The Use of DNA Barcoding to Assess Phylogenetic β-Diversity in Mid-Subtropical Evergreen Broad-Leaved Forests of China


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Materials
2.2. Measures of Phylogenetic β-Diversity
2.3. Patterns of Phylogenetic Dissimilarity in Different Mountains
2.4. Quantifying the Relative Importance of Explanatory Variables
3. Results
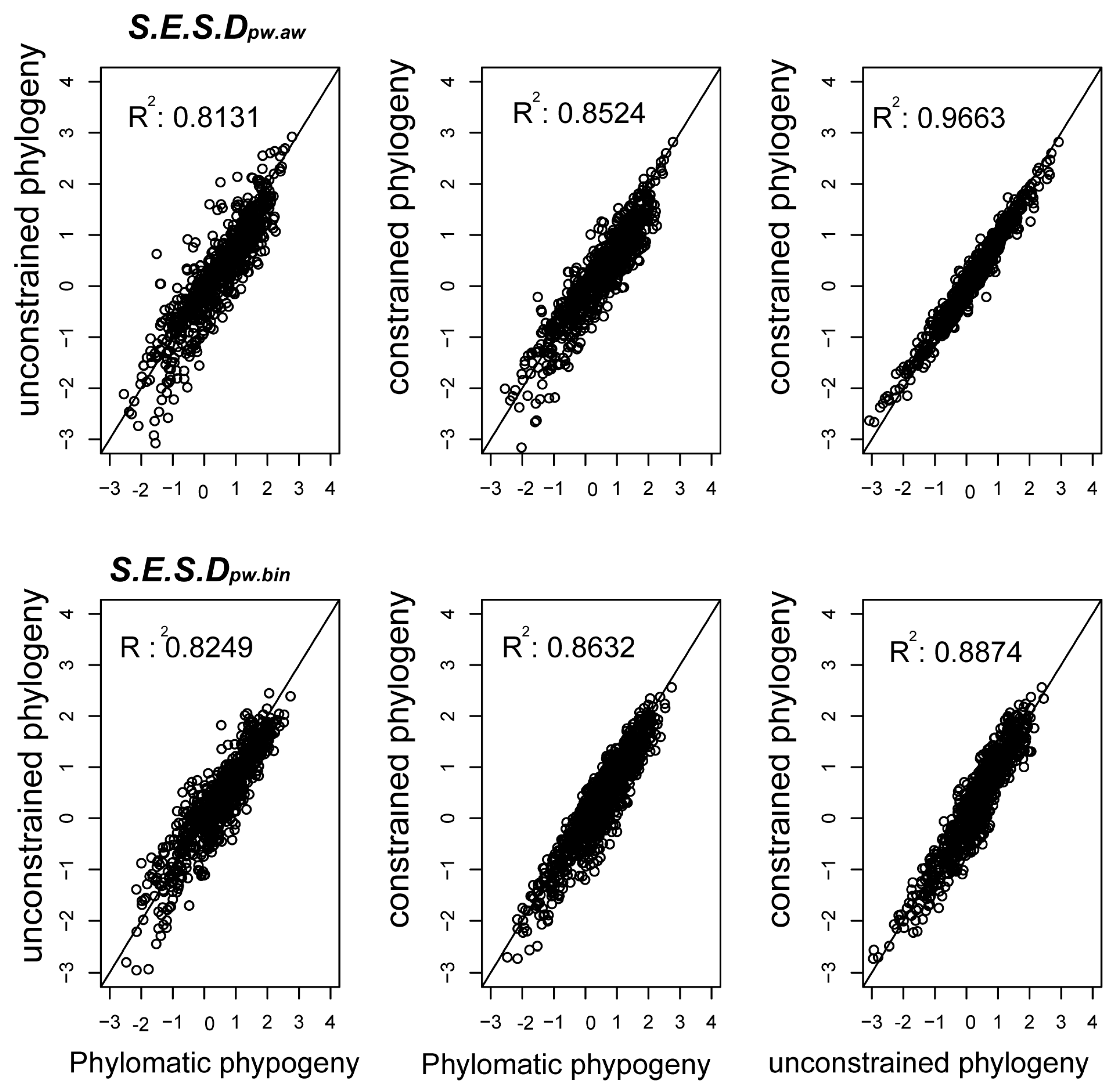
3.1. Assessment of Phylogenetic β-Diversity
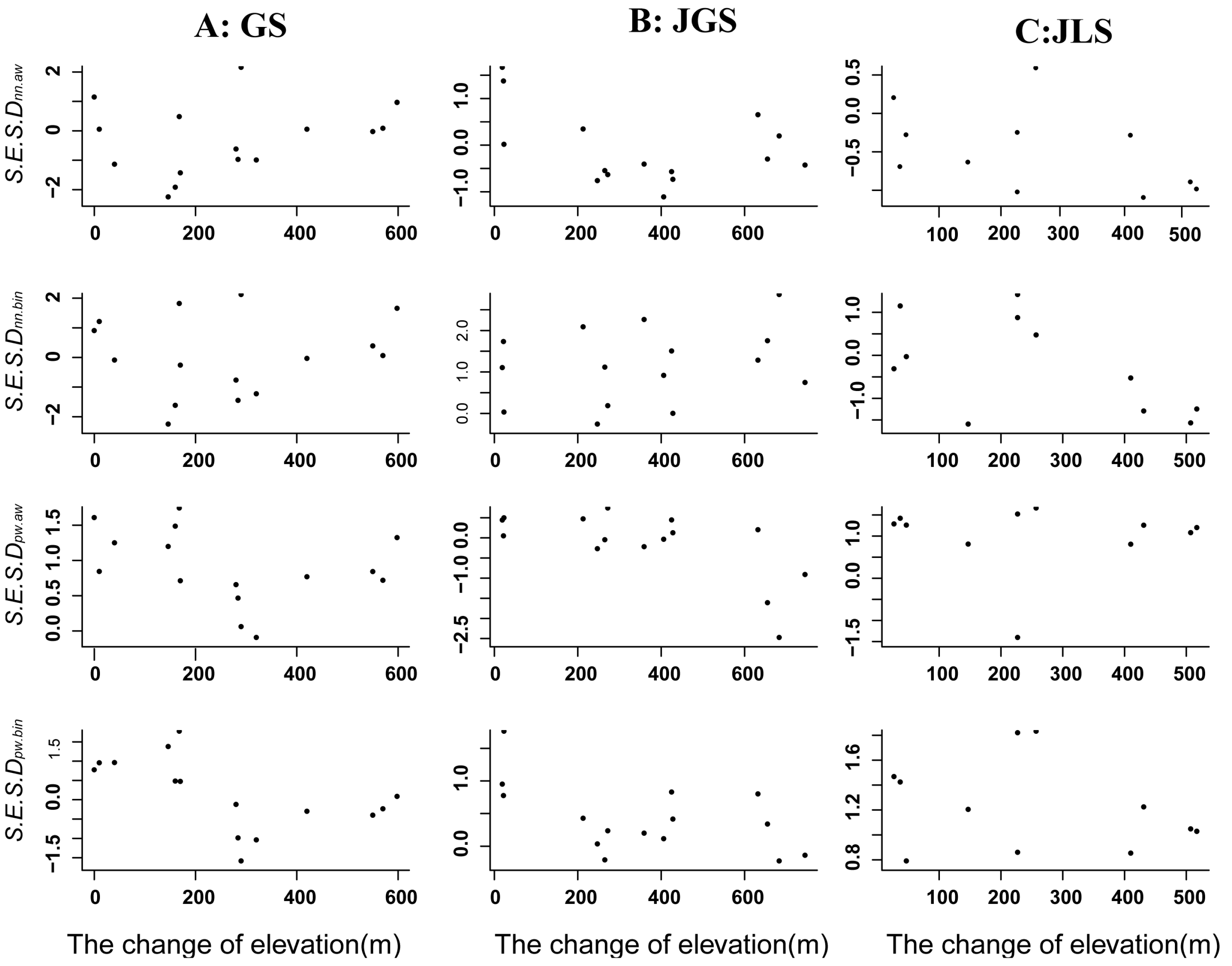
3.2. Patterns of Phylogenetic β-Diversity of Evergreen Broad Assemblages along Latitudinal and Altitudinal Gradients in Sub-Tropical Forests
3.3. Relative Importance of Environmental Distance, Climatic Distance, and Geographical Distance on Phylogenetic β-Diversity
4. Discussion
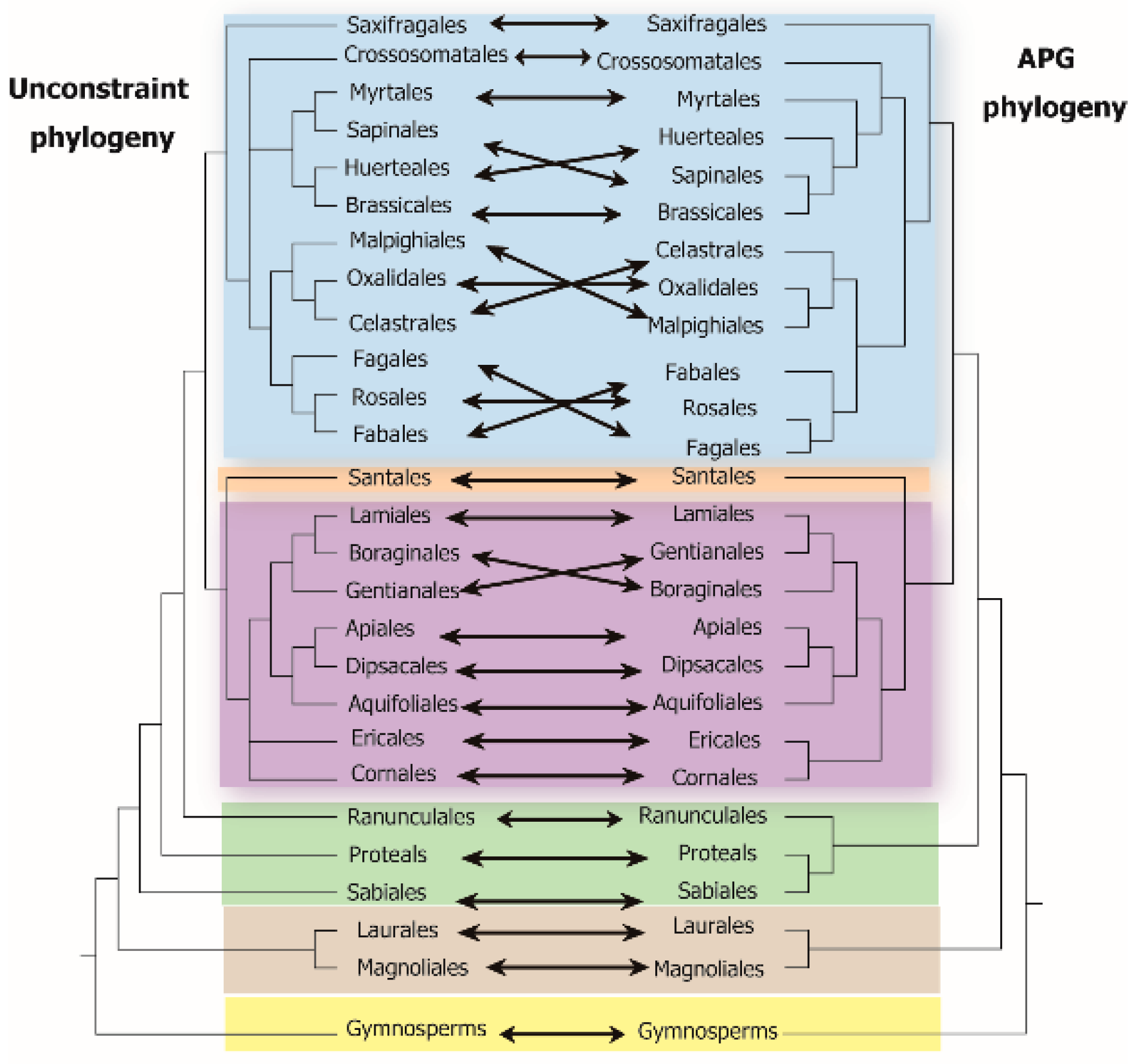
4.1. Assessment of Phylogenetic β-Diversity Based on Different Phylogenetic Methods
4.2. Patterns of Phylogenetic Turnover along Latitudinal and Altitudinal Gradients in Subtropical Forests
4.3. Relative Contribution of Geographical and Environmental Distances to Phylogenetic β-Diversity
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keddy, P.A. Assembly and response rules: Two goals for predictive community ecology. J. Veg. Sci. 1992, 3, 157–164. [Google Scholar] [CrossRef]
- Kubota, Y.; Hirao, T.; Fujii, S.J.; Shiono, T.; Kusumoto, B. Beta diversity of woody plants in the Japanese archipelago: The roles of geohistorical and ecological processes. J. Biogeogr. 2014, 41, 1267–1276. [Google Scholar] [CrossRef]
- Hawkins, B.A.; Porter, E.E. Relative influences of current and historical factors on mammal and bird diversity patterns in deglaciated North America. Glob. Ecol. Biogeogr. 2003, 12, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Willis, K.J.; Niklas, K.J. The Role of Quaternary Environmental Change in Plant Macroevolution: The Exception or the Rule? Philos. Trans. R. Soc. B 2004, 359, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, M.; Alessio, B.; Enzo, B.; Germano, F.; Fabrizio, M.; Filippo, P.; Riccardo, B. Beta-diversity patterns elucidate mechanisms of alien plant invasion in mountains. Glob. Ecol. Biogeogr. 2013, 22, 450–460. [Google Scholar]
