



The Highly Accurate Interatomic Potential of CsPbBr3 Perovskite with Temperature Dependence on the Structure and Thermal Properties
Abstract
:1. Introduction
2. Methodology
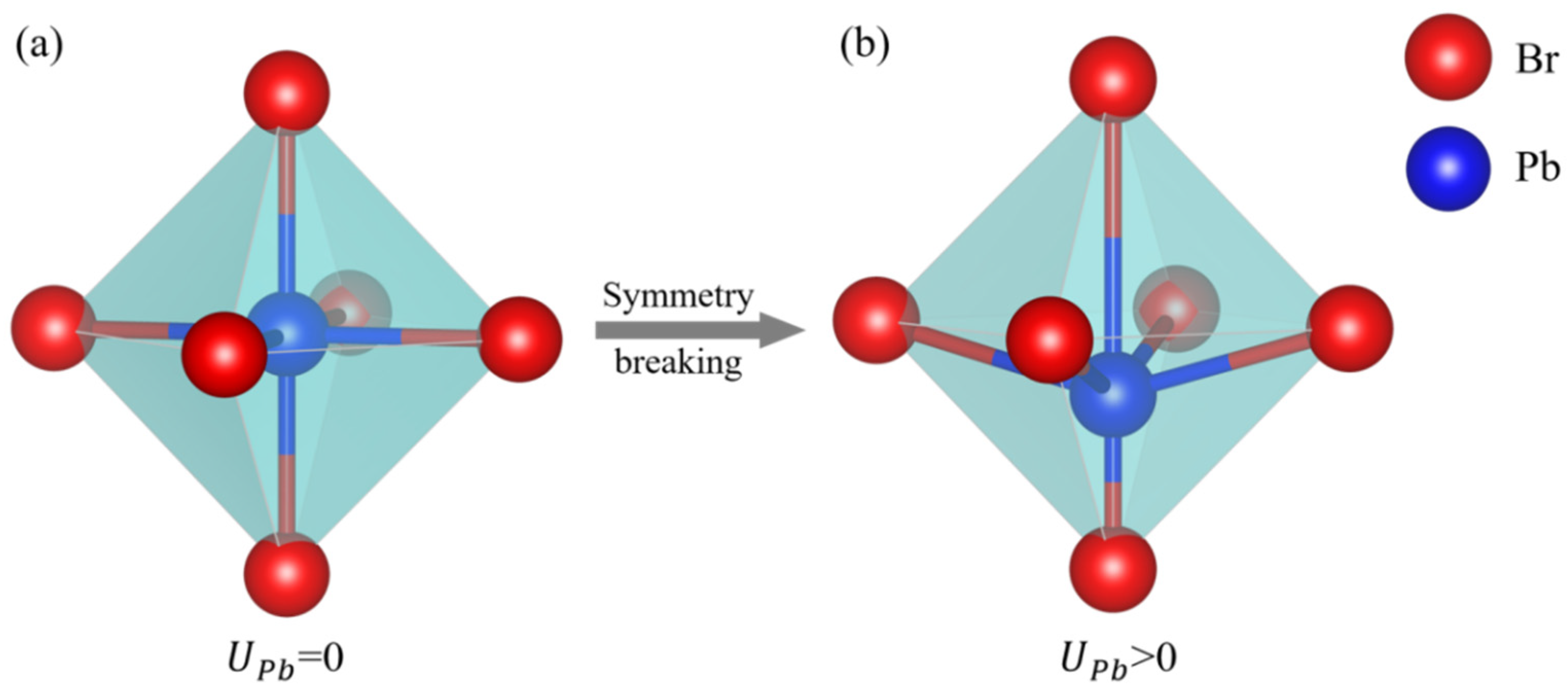
2.1. Potential Model
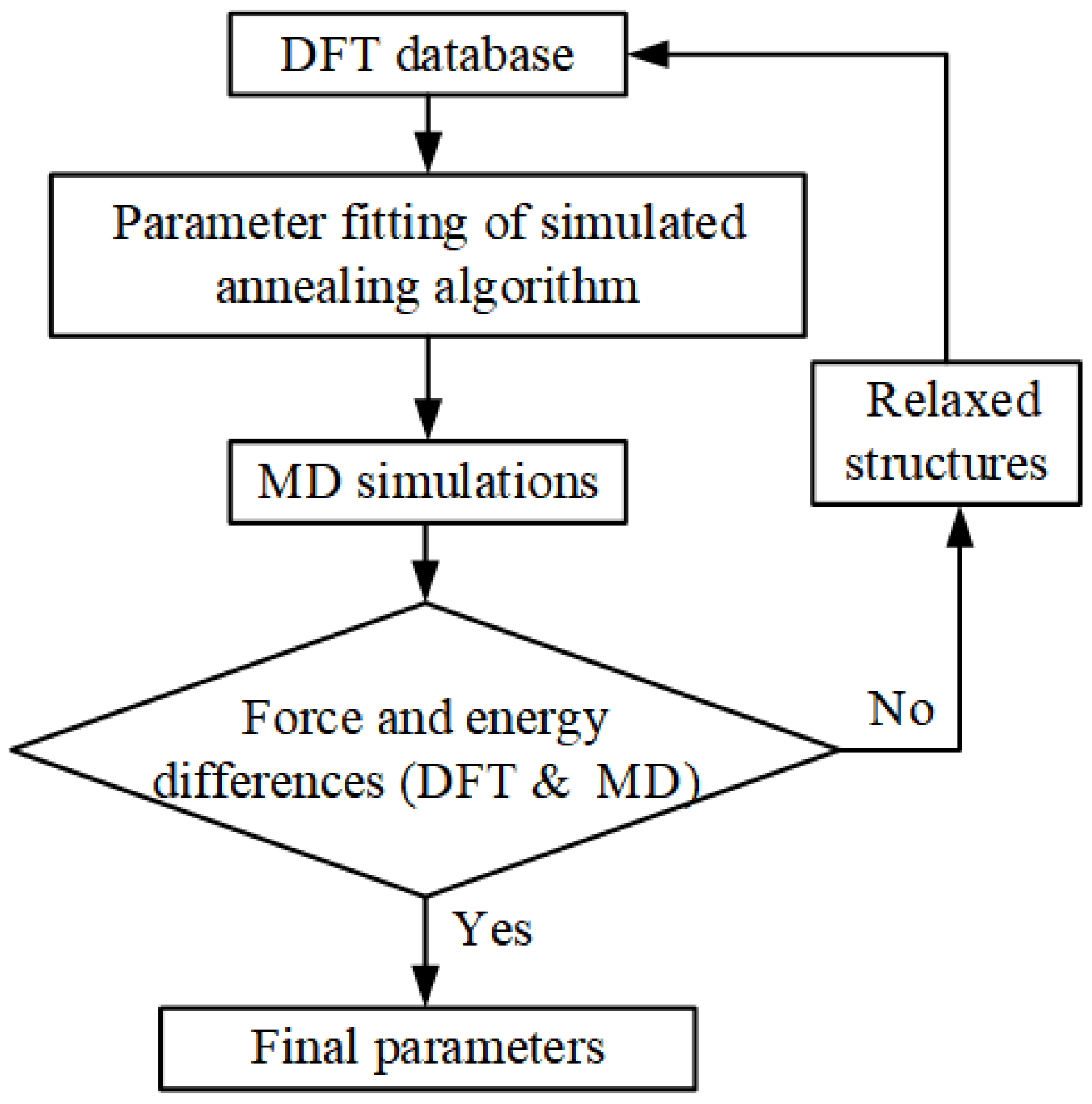
2.2. Method of BV Potential Parameters
3. Results and Discussion
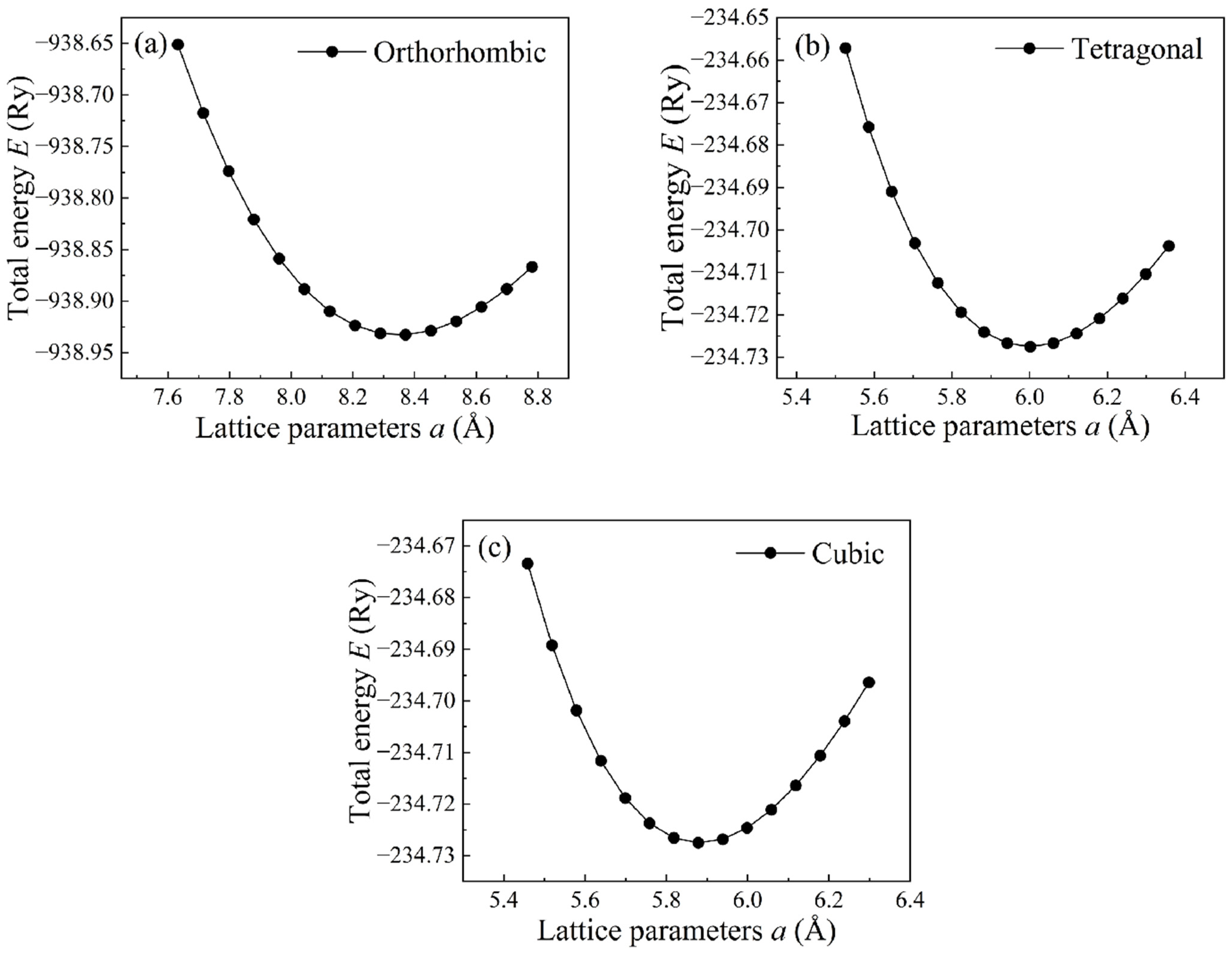
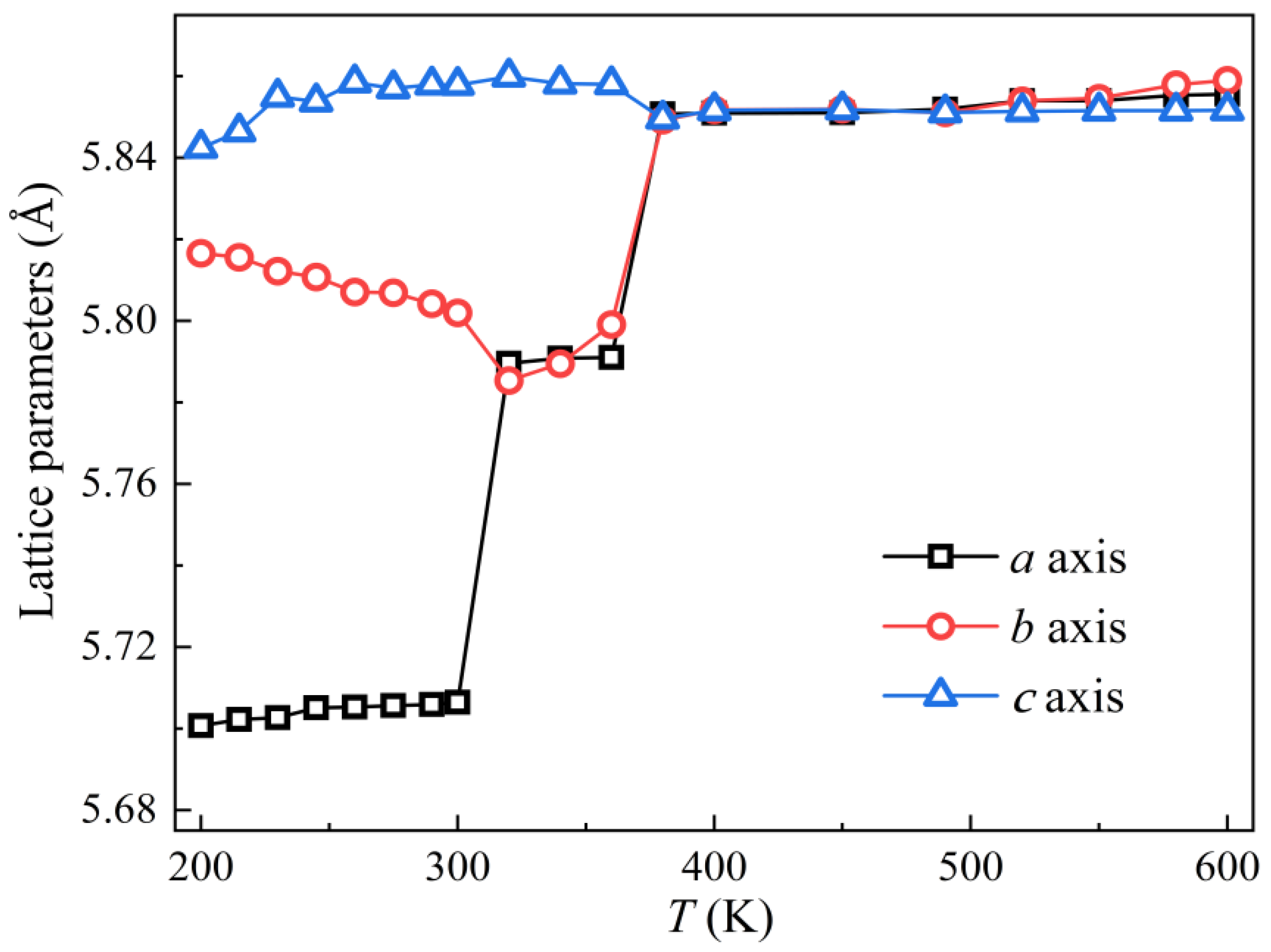
3.1. Lattice Parameters and Elastic Constants
