
Hydrogen Dissociation Reaction on First-Row Transition Metal Doped Nanobelts
 , ,
, ,
Abstract
:1. Introduction
2. Methodology
3. Results and Discussion
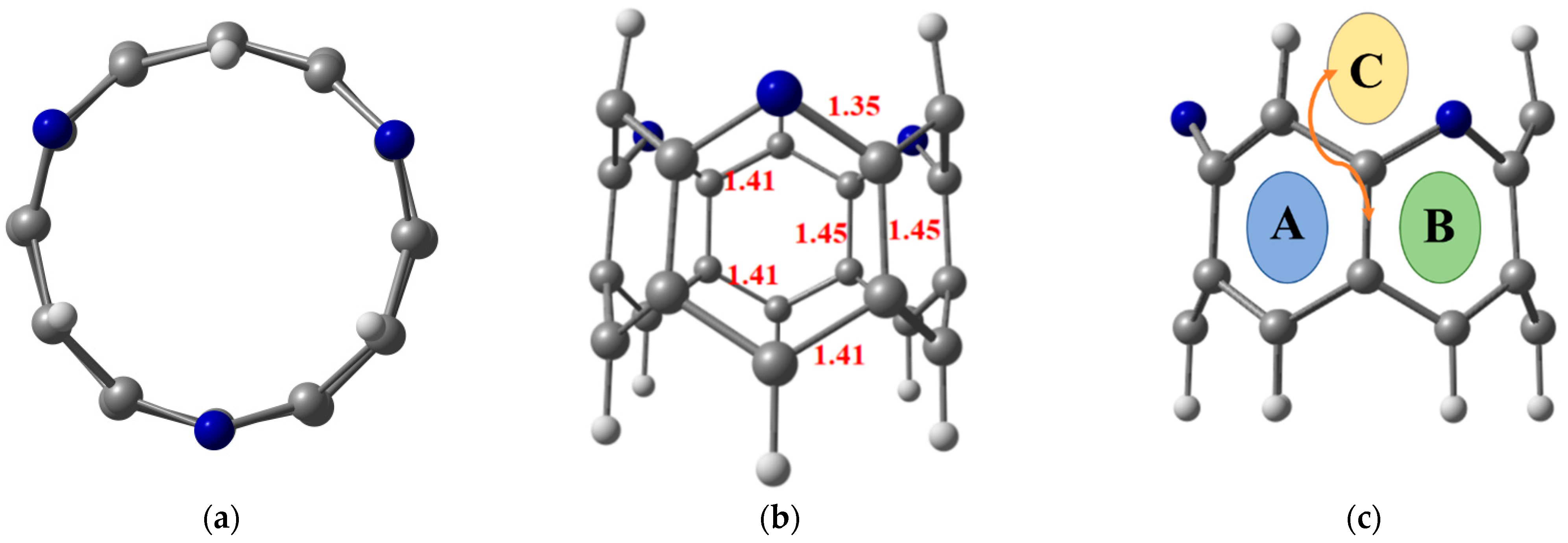
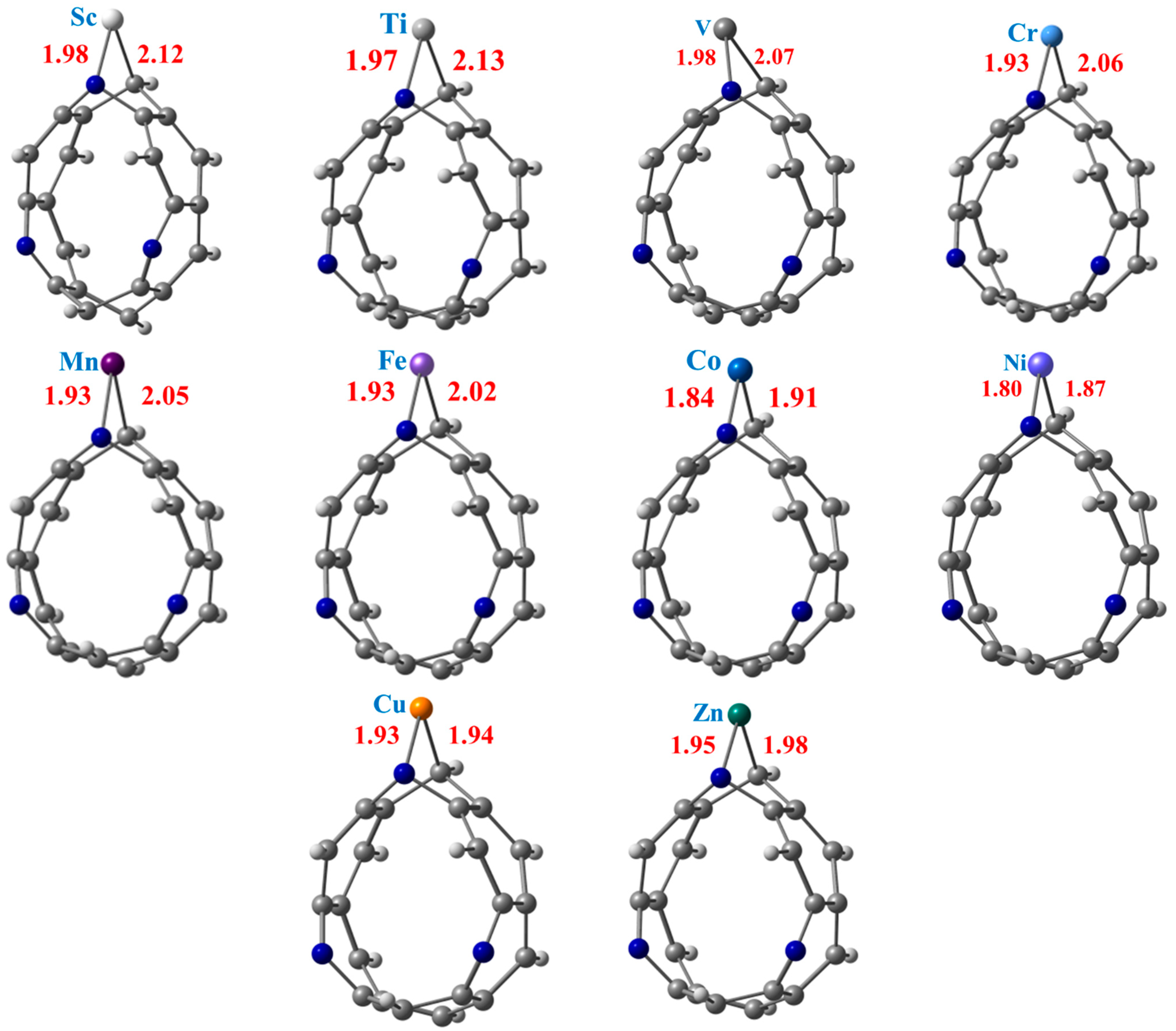
3.1. Geometry Optimization and Adsorption Energy
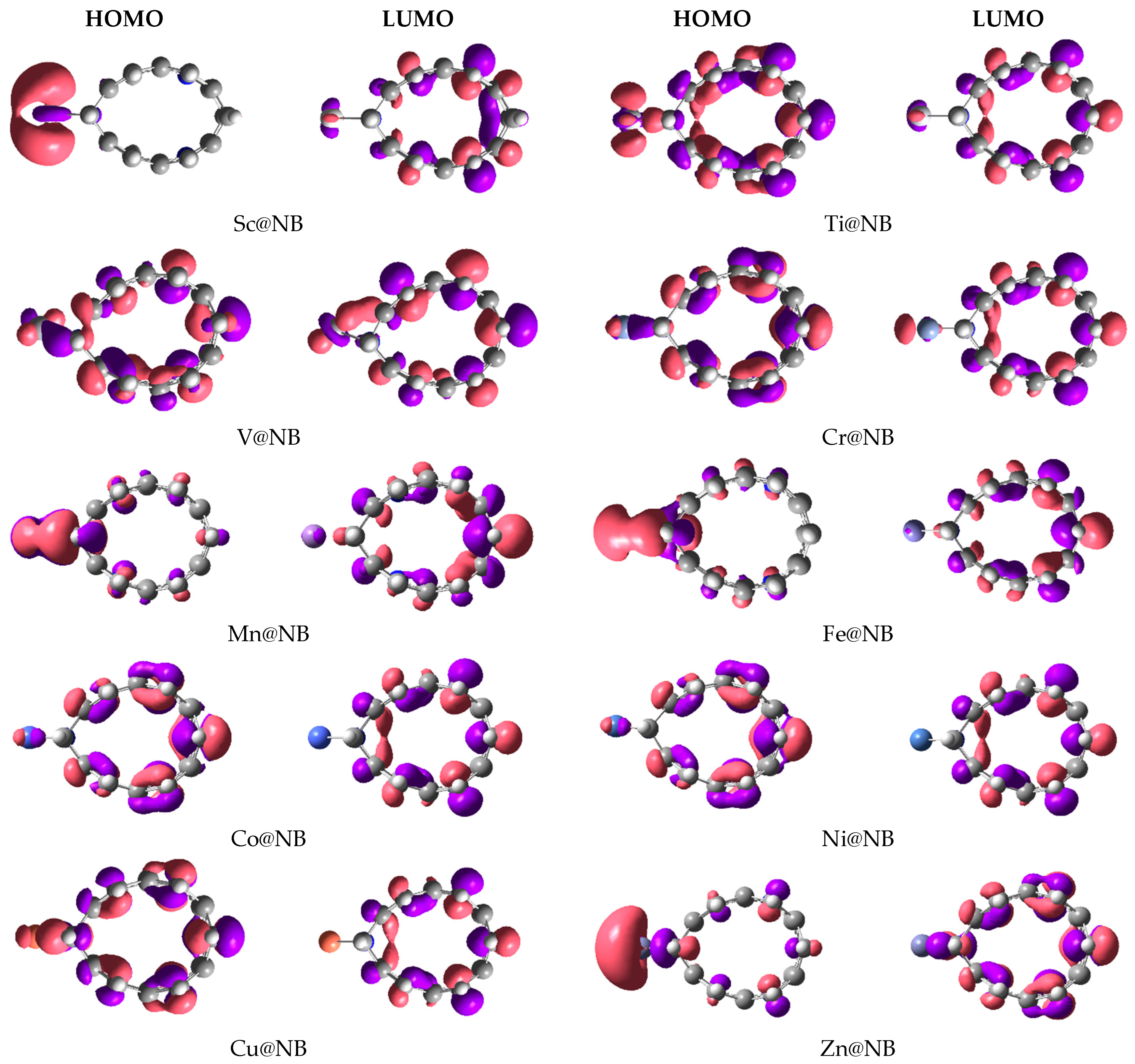
3.2. Electronic Properties of TM@NB Complexes
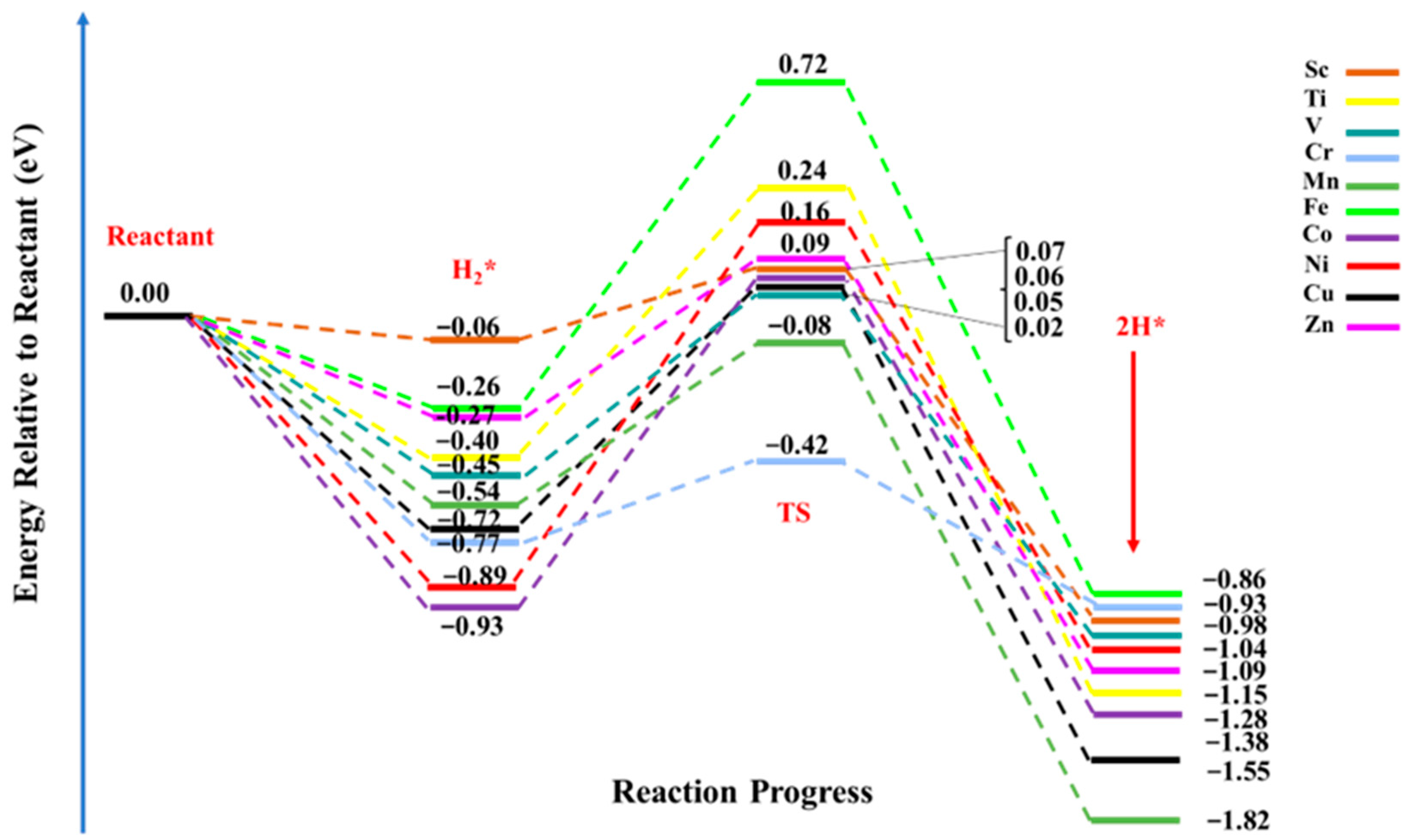
3.3. Hydrogen Molecule Adsorption over TM@NB Complexes
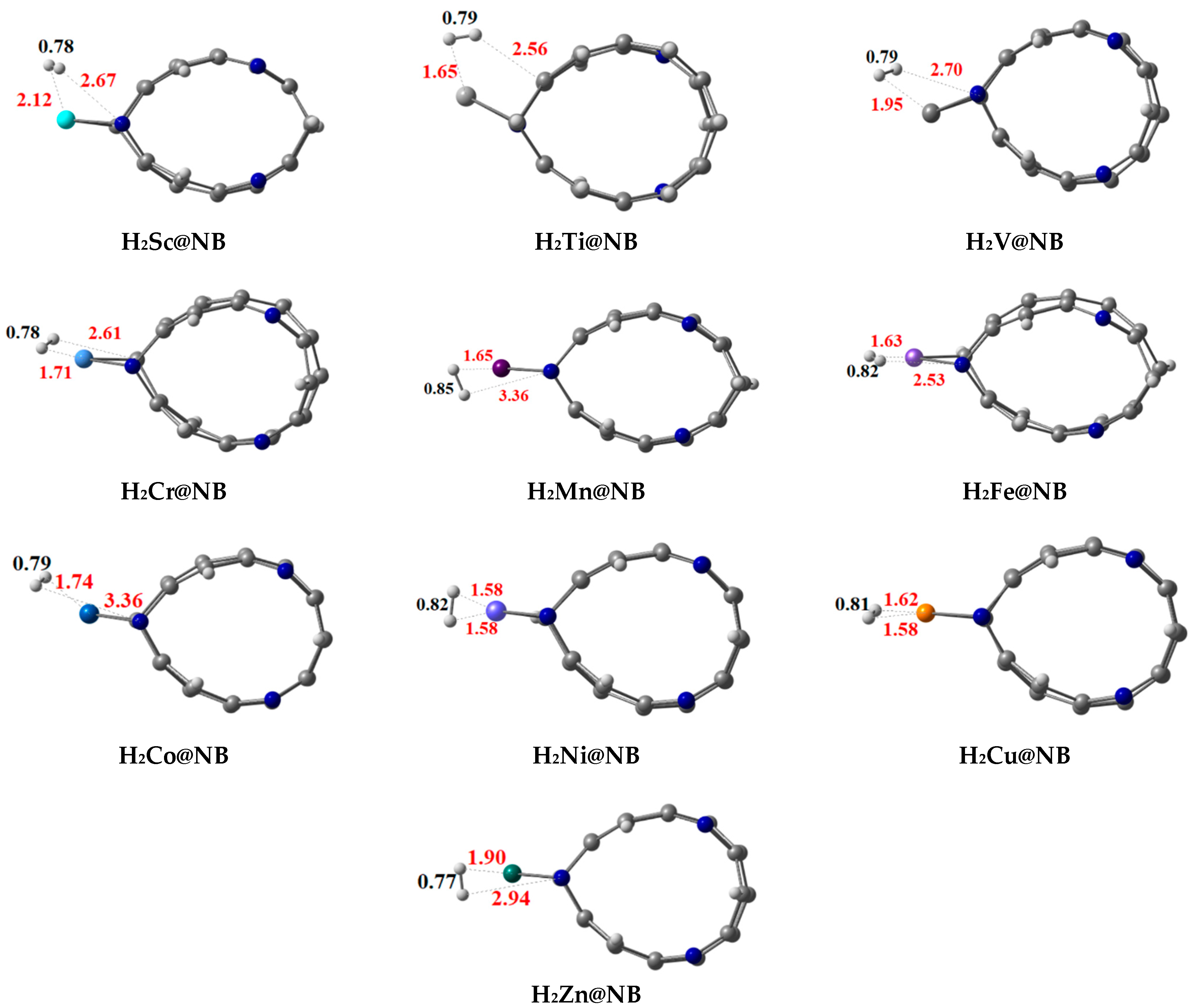
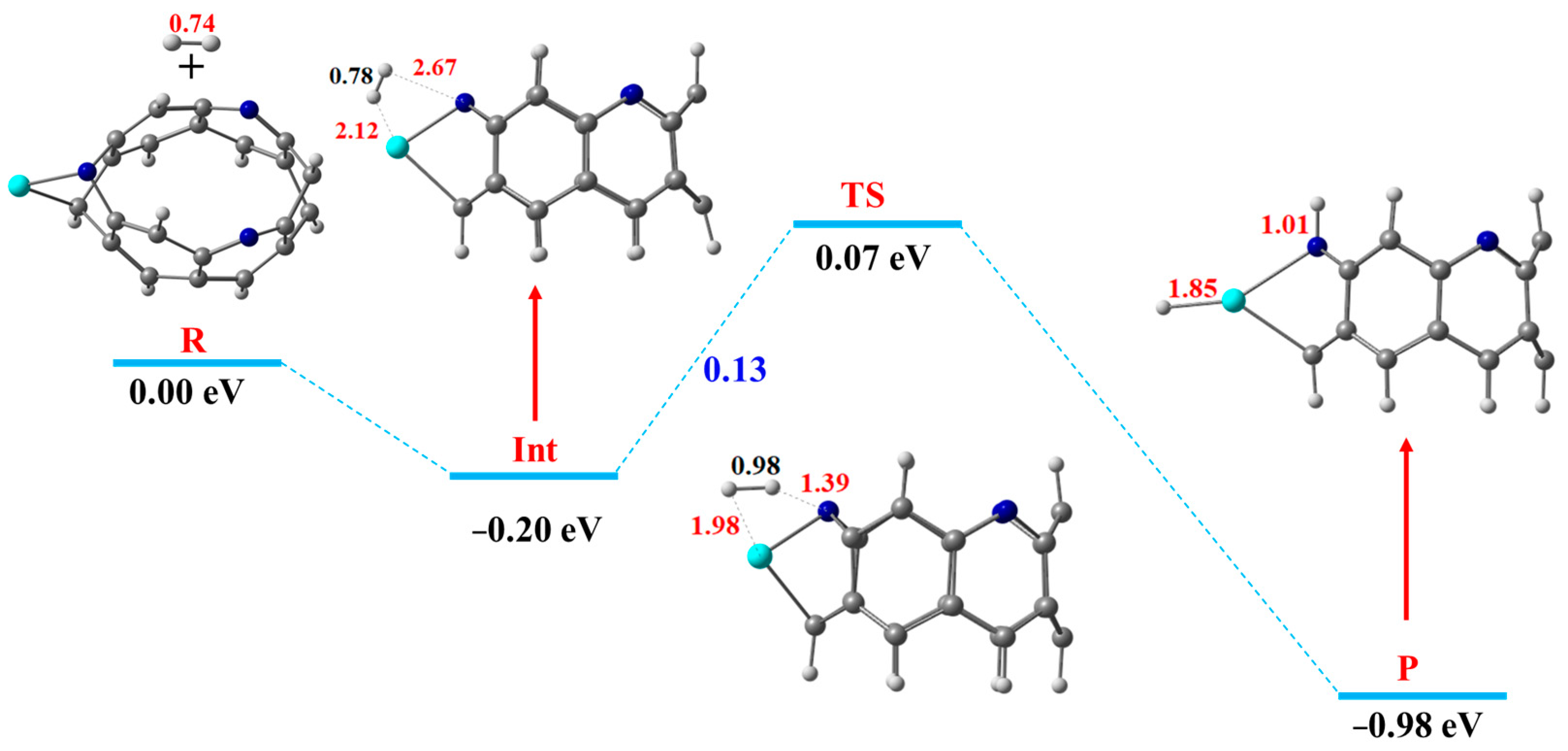
3.4. Dissociation of the Hydrogen Molecule over TM@NB Complexes
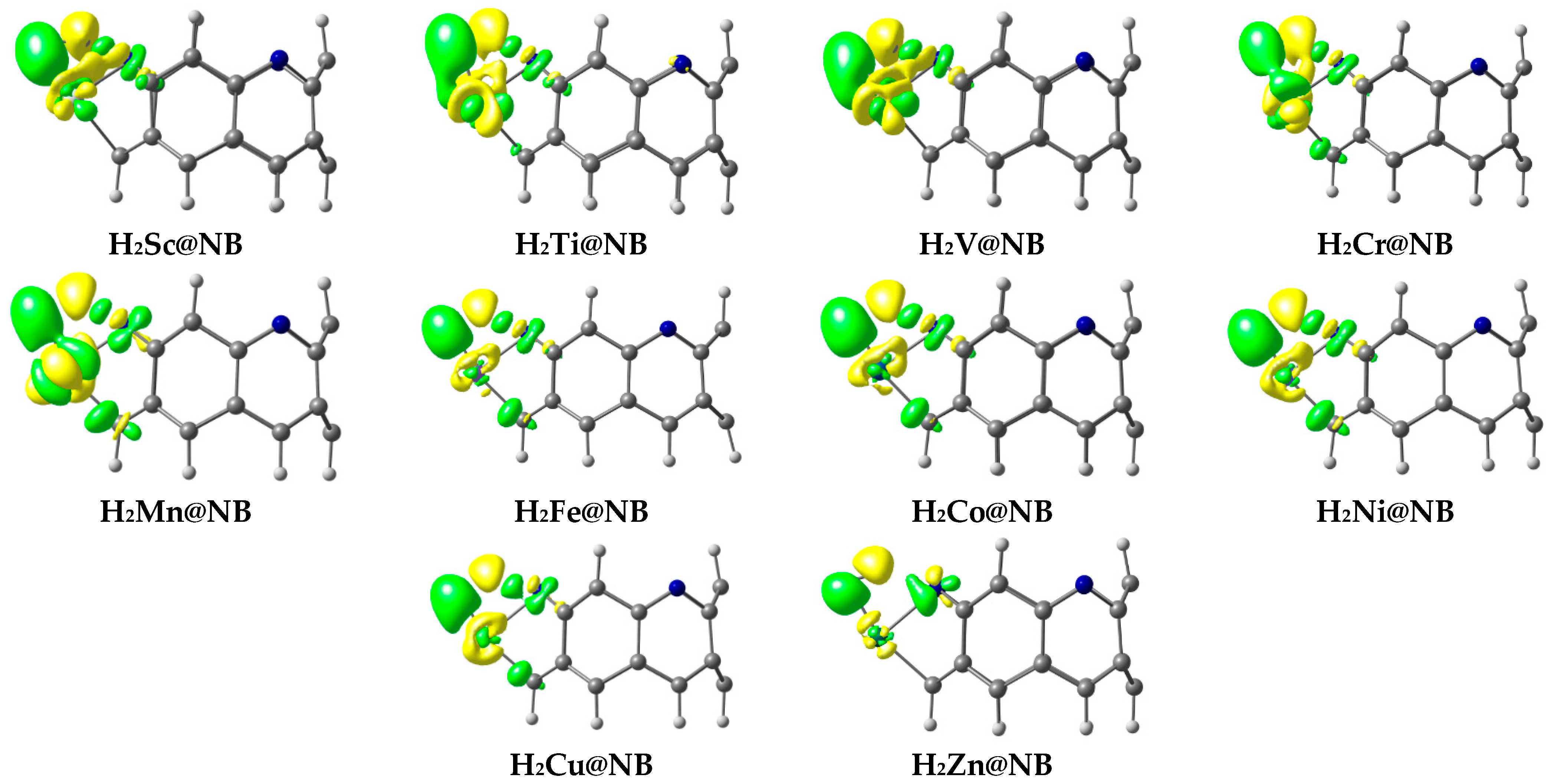
3.5. NBO and EDD Analyses
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arsad, A.Z.; Hannan, M.; Al-Shetwi, A.Q.; Mansur, M.; Muttaqi, K.; Dong, Z.; Blaabjerg, F. Hydrogen energy storage integrated hybrid renewable energy systems: A review analysis for future research directions. Int. J. Hydrogen Energy 2022, 47, 17285–17312. [Google Scholar] [CrossRef]
- Rosen, M.A.; Koohi-Fayegh, S. The prospects for hydrogen as an energy carrier: An overview of hydrogen energy and hydrogen energy systems. Energy Ecol. Environ. 2016, 1, 10–29. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zhao, P.; Niu, M.; Maddy, J. The survey of key technologies in hydrogen energy storage. Int. J. Hydrogen Energy 2016, 41, 14535–14552. [Google Scholar] [CrossRef]
- Yanxing, Z.; Maoqiong, G.; Yuan, Z.; Xueqiang, D.; Jun, S. Thermodynamics analysis of hydrogen storage based on compressed gaseous hydrogen, liquid hydrogen and cryo-compressed hydrogen. Int. J. Hydrogen Energy 2019, 44, 16833–16840. [Google Scholar] [CrossRef]
- Niaz, S.; Manzoor, T.; Pandith, A.H. Hydrogen storage: Materials, methods and perspectives. Renew. Sustain. Energy Rev. 2015, 50, 457–469. [Google Scholar] [CrossRef]
- Rusman, N.A.A.; Dahari, M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications. Int. J. Hydrogen Energy 2016, 41, 12108–12126. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, C.; McClain, M.J.; Manjavacas, A.; Krauter, C.M.; Tian, S.; Berg, F.; Everitt, H.O.; Carter, E.A.; Nordlander, P. Aluminum nanocrystals as a plasmonic photocatalyst for hydrogen dissociation. Nano Lett. 2016, 16, 1478–1484. [Google Scholar] [CrossRef]
- Sun, M.; Nelson, A.E.; Adjaye, J. Ab initio DFT study of hydrogen dissociation on MoS2, NiMoS, and CoMoS: Mechanism, kinetics, and vibrational frequencies. J. Catal. 2005, 233, 411–421. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Akita, T.; Okumura, M.; Haruta, M. Hydrogen dissociation by gold clusters. Angew. Chem. Int. Ed. 2009, 48, 9515–9518. [Google Scholar] [CrossRef]