- Qian, H.; Ricklefs, R.E. A latitudinal gradient in large-scale beta diversity for vascular plants in North America. Ecol. Lett. 2007, 10, 737–744. [Google Scholar] [CrossRef]
- Legendre, P.; Borcard, D.; Peres-Neto, P.R. Analyzing beta diversity: Partitioning the spatial variation of community composition data. Ecol. Monogr. 2005, 75, 435–450. [Google Scholar] [CrossRef]
- Swenson, N.G. Phylogenetic beta diversity metrics, trait evolution and inferring the functional beta diversity of communities. PLoS ONE 2011, 6, e21264. [Google Scholar] [CrossRef]
- Xing, D.; He, F. Environmental filtering explains a U-shape latitudinal pattern in regional β-deviation for eastern North American trees. Ecol. Lett. 1111, 22, 284–291. [Google Scholar] [CrossRef]
- Webb, C.O. Exploring the phylogenetic structure of ecological communities: An example for rain forest trees. Am. Nat. 2000, 156, 145–155. [Google Scholar] [CrossRef]
- Webb, C.O.; Ackerly, D.D.; McPeek, M.A.; Donoghue, M.J. Phylogenies and community ecology. Annu. Rev. Ecol. Evol. Syst. 2002, 33, 475–505. [Google Scholar] [CrossRef]
- Qian, H.; Zhang, Y.; Zhang, J.; Wang, X. Latitudinal gradients in phylogenetic relatedness of angiosperm trees in North America. Glob. Ecol. Biogeogr. 2013, 22, 1183–1191. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, G.; Ci, X.; Swenson, N.G.; Cao, M.; Sha, L.; Li, J.; Baskin, C.C.; Slik, J.; Lin, L. Functional and phylogenetic assembly in a Chinese tropical tree community across size classes, spatial scales and habitats. Funct. Ecol. 2014, 28, 520–529. [Google Scholar] [CrossRef]
- McFadden, L.R.; Sandel, B.; Tsirogiannis, C.; Morueta-Holme, N.; Svenning, J.C.; Enquist, B.J.; Kraft, N.J.B. Temperature shapes opposing latitudinal gradients of plant taxonomic and phylogenetic beta diversity. Ecol. Lett. 2019, 22, 112–1135. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Hao, Z.; Zhang, J. Phylogenetic structure and phylogenetic diversity of angiosperm assemblages in forests along an elevational gradient in Changbaishan, China. J. Plant Ecol. 2014, 7, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Swenson, N.G.; Zhang, J.; Ma, K. The environment and space, not phylogeny, determine trait dispersion in a subtropical forest. Funct. Ecol. 2013, 27, 264–272. [Google Scholar] [CrossRef]