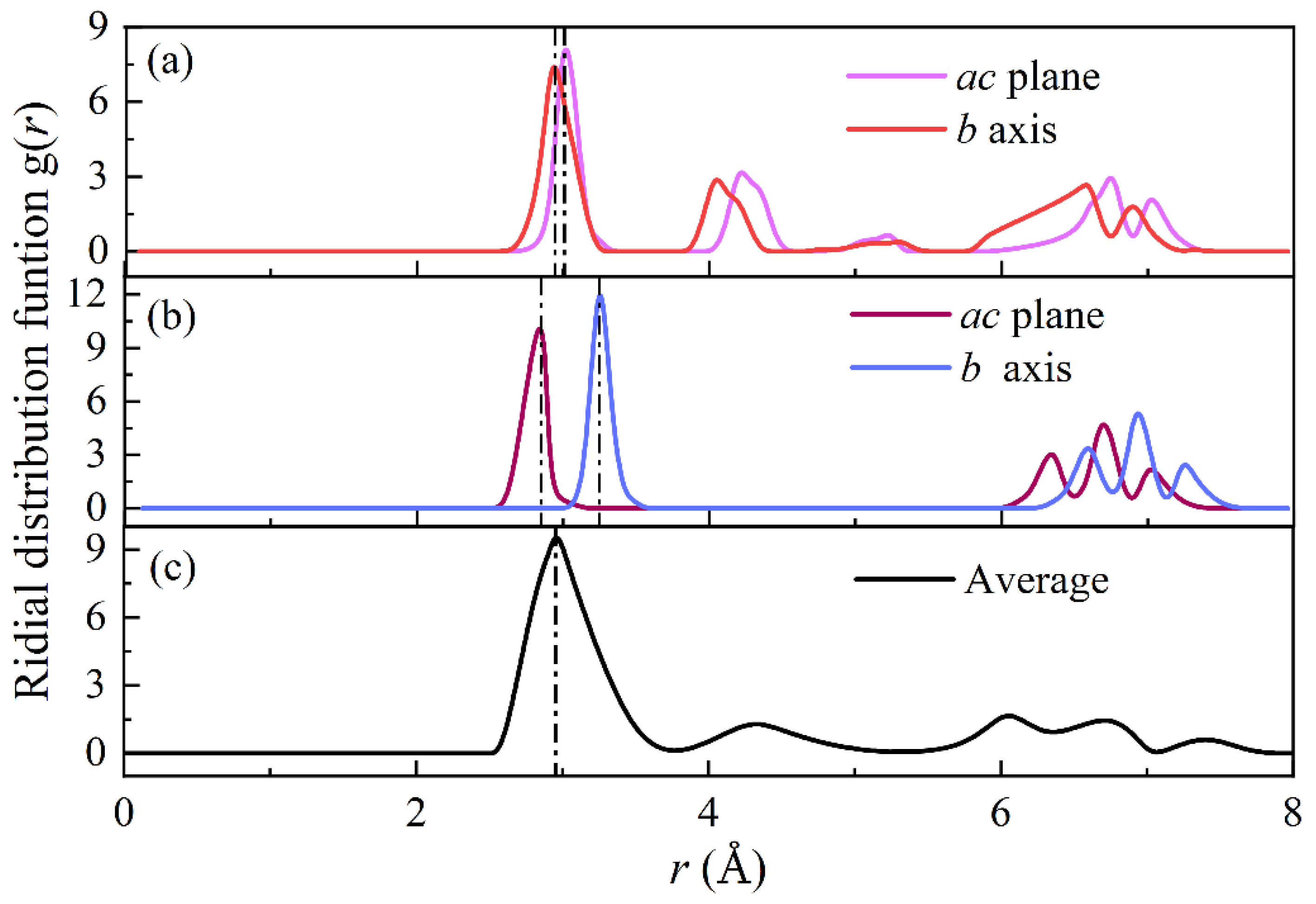
3.2. Bond Lengths
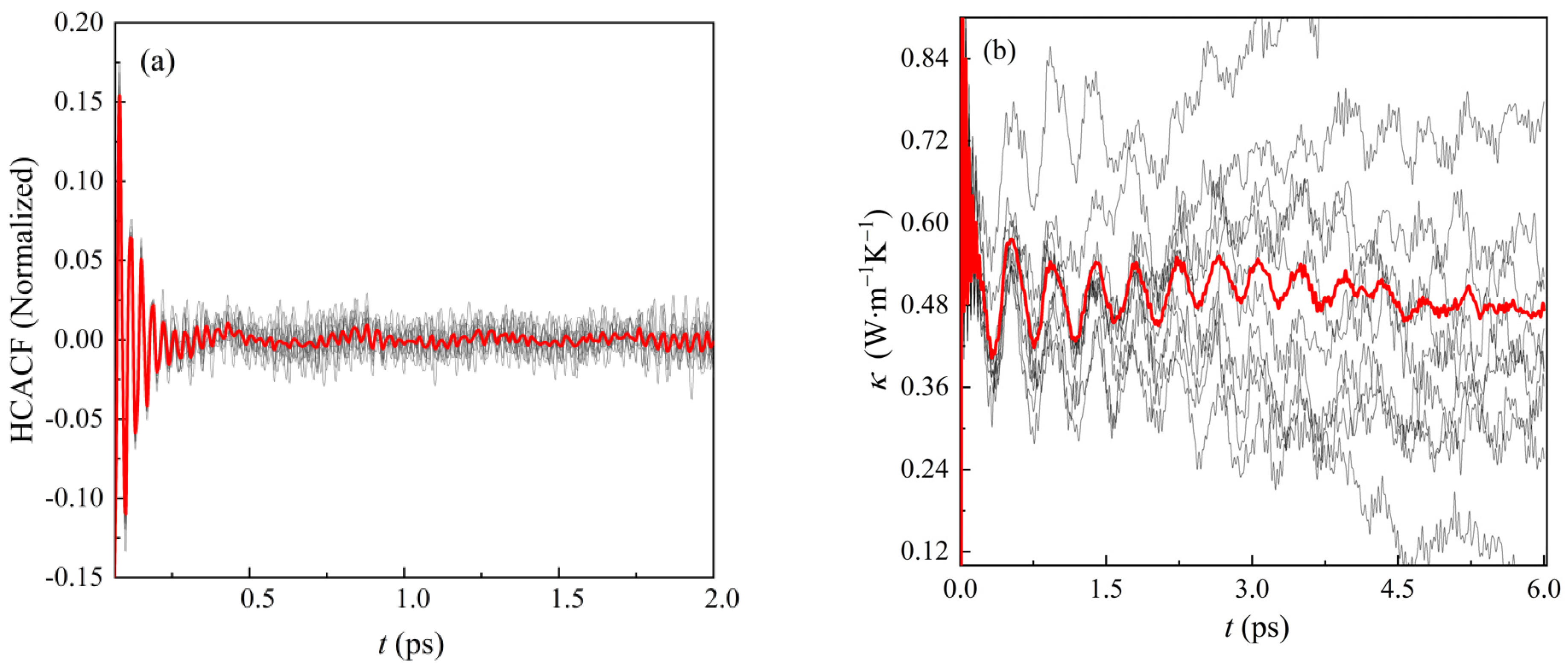
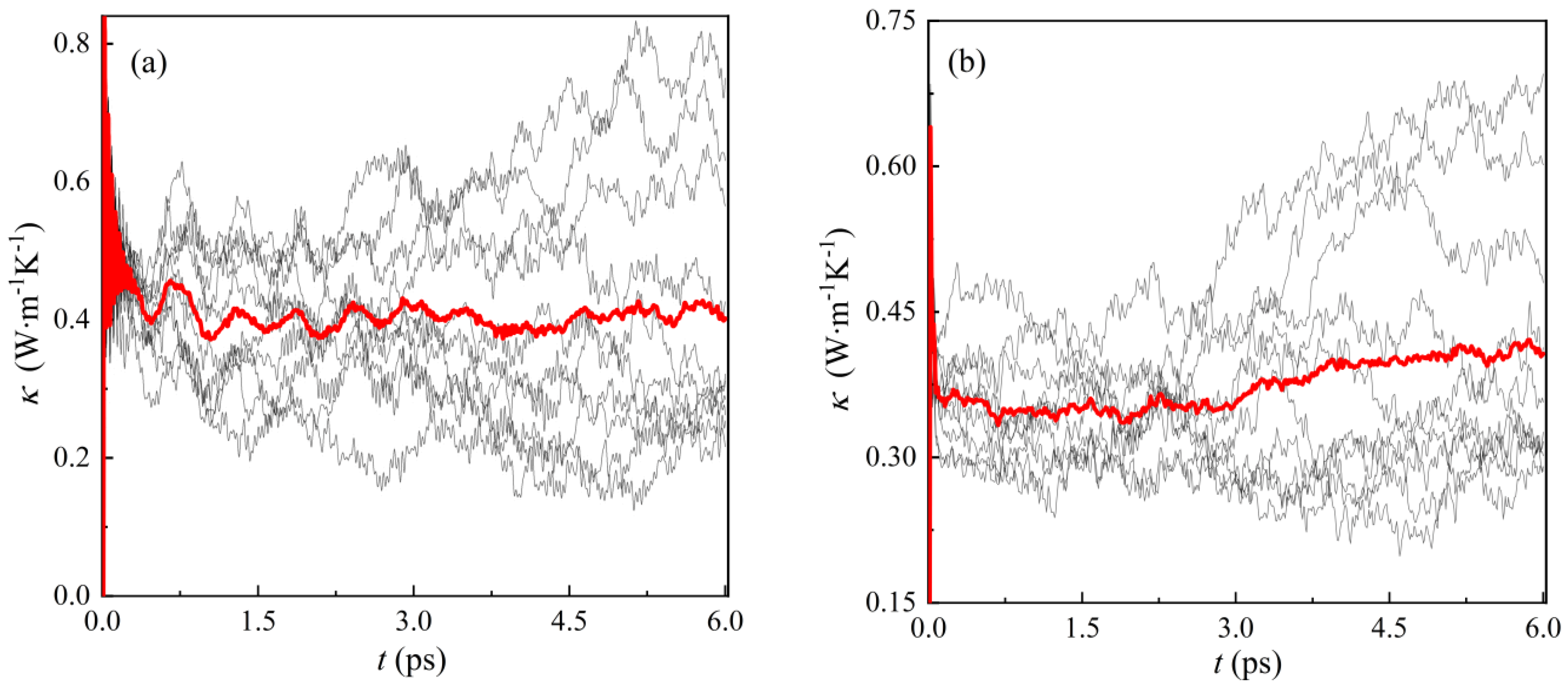
3.3. Thermal Conductivity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gao, W.; Zhang, Z.; Xu, R.; Chan, E.M.; Yuan, G.; Liu, J. Development and Prospects of Halide Perovskite Single Crystal Films. Adv. Electron. Mater. 2022, 8, 2100980. [Google Scholar] [CrossRef]