- Allangawi, A.; Gilani, M.A.; Ayub, K.; Mahmood, T. First row transition metal doped B12P12 and Al12P12 nanocages as excellent single atom catalysts for the hydrogen evolution reaction. Int. J. Hydrogen Energy 2023, in press. [Google Scholar] [CrossRef]
- Mehboob, M.Y.; Hussain, R.; Younas, F.; Jamil, S.; Iqbal, M.M.A.; Ayub, K.; Sultana, N.; Janjua, M.R.S.A. Computation assisted design and prediction of alkali-metal-centered B12N12 nanoclusters for efficient H2 adsorption: New hydrogen storage materials. J. Clust. Sci. 2022, 1–11. [Google Scholar] [CrossRef]
- Mehboob, M.Y.; Hussain, F.; Hussain, R.; Ali, S.; Irshad, Z.; Adnan, M.; Ayub, K. Designing of Inorganic Al12N12 Nanocluster with Fe, Co, Ni, Cu and Zn Metals for Efficient Hydrogen Storage Materials. J. Comput. Biophys. Chem. 2021, 20, 359–375. [Google Scholar] [CrossRef]
- Jing, S.; Zhang, L.; Luo, L.; Lu, J.; Yin, S.; Shen, P.K.; Tsiakaras, P. N-doped porous molybdenum carbide nanobelts as efficient catalysts for hydrogen evolution reaction. Appl. Catal. B Environ. 2018, 224, 533–540. [Google Scholar] [CrossRef]
- Chen, J.L.; Zhang, X.G.; Wu, D.Y. Dissociation reactions of hydrogen molecules at active sites on gold clusters: A DFT study. J. Chin. Chem. Soc. 2022. [Google Scholar] [CrossRef]
- Tierney, H.L.; Baber, A.E.; Kitchin, J.R.; Sykes, E.C.H. Hydrogen dissociation and spillover on individual isolated palladium atoms. Phys. Rev. Lett. 2009, 103, 246102. [Google Scholar] [CrossRef]
- van Steen, E.; van Helden, P. A DFT study of hydrogen dissociation on CO-and C-precovered Fe (100) surfaces. J. Phys. Chem. C 2010, 114, 5932–5940. [Google Scholar] [CrossRef]
- Zha, H.; Dong, X.; Yu, Y.; Zhang, M. Hydrogen-assisted versus hydroxyl-assisted CO dissociation over Co-doped Cu (111): A DFT study. Surf. Sci. 2018, 669, 114–120. [Google Scholar] [CrossRef]
- Weng, M.H.; Chen, H.-T.; Wang, Y.-C.; Ju, S.-P.; Chang, J.-G.; Lin, M.-C. Kinetics and mechanisms for the adsorption, dissociation, and diffusion of hydrogen in Ni and Ni/YSZ slabs: A DFT study. Langmuir 2012, 28, 5596–5605. [Google Scholar] [CrossRef]
- Liu, D.; Barbar, A.; Najam, T.; Javed, M.S.; Shen, J.; Tsiakaras, P.; Cai, X. Single noble metal atoms doped 2D materials for catalysis applications. Appl. Catal. B Environ. 2021, 297, 120389. [Google Scholar] [CrossRef]
- Liang, S.; Hao, C.; Shi, Y. The power of single-atom catalysis. ChemCatChem 2015, 7, 2559–2567. [Google Scholar] [CrossRef]
- Chen, F.; Jiang, X.; Zhang, L.; Lang, R.; Qiao, B. Single-atom catalysis: Bridging the homo-and heterogeneous catalysis. Chin. J. Catal. 2018, 39, 893–898. [Google Scholar] [CrossRef]
- Cheng, N.; Sun, X. Single atom catalyst by atomic layer deposition technique. Chin. J. Catal. 2017, 38, 1508–1514. [Google Scholar] [CrossRef]