- Chi, X.; Tang, Z.; Fang, J. Patterns of phylogenetic beta diversity in China’s grasslands in relation to geographical and environmental distance. Basic Appl. Ecol. 2014, 15, 416–425. [Google Scholar] [CrossRef]
- González-Caro, S.; Umaña, M.N.; Álvarez, E.; Stevenson, P.R.; Swenson, N.G. Phylogenetic alpha and beta diversity in tropical tree assemblages along regional-scale environmental gradients in northwest South America. J. Plant Ecol. 2014, 7, 145–153. [Google Scholar] [CrossRef]
- Zhang, J.L.; Swenson, N.G.; Chen, S.B.; Liu, X.J.; Li, Z.S.; Huang, J.H.; Mi, X.C.; Ma, K.P. Phylogenetic beta diversity in tropical forests: Implications for the roles of geographical and environmental distance. J. Syst. Evol. 2013, 51, 71–85. [Google Scholar] [CrossRef]
- Lahaye, R.; van der Bank, M.; Bogarin, D.; Warner, J.; Pupulin, F.; Gigot, G.; Maurin, O.; Duthoit, S.; Barraclough, T.G.; Savolainen, V. From the Cover: DNA barcoding the floras of biodiversity hotspots. Proc. Natl. Acad. Sci. USA 2008, 105, 2923–2928. [Google Scholar] [CrossRef]
- Pei, N.C.; Lian, J.Y.; Erickson, D.L.; Swenson, N.G.; Kress, W.J.; Ye, W.H.; Ge, X.J. Exploring tree-habitat associations in a Chinese subtropical forest plot using a molecular phylogeny generated from DNA barcode loci. PLoS ONE 2011, 6, e21273. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L.; Jones, F.A.; Swenson, N.G.; Perez, R.; Sanjur, O.; Bermingham, E. Plant DNA barcodes and a community phylogeny of a tropical forest dynamics plot in Panama. Proc. Natl. Acad. Sci. USA 2009, 106, 18621–18626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costion, C.; Ford, A.; Cross, H.; Crayn, D.; Harrington, M.; Lowe, A. Plant DNA barcodes can accurately estimate species richness in poorly known floras. PLoS ONE 2011, 6, e26841. [Google Scholar] [CrossRef] [PubMed]
