta-amylase (3.2.1.2) | Hydrolysis of (1->4)-alpha-d-glucosidic linkages in polysaccharides | Carbon cycling | R4 |
| Sulfate adenylyltransferase (2.7.7.4) | Participates in assimilatory sulfate reduction | Sulfur cycling | R4 |
| Cellulase (3.2.1.4) | Cellulose degradation with release of glucose | Carbon cycling | R4, NS |
| Alpha-glucosidase (3.2.1.20) | Catalyzes the hydrolysis of o-glycosyl bonds in hemicellulose | Carbon cycling | R4 |
| Glyceraldehyde-3-phosphate dehydrogenase (1.2.1.12) | Transformation of organic matter | Transformation of organic matter through microbial metabolism | R12 |
| Sphingomyelin phosphodiesterase (3.1.4.12) | Phospholipase activity | Phosphorus cycling | R12 |
| Malate dehydrogenase (1.1.1.40) | Transformation of organic matter | Transformation of organic matter through microbial metabolism | R14 |
| Exoribonuclease II (3.1.13.1) | Exonucleolytic cleavage to yield nucleoside 5′-phosphates | Phosphorus cycling | R14, NS |
| Phosphodiesterase I (3.1.4.1) | Hydrolyses both ribonucleotides and deoxyribonucleotides | Phosphorus cycling | R14 |
| Arabinogalactan endo-beta-1,4-galactanase (3.2.1.89) | Hydrolyses (1->4)-beta-d-galactosidic linkages | Carbon cycling | NS |
| Urease (3.5.1.5) | Organic N mineralization to ammonia | Nitrogen cycling | NS |
| Beta-glucosidase (3.2.1.21) | Hydrolyzes maltose and cellobiose to glucose | Carbon cycling | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trindade, F.C.; Gastauer, M.; Ramos, S.J.; Caldeira, C.F.; Araújo, J.F.d.; Oliveira, G.; Valadares, R.B.d.S. Soil Metaproteomics as a Tool for Environmental Monitoring of Minelands. Forests 2021, 12, 1158. https://doi.org/10.3390/f12091158
Trindade FC, Gastauer M, Ramos SJ, Caldeira CF, Araújo JFd, Oliveira G, Valadares RBdS. Soil Metaproteomics as a Tool for Environmental Monitoring of Minelands. Forests. 2021; 12(9):1158. https://doi.org/10.3390/f12091158
Chicago/Turabian StyleTrindade, Felipe Costa, Markus Gastauer, Silvio Junio Ramos, Cecílio Frois Caldeira, Josiney Farias de Araújo, Guilherme Oliveira, and Rafael Borges da Silva Valadares. 2021. "Soil Metaproteomics as a Tool for Environmental Monitoring of Minelands" Forests 12, no. 9: 1158. https://doi.org/10.3390/f12091158