- Chen, Y.; Ji, S.; Chen, C.; Peng, Q.; Wang, D.; Li, Y. Single-atom catalysts: Synthetic strategies and electrochemical applications. Joule 2018, 2, 1242–1264. [Google Scholar] [CrossRef] [Green Version]
- Righi, G.; Magri, R.; Selloni, A. H2 Dissociation on Noble Metal Single Atom Catalysts Adsorbed on and Doped into CeO2 (111). J. Phys. Chem. C 2019, 123, 9875–9883. [Google Scholar] [CrossRef]
- Guo, Y.; Lang, R.; Qiao, B. Highlights of major progress on single-atom catalysis in 2017. Catalysts 2019, 9, 135. [Google Scholar] [CrossRef] [Green Version]
- Qiao, B.; Wang, A.; Yang, X.; Allard, L.F.; Jiang, Z.; Cui, Y.; Liu, J.; Li, J.; Zhang, T. Single-atom catalysis of CO oxidation using Pt 1/FeOx. Nat. Chem. 2011, 3, 634–641. [Google Scholar] [CrossRef]
- Ghosh, T.K.; Nair, N.N. Rh1/γ-Al2O3 Single-Atom Catalysis of O2 Activation and CO Oxidation: Mechanism, Effects of Hydration, Oxidation State, and Cluster Size. ChemCatChem 2013, 5, 1811–1821. [Google Scholar] [CrossRef]
- Parkinson, G.S. Single-atom catalysis: How structure influences catalytic performance. Catal. Lett. 2019, 149, 1137–1146. [Google Scholar] [CrossRef]
- Cheng, N.; Zhang, L.; Doyle-Davis, K.; Sun, X. Single-atom catalysts: From design to application. Electrochem. Energy Rev. 2019, 2, 539–573. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.-X.; Yang, X.-F.; Wang, A.; Zhang, T.; Li, J. Theoretical investigations of non-noble metal single-atom catalysis: Ni 1/FeOx for CO oxidation. Catal. Sci. Technol. 2016, 6, 6886–6892. [Google Scholar] [CrossRef]
- Thiel, W. Computational catalysis—Past, present, and future. Angew. Chem. Int. Ed. 2014, 53, 8605–8613. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, M.; Alfe, D. Hydrogen dissociation and diffusion on transition metal (= Ti, Zr, V, Fe, Ru, Co, Rh, Ni, Pd, Cu, Ag)-doped Mg (0001) surfaces. Int. J. Hydrogen Energy 2009, 34, 1922–1930. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Li, T.; Wang, Q.; Yang, G.; He, C.; Ma, B.; Lu, Z. Graphyne as a promising substrate for the noble-metal single-atom catalysts. Carbon 2015, 95, 756–765. [Google Scholar] [CrossRef]
- Ullah, F.; Ayub, K.; Mahmood, T. High performance SACs for HER process using late first-row transition metals anchored on graphyne support: A DFT insight. Int. J. Hydrogen Energy 2021, 46, 37814–37823. [Google Scholar] [CrossRef]
- Sun, T.; Xu, L.; Wang, D.; Li, Y. Metal organic frameworks derived single atom catalysts for electrocatalytic energy conversion. Nano Res. 2019, 12, 2067–2080. [Google Scholar] [CrossRef]
- Fu, J.; Wang, S.; Wang, Z.; Liu, K.; Li, H.; Liu, H.; Hu, J.; Xu, X.; Li, H.; Liu, M. Graphitic carbon nitride based single-atom photocatalysts. Front. Phys. 2020, 15, 33201. [Google Scholar] [CrossRef]