- Erickson, D.L.; Jones, F.A.; Swenson, N.G.; Pei, N.C.; Bourg, N.A.; Chen, W.; Davies, S.J.; Ge, X.J.; Hao, Z.; Howe, R.W. Comparative evolutionary diversity and phylogenetic structure across multiple forest dynamics plots: A mega-phylogeny approach. Front. Genet. 2014, 5, 358. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yan, H.F.; Newmaster, S.G.; Pei, N.C.; Ragupathy, S.; Ge, X.J. The use of DNA barcoding as a tool for the conservation biogeography of subtropical forests in China. Divers. Distrib. 2015, 21, 188–199. [Google Scholar] [CrossRef]
- Huang, X.C.; Ci, X.Q.; Conran, J.G.; Li, J. Application of DNA barcodes in Asian tropical trees–A case study from Xishuangbanna nature reserve, Southwest China. PLoS ONE 2015, 10, e0129295. [Google Scholar] [CrossRef]
- Tripathi, A.M.; Tyagi, A.; Kumar, A.; Singh, A.; Singh, S.; Chaudhary, L.B.; Roy, S. The internal transcribed spacer (its) region and trnH-psbA are suitable candidate loci for DNA barcoding of tropical tree species of India. PLoS ONE 2013, 8, e57934. [Google Scholar] [CrossRef]
- Liu, J.; Yan, H.F.; Ge, X.J. The use of DNA barcoding on recently diverged species in the genus Gentiana (Gentianaceae) in China. PLoS ONE 2016, 11, e0153008. [Google Scholar] [CrossRef]
- Li, D.Z.; Gao, L.M.; Li, H.T.; Wang, H.; Ge, X.J.; Liu, J.Q.; Chen, Z.D.; Zhou, S.L.; Chen, S.L.; Yang, J.B.; et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19641–19646. [Google Scholar] [Green Version]
- Yan, L.J.; Liu, J.; Möller, M.; Zhang, L.; Zhang, X.M.; Li, D.Z.; Gao, L.M. DNA barcoding of Rhododendron (Ericaceae), the largest Chinese plant genus in biodiversity hotspots of the Himalaya–Hengduan Mountains. Mol. Ecol. Resour. 2014, 15, 932–944. [Google Scholar] [CrossRef]
- Wang, X.H.; Kent, M.; Fang, X.F. Evergreen broad-leaved forest in Eastern China: Its ecology and conservation and the importance of resprouting in forest restoration. For. Ecol. Manag. 2007, 245, 76–87. [Google Scholar] [CrossRef]