- Rad, A.S.; Ayub, K. Enhancement in hydrogen molecule adsorption on B12N12 nano-cluster by decoration of nickel. Int. J. Hydrogen Energy 2016, 41, 22182–22191. [Google Scholar] [CrossRef]
- Papa, V.; Cao, Y.; Spannenberg, A.; Junge, K.; Beller, M. Development of a practical non-noble metal catalyst for hydrogenation of N-heteroarenes. Nat. Catal. 2020, 3, 135–142. [Google Scholar] [CrossRef]
- Franco, F.; Rettenmaier, C.; Jeon, H.S.; Cuenya, B.R. Transition metal-based catalysts for the electrochemical CO2 reduction: From atoms and molecules to nanostructured materials. Chem. Soc. Rev. 2020, 49, 6884–6946. [Google Scholar] [CrossRef]
- Ayub, K. Transportation of hydrogen atom and molecule through X12Y12 nano-cages. Int. J. Hydrogen Energy 2017, 42, 11439–11451. [Google Scholar] [CrossRef]
- Shah, A.B.; Sarfaraz, S.; Yar, M.; Sheikh, N.S.; Hammud, H.H.; Ayub, K. Remarkable Single Atom Catalyst of Transition Metal (Fe, Co & Ni) Doped on C2N Surface for Hydrogen Dissociation Reaction. Nanomaterials 2023, 13, 29. [Google Scholar]
- Shi, T.-H.; Tong, S.; Jiao, L.; Wang, M.-X. A Theoretical Study on the Macrocyclic Strain of Zigzag Molecular Belts. Org. Mater. 2020, 2, 300–305. [Google Scholar] [CrossRef]
- Shi, T.-H.; Wang, M.-X. Zigzag hydrocarbon belts. CCS Chem. 2021, 3, 916–931. [Google Scholar] [CrossRef]
- Zhang, Y.-E.; Tong, S.; Wang, M.-X. Selective Oxidation of Belt [4] arene [4] tropilidene and Its Application to Construct Hydrocarbon Belts of Truncated Cone Structure with Expand Cavity. Org. Lett. 2021, 23, 7259–7263. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, Y.-E.; Tong, S.; Wang, M.-X. Hydrocarbon belts with truncated cone structures. J. Am. Chem. Soc. 2020, 142, 1196–1199. [Google Scholar] [CrossRef]
- Cheung, K.Y.; Watanabe, K.; Segawa, Y.; Itami, K. Synthesis of a zigzag carbon nanobelt. Nat. Chem. 2021, 13, 255–259. [Google Scholar] [CrossRef]
- Zhang, Y.; Tong, S.; Wang, M.X. Synthesis and structure of functionalized zigzag hydrocarbon belts. Angew. Chem. 2020, 132, 18308–18312. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Li, N.; Zhuang, Y.; Sui, L.; Xiao, Z.; Fan, D.; Aravindan, V.; Bowen, C.; Lu, H.; Liu, Y. Interface charge density modulation of a lamellar-like spatially separated Ni9S8 nanosheet/Nb2O5 nanobelt heterostructure catalyst coupled with nitrogen and metal (M = Co, Fe, or Cu) atoms to accelerate acidic and alkaline hydrogen evolution reactions. Chem. Eng. J. 2022, 431, 134073. [Google Scholar] [CrossRef]
- Xu, H.; Jia, H.; Fei, B.; Ha, Y.; Li, H.; Guo, Y.; Liu, M.; Wu, R. Charge Transfer Engineering via Multiple Heteroatom Doping in Dual Carbon-Coupled Cobalt Phosphides for Highly Efficient Overall Water Splitting. Appl. Catal. B Environ. 2020, 268, 118404. [Google Scholar] [CrossRef]
- Liu, Y.; Jiang, S.; Li, S.; Zhou, L.; Li, Z.; Li, J.; Shao, M. Interface engineering of (Ni, Fe)S2@MoS2 heterostructures for synergetic electrochemical water splitting. Appl. Catal. B Environ. 2019, 247, 107–114. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Gu, C.; Zhang, L.; Wang, X.; Tu, J. Active Co@CoO core/shell nanowire arrays as efficient electrocatalysts for hydrogen evolution reaction. Chem. Eng. J. 2022, 429, 132226. [Google Scholar] [CrossRef]
- Huang, Z.-F.; Song, J.; Du, Y.; Xi, S.; Dou, S.; Nsanzimana, J.M.V.; Wang, C.; Xu, Z.J.; Wang, X. Chemical and structural origin of lattice oxygen oxidation in Co–Zn oxyhydroxide oxygen evolution electrocatalysts. Nat. Energy 2019, 4, 329–338. [Google Scholar] [CrossRef]
- Hussain, S.; Shahid Chatha, S.A.; Hussain, A.I.; Hussain, R.; Mehboob, M.Y.; Gulzar, T.; Mansha, A.; Shahzad, N.; Ayub, K. Designing novel Zn-decorated inorganic B12P12 nanoclusters with promising electronic properties: A step forward toward efficient CO2 sensing materials. ACS Omega 2020, 5, 15547–15556. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.; Ullah, H.; Ullah, Z.; Tariq, M.; Ayub, K. External stimulus controlled recombination of hydrogen in photochromic dithienylethene frustrated lewis pairs. Int. J. Hydrogen Energy 2019, 44, 31141–31152. [Google Scholar]
- Sajid, H.; Malik, S.; Rashid, U.; Mahmood, T.; Ayub, K. Hydrogen adsorption on Ge52−, Ge92− and Sn92− Zintl clusters: A DFT study. Comput. Theor. Chem. 2021, 1199, 113191. [Google Scholar] [CrossRef]
- Frisch, M.; Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. Gaussian 09, Revision D. 01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView, Version 5; Semichem Inc.: Shawnee Mission, KS, USA, 2009. [Google Scholar]
- Sarfaraz, S.; Yar, M.; Ans, M.; Gilani, M.A.; Ludwig, R.; Hashmi, M.A.; Hussain, M.; Muhammad, S.; Ayub, K. Computational investigation of a covalent triazine framework (CTF-0) as an efficient electrochemical sensor. RSC Adv. 2022, 12, 3909–3923. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Yar, M.; Ayub, K. Covalent triazine framework (CTF-0) surface as a smart sensing material for the detection of CWAs and industrial pollutants. Mater. Sci. Semicond. Process. 2022, 139, 106334. [Google Scholar] [CrossRef]
- Sarfaraz, S.; Yar, M.; Khan, A.A.; Ahmad, R.; Ayub, K. DFT investigation of adsorption of nitro-explosives over C2N surface: Highly selective towards trinitro benzene. J. Mol. Liq. 2022, 352, 118652. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Jindal, R.; Sharma, V.; Shukla, A. Density functional theory study of the hydrogen evolution reaction in haeckelite boron nitride quantum dots. Int. J. Hydrogen Energy 2022, 47, 41783–41794. [Google Scholar] [CrossRef]