- Condit, R.; Pitman, N.; Leigh, E.G.; Chave, J.; Terborgh, J.; Foster, R.B.; Nunez, V.P.; Aguilar, S.; Valencia, R.; Villa, G.; et al. Beta-diversity in tropical forest trees. Science 2002, 925, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Legendre, P.; Mi, X.C.; Ren, H.B.; Ma, K.P.; Yu, M.J.; Sun, I.F.; He, F.L. Partitioning beta diversity in a subtropical broad-leaved forest of China. Ecology 2009, 90, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Qiao, X.J.; Jiang, Q.H.; Jiang, Q.H.; Lu, J.M.; Franklin, S.; Tang, Z.Y.; Wang, Q.G.; Zhang, J.X.; Lu, Z.J.; Bao, D.C.; et al. Beta diversity determinants in Badagongshan, a subtropical forest in central China. Sci. Rep. 2015, 5, 17403. [Google Scholar] [CrossRef]
- Tian, S.; Kou, Y.X.; Zhang, Z.R.; Yuan, L.; Li, D.R.; López-Pujol, J.; Fan, D.M.; Zhang, Z.Y. Phylogeography of Eomecon chionantha in subtropical China: The dual roles of the Nanling Mountains as a glacial refugium and a dispersal corridor. BMC Evol. Biol. 2018, 18, 20. [Google Scholar] [CrossRef]
- Muellner-Riehl, A.N. Mountains as evolutionary arenas: Patterns, emerging approaches, paradigm shifts, and their implications for plant phylogeographic research in the Tibeto-Himalayan region. Front. Plant Sci. 2019, 10, 195. [Google Scholar] [CrossRef]
- Song, Y.C.; Da, L.J. Evergreen broad-leaved forest of East Asia. In Vegetation Structure and Function at Multiple Spatial, Temporal and Conceptual Scales; Box, E.O., Ed.; Springer International: Switzerland, The Netherlands, 2016; Volume 1, pp. 101–128. [Google Scholar] [CrossRef]
- Fang, J.Y.; Wang, X.P.; Tang, Z.Y.; Shen, Z.H.; Zheng, C.Y. Exploring patterns of plant diversity in China’s mountains. In Data Mining for Global Trends in Mountain Biodiversity; Box, E.O., Ed.; Springer International: Switzerland, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Wan, S.; Zhang, C.; Chen, Y.; Zhao, J.; Wang, X.; Wu, J.; Zhou, L.; Lin, Y.; Liu, Z.; Fu, S. The understory fern Dicranopteris dichotomafacilitates the overstory Eucalyptus trees in subtropical plantations. Ecosphere 2014, 5, 51. [Google Scholar] [CrossRef]
- Doyle, J.L.; Doyle, J.L. A rapid DNA isolate procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Zane, A.E.; Tank, D.C.; Cornwell, W.K. Eastman, J.M.; Smith, S.A.; FitzJohh, R.G.; McGlinn, D.J.; O’Meara, B.C.; Moles, A.T.; Reich, P.B.; et al. Three keys to the radiation of angiosperms into freezing environment. Nature 2014, 506, 89–92. [Google Scholar] [CrossRef]