- Ans, M.; Iqbal, J.; Ahmad, Z.; Muhammad, S.; Hussain, R.; Eliasson, B.; Ayub, K. Designing three-dimensional (3D) non-fullerene small molecule acceptors with efficient photovoltaic parameters. ChemistrySelect 2018, 3, 12797–12804. [Google Scholar]
- Ans, M.; Iqbal, J.; Ayub, K.; Ali, E.; Eliasson, B. Spirobifluorene based small molecules as an alternative to traditional fullerene acceptors for organic solar cells. Mater. Sci. Semicond. Process. 2019, 94, 97–106. [Google Scholar] [CrossRef]
- Minenkov, Y.; Singstad, Å.; Occhipinti, G.; Jensen, V.R. The accuracy of DFT-optimized geometries of functional transition metal compounds: A validation study of catalysts for olefin metathesis and other reactions in the homogeneous phase. Dalton Trans. 2012, 41, 5526–5541. [Google Scholar] [CrossRef]
- Zara, Z.; Iqbal, J.; Ayub, K.; Irfan, M.; Mahmood, A.; Khera, R.A.; Eliasson, B. A comparative study of DFT calculated and experimental UV/Visible spectra for thirty carboline and carbazole based compounds. J. Mol. Struct. 2017, 1149, 282–298. [Google Scholar] [CrossRef]
- Rad, A.S.; Ayub, K. Adsorption properties of acetylene and ethylene molecules onto pristine and nickel-decorated Al12N12 nanoclusters. Mater. Chem. Phys. 2017, 194, 337–344. [Google Scholar] [CrossRef]
- Allangawi, A.; Mahmood, T.; Ayub, K.; Gilani, M.A. Anchoring the late first row transition metals with B12P12 nanocage to act as single atom catalysts toward oxygen evolution reaction (OER). Mater. Sci. Semicond. Process. 2023, 153, 107164. [Google Scholar] [CrossRef]
- Mukhtar, A.; Sarfaraz, S.; Ayub, K. Organic transformations in the confined space of porous organic cage CC2; catalysis or inhibition. RSC Adv. 2022, 12, 24397–24411. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, B.; Liu, S.; Zhu, D.; Huang, F.; Yang, H.; Guan, H.; Song, A.; Xu, D.; Sun, L. Catalytic pyrolysis of biomass with Ni/Fe-CaO-based catalysts for hydrogen-rich gas: DFT and experimental study. Energy Convers. Manag. 2022, 254, 115246. [Google Scholar]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Kosar, N.; Tahir, H.; Ayub, K.; Gilani, M.A.; Imran, M.; Mahmood, T. Remarkable nonlinear optical response of Mn@C20 (M = Na & K and n = 1–6); a DFT outcome. Mater. Sci. Semicond. Process. 2022, 138, 106269. [Google Scholar] [CrossRef]
- Zhu, C.; Xia, H. Carbolong chemistry: A story of carbon chain ligands and transition metals. Acc. Chem. Res. 2018, 51, 1691–1700. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Pakhira, S.; Fujisawa, K.; Wang, X.; Iyiola, O.O.; Perea López, N.s.; Laura Elías, A.; Pulickal Rajukumar, L.; Zhou, C.; Kabius, B. Low-temperature synthesis of heterostructures of transition metal dichalcogenide alloys (Wx Mo1–x S2) and graphene with superior catalytic performance for hydrogen evolution. ACS Nano 2017, 11, 5103–5112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, X.; Liu, C.; Guo, J.; Ning, H. Hydrogen adsorption and dissociation on nickel-adsorbed and -substituted Mg17Al12 (100) surface: A density functional theory study. Int. J. Hydrogen Energy 2018, 43, 793–800. [Google Scholar] [CrossRef]
- Sun, K.; Kohyama, M.; Tanaka, S.; Takeda, S. A study on the mechanism for H2 dissociation on Au/TiO2 catalysts. J. Phys. Chem. C 2014, 118, 1611–1617. [Google Scholar] [CrossRef]
- Du, A.; Smith, S.C.; Yao, X.; Lu, G. The role of Ti as a catalyst for the dissociation of hydrogen on a Mg (0001) surface. J. Phys. Chem. B 2005, 109, 18037–18041. [Google Scholar] [CrossRef] [PubMed]
- Noreen, M.; Rasool, N.; Gull, Y.; Zubair, M.; Mahmood, T.; Ayub, K.; Nasim, F.-u.-H.; Yaqoob, A.; Zia-Ul-Haq, M.; De Feo, V. Synthesis, density functional theory (DFT), urease inhibition and antimicrobial activities of 5-aryl thiophenes bearing sulphonylacetamide moieties. Molecules 2015, 20, 19914–19928. [Google Scholar] [CrossRef] [Green Version]
- Salman, G.A.; Nisa, R.U.; Iaroshenko, V.O.; Iqbal, J.; Ayub, K.; Langer, P. Pyrrole versus quinoline formation in the palladium catalyzed reaction of 2-alkynyl-3-bromothiophenes and 2-alkynyl-3-bromofurans with anilines. A combined experimental and computational study. Org. Biomol. Chem. 2012, 10, 9464–9473. [Google Scholar] [CrossRef]
- Ahmad, G.; Rasool, N.; Ikram, H.M.; Gul Khan, S.; Mahmood, T.; Ayub, K.; Zubair, M.; Al-Zahrani, E.; Ali Rana, U.; Akhtar, M.N. Efficient synthesis of novel pyridine-based derivatives via Suzuki cross-coupling reaction of commercially available 5-bromo-2-methylpyridin-3-amine: Quantum mechanical investigations and biological activities. Molecules 2017, 22, 190. [Google Scholar] [CrossRef] [Green Version]
- Islam, M.M.; Alam, M.T.; Ohsaka, T. Electrical double-layer structure in ionic liquids: A corroboration of the theoretical model by experimental results. J. Phys. Chem. C 2008, 112, 16568–16574. [Google Scholar] [CrossRef]
- Hassan, J.; Naz, S.; Haider, A.; Raza, A.; Ul-Hamid, A.; Qumar, U.; Haider, J.; Goumri-Said, S.; Kanoun, M.B.; Ikram, M. h-BN nanosheets doped with transition metals for environmental remediation; a DFT approach and molecular docking analysis. Mater. Sci. Eng. B 2021, 272, 115365. [Google Scholar] [CrossRef]
- DiLabio, G.A.; Koleini, M. Dispersion-correcting potentials can significantly improve the bond dissociation enthalpies and noncovalent binding energies predicted by density-functional theory. J. Chem. Phys. 2014, 140, 18A542. [Google Scholar] [CrossRef] [PubMed]