- Kress, W.J.; Erickson, D.L.; Swenson, N.G.; Thompson, J.; Uriarte, M.; Zimmerman, J.K.; Chave, J. Advances in the use of DNA barcodes to build a community phylogeny for tropical trees in a Puerto Rican forest dynamics plot. PLoS ONE 2010, 5, e15409. [Google Scholar] [CrossRef]
- Miller, A.M.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LO, USA, 14 November 2010. [Google Scholar] [CrossRef]
- Dariba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Britton, T.; Oxelman, B.; Vinnersten, A.; Bremer, K. Phylogenetic dating with confidence intervals using mean path lengths. Mol. Phylogenet. Evol. 2012, 24, 58–65. [Google Scholar] [CrossRef]
- Swenson, N.G.; Erickson, D.L.; Mi, X.; Bourg, N.A.; Forero-Montaña, J.; Ge, X.; Howe, R.; Lake, J.K.; Liu, X.; Ma, K. Phylogenetic and functional alpha and beta diversity in temperate and tropical tree communities. Ecology 2012, 93, S112–S125. [Google Scholar] [CrossRef] [Green Version]
- Kubota, Y.; Kusumoto, B.; Shiono, T.; Ulrich, W. Environmental filters shaping angiosperm trees assembly along climatic and geographical gradients. J. Veg. Sci. 2018, 29, 1–12. [Google Scholar] [CrossRef]
- Massante, J.C.; Götzenberger, L.; Takkis, K.; Hallikma, T.; Kaasik, A.; Laanisto, L.; Hutchings, M.; Gerhold, P. Contrasting latitudinal patterns in phylogenetic diversity between woody and herbaceous communities. Sci. Rep. 2019, 9, 6443. [Google Scholar] [CrossRef]
- Heckenhauer, J.; Salim, K.A.; Chase, M.W.; Dexter, K.G.; Pennington, R.T.; Tan, S.; Kaye, M.E.; Samuel, R. Plant DNA barcodes and assessment of phylogenetic community structure of a tropical mixed dipterocarp forest in Brunei Darussalam (Borneo). PLoS ONE 2017, 12, e0185861. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, J.; Shan, Y.X.; Ge, X.J.; Burgess, K.S. The use of DNA barcodes to estimate phylogenetic diversity in forest communities of southern China. Ecol. Evol. 2019, 9, 5372–5379. [Google Scholar] [CrossRef]
- Qian, H.; Ricklefs, R.E.; White, P.S. Beta diversity of angiosperms in temperate floras of eastern Asia and eastern North America. Ecol. Lett. 2005, 8, 15–22. [Google Scholar] [CrossRef]
- Alexander, J.M.; Kueffer, C.; Daehler, C.C.; Edwards, P.J.; Pauchard, A.; Seipel, T.; Consortium, M.; Arévalo, J.; Cavieres, L.; Dietz, H. Assembly of nonnative floras along elevational gradients explained by directional ecological filtering. Proc. Natl. Acad. Sci. USA 2011, 108, 656–661. [Google Scholar] [CrossRef]
- Haider, S.; Kueffer, C.; Bruelheide, H.; Seipel, T.; Alexander, J.M.; Rew, L.J.; Arévalo, J.R.; Cavieres, L.A.; McDougall, K.L.; Milbau, A.; et al. Mountain roads and non-native species modify elevational patterns of plant diversity. Global Ecol. Biogeogr. 2018, 27, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Fang, J.; Chi, X.; Feng, J.; Liu, Y.; Shen, Z.; Wang, X.; Wang, Z.; Wu, X.; Zheng, C. Patterns of plant beta-diversity along elevational and latitudinal gradients in mountain forests of China. Ecography 2012, 35, 1083–1091. [Google Scholar] [CrossRef]
- Tang, Z.Y.; Fang, J.Y. Temperature variation along the northern and southern slopes of Mt. Taibai, China. Agric. For. Meteorol. 2006, 139, 200–207. [Google Scholar] [CrossRef]
- Deng, T.; Zhou, Z.; Sun, H.; Chen, Y.-S. Is the East Asian flora ancient or not? Natl. Sci. Rev. 2018, 5, 920–932. [Google Scholar] [CrossRef]
- Swenson, N.G.; Enquist, B.J.; Pither, J.; Thompson, J.; Zimmerman, J.K. The problem and promise of scale dependency in community phylogenetics. Ecology 2006, 87, 2418–2424. [Google Scholar] [CrossRef]
- Kraft, N.J.; Comita, L.S.; Chase, J.M.; Sanders, N.J.; Swenson, N.G.; Crist, T.O.; Stegen, J.C.; Vellend, M.; Boyle, B.; Anderson, M.J. Disentangling the drivers of β-diversity along latitudinal and elevational gradients. Science 2011, 333, 1755–1758. [Google Scholar] [CrossRef]
- Wang, Z.; Fang, J.; Tang, Z.; Shi, L. Geographical patterns in the beta diversity of China’s woody plants: The influence of space, environment and range size. Ecography 2012, 35, 1092–1102. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Climate | Soil | |||||