- Feyereisen, M.W.; Feller, D.; Dixon, D.A. Hydrogen Bond Energy of the Water Dimer. J. Phys. Chem. 1996, 100, 2993–2997. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complexes | ΔEint (eV) | HOMO (eV) | LUMO (eV) | Eg (eV) | QTM |e| |
|---|---|---|---|---|---|
| Sc@NB | −3.54 | −6.06 | −0.52 | 5.53 | 1.211 |
| Ti@NB | −3.57 | −6.28 | −0.86 | 5.42 | 1.131 |
| V@NB | −3.31 | −6.47 | −0.81 | 5.66 | 1.124 |
| Cr@NB | −3.68 | −6.42 | −1.31 | 5.11 | 1.097 |
| Mn@NB | −4.38 | −5.33 | −2.10 | 3.23 | 0.991 |
| Fe@NB | −2.10 | −6.20 | −1.07 | 5.13 | 0.971 |
| Co@NB | −4.59 | −6.41 | −0.79 | 5.62 | 0.939 |
| Ni@NB | −4.97 | −6.34 | −0.89 | 5.45 | 0.779 |
| Cu@NB | −3.01 | −6.27 | −0.95 | 5.32 | 0.939 |
| Zn@NB | −0.18 | −6.20 | −1.44 | 4.76 | 1.241 |
| Nanobelt (NB) | -- | −6.94 | −1.28 | 5.66 | -- |
| Complexes | ΔEads (eV) | ΔE | Ea |
|---|---|---|---|
| H2Sc@NB | −0.06 | −0.98 | 0.13 |
| H2Ti@NB | −0.40 | −1.28 | 0.65 |
| H2V@NB | −0.45 | −1.04 | 0.47 |
| H2Cr@NB | −0.77 | −0.93 | 0.35 |
| H2Mn@NB | −0.54 | −1.82 | 0.46 |
| H2Fe@NB | −0.26 | −0.86 | 0.98 |
| H2Co@NB | −0.93 | −1.38 | 0.99 |
| H2Ni@NB | −0.89 | −1.09 | 1.05 |
| H2Cu@NB | −0.72 | −1.55 | 0.77 |
| H2Zn@NB | −0.27 | −1.15 | 0.36 |
| Complexes | H1 (TM Side) |e| | TM (|e|) | H2 (N Side) |e| | N (|e|) |
|---|---|---|---|---|
| H2Sc@NB | −0.257 | 1.444 | 0.244 | −0.764 |
| H2Ti@NB | −0.315 | 1.254 | 0.252 | −0.809 |
| H2V@NB | −0.284 | 1.106 | 0.252 | −0.774 |
| H2Cr@NB | −0.315 | 1.235 | 0.245 | −0.789 |
| H2Mn@NB | −0.317 | 1.049 | 0.273 | −0.800 |
| H2Fe@NB | −0.323 | 1.111 | 0.271 | −0.755 |
| H2Co@NB | −0.327 | 1.050 | 0.281 | −0.743 |
| H2Ni@NB | −0.309 | 0.916 | 0.295 | −0.711 |
| H2Cu@NB | −0.295 | 0.975 | 0.238 | −0.705 |
| H2Zn@NB | −0.218 | 1.351 | 0.176 | −0.982 |
| Catalysts for HDR | Energy Barrier (eV) | Reference |
|---|---|---|
| Sc@NB | 0.13 eV | Our work |
| Fe@C2N | 0.36 eV | Shah et al. [41] |
| Mg15Ni2Al12 | 0.53 eV | Zhang et al. [75] |
| Au/TiO2 system | 0.54 eV | Sun et al. [76] |
| Ti-doped Mg Surface | 0.35 eV | Du et al. [77] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bayach, I.; Sarfaraz, S.; Sheikh, N.S.; Alamer, K.; Almutlaq, N.; Ayub, K. Hydrogen Dissociation Reaction on First-Row Transition Metal Doped Nanobelts. Materials 2023, 16, 2792. https://doi.org/10.3390/ma16072792
Bayach I, Sarfaraz S, Sheikh NS, Alamer K, Almutlaq N, Ayub K. Hydrogen Dissociation Reaction on First-Row Transition Metal Doped Nanobelts. Materials. 2023; 16(7):2792. https://doi.org/10.3390/ma16072792
Chicago/Turabian StyleBayach, Imene, Sehrish Sarfaraz, Nadeem S. Sheikh, Kawther Alamer, Nadiah Almutlaq, and Khurshid Ayub. 2023. "Hydrogen Dissociation Reaction on First-Row Transition Metal Doped Nanobelts" Materials 16, no. 7: 2792. https://doi.org/10.3390/ma16072792