|---|---|---|---|---|---|---|
| PC1 | PC2 | PC3 | PC1 | PC2 | PC3 | |
| Eigenvalue | 3.545 | 1.362 | 0.089 | 2.657 | 1.566 | 0.3903 |
| Proportion of variation | 70.89 | 27.24 | 1.799 | 53.13 | 31.33 | 7.810 |
| Cumulative % of variance | 70.89 | 98.13 | 99.93 | 53.13 | 84.46 | 92.26 |
| Geographical Distance | Change of Soil | Change of Elevation | Change of Climate | |
|---|---|---|---|---|
| Dnn abundance-weighted | 0.0175 ** | 0.2870 | 0.0927 | 0.1534 * |
| Dnn presence-absence | 0.1449 ** | 0.1953 * | 0.0895 | 0.1701 ** |
| Dpw abunance weighted | 0.1620 ** | 0.1766 * | 0.1420 * | 0.1593 ** |
| Dpw presence-absence | 0.2301 ** | 0.2870 * | 0.0855 | 0.1626 ** |
| S.E.S. Dnn.aw | S.E.S. Dnn.bin | S.E.S. Dpw.aw | S.E.S. Dpw.bin | |
|---|---|---|---|---|
| Geographical distance | 6.100 ** | 2.706 ** | 0.260 | 0.579 * |
| Edaphic distance | 0.2280 | 0.067 | 2.480 ** | 1.857 ** |
| Climatic distance | 3.445 ** | 0.824 ** | 0.039 | 1.485 ** |
| Geographical distance + edaphic distance | 0.39 | 0.101 | 0.793 ** | 0.044 |
| Geographical distance + climatic distance | 0.008 | 0.036 | 1.812 ** | 0.895 ** |
| Edaphic distance + climatic distance | 1.885 ** | 1.046 * | 0.624 * | 0.420 * |
| Geographical distance + edaphic distance + climatic distance | 0.03 | 0.297 | 0.133 | 0.032 |
| Residuals | 88.02 | 94.922 | 93.86 | 94.69 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, J.; Liu, J.; Ge, X.; Huang, G.; Zhou, Z.; Wan, S. The Use of DNA Barcoding to Assess Phylogenetic β-Diversity in Mid-Subtropical Evergreen Broad-Leaved Forests of China. Forests 2019, 10, 923. https://doi.org/10.3390/f10100923
Liu J, Liu J, Ge X, Huang G, Zhou Z, Wan S. The Use of DNA Barcoding to Assess Phylogenetic β-Diversity in Mid-Subtropical Evergreen Broad-Leaved Forests of China. Forests. 2019; 10(10):923. https://doi.org/10.3390/f10100923
Chicago/Turabian StyleLiu, Juan, Jiajia Liu, Xuejun Ge, Guomin Huang, Zengliang Zhou, and Songze Wan. 2019. "The Use of DNA Barcoding to Assess Phylogenetic β-Diversity in Mid-Subtropical Evergreen Broad-Leaved Forests of China" Forests 10, no. 10: 923. https://doi.org/10.3390/f10100923