- Dey, A.; Ye, J.; De, A.; Debroye, E.; Ha, S.K.; Bladt, E.; Kshirsagar, A.S.; Wang, Z.; Yin, J.; Wang, Y.; et al. State of the Art and Prospects for Halide Perovskite Nanocrystals. ACS Nano 2021, 15, 10775–10981. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Huang, T.; Xue, J.; Tong, J.; Zhu, K.; Yang, Y. Prospects for metal halide perovskite-based tandem solar cells. Nat. Photon. 2021, 15, 411–425. [Google Scholar] [CrossRef]
- Haque, M.; Kee, S.; Villalva, D.R.; Ong, W.L.; Baran, D. Halide perovskites: Thermal transport and prospects for thermoelectricity. Adv. Sci. 2020, 7, 1903389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Li, J.; Yang, Z.; Wang, M. All-inorganic lead halide perovskites: A promising choice for photovoltaics and detectors. J. Mater. Chem. C 2019, 7, 12415–12440. [Google Scholar] [CrossRef]
- Gao, Y.; Wu, Y.; Lu, H.; Chen, C.; Liu, Y.; Bai, X.; Yang, L.; Yu, W.W.; Dai, Q.; Zhang, Y. CsPbBr3 perovskite nanoparticles as additive for environmentally stable perovskite solar cells with 20.46% efficiency. Nano Energy 2019, 59, 517–526. [Google Scholar] [CrossRef]
- Ma, X.; Yang, L.; Zheng, S. All-Inorganic perovskite solar cells: Status and future. Prog. Chem. 2020, 32, 1608–1632. [Google Scholar]
- Brennan, M.C.; Kuno, M.; Rouvimov, S. Crystal Structure of Individual CsPbBr3 Perovskite Nanocubes. Inorg. Chem. 2019, 58, 1555–1560. [Google Scholar] [CrossRef]
- Dastidar, S.; Hawley, C.J.; Dillon, A.D.; Gutierrez-Perez, A.D.; Spanier, J.E.; Fafarman, A.T. Quantitative Phase-Change Thermodynamics and Metastability of Perovskite-Phase Cesium Lead Iodide. J. Phys. Chem. Lett. 2017, 8, 1278–1282. [Google Scholar] [CrossRef]
- Nagal, V.; Kumar, V.; Kumar, R.; Singh, K.; Khosla, A.; Ahmad, R.; Hafiz, A.K. CsPbBr3 nanoplatelets: Synthesis and understanding of ultraviolet light-induced structural phase change and luminescence degradation. ECS J. Solid State Sci. Technol. 2021, 10, 096002. [Google Scholar] [CrossRef]
- Svirskas, Š.; Balčiūnas, S.; Šimėnas, M.; Usevičius, G.; Kinka, M.; Velička, M.; Kubicki, D.; Castillo, M.E.; Karabanov, A.; Shvartsman, V.V.; et al. Phase transitions, screening and dielectric response of CsPbBr3. J. Mater. Chem. A 2020, 8, 14015–14022. [Google Scholar] [CrossRef]
- Ahmad, M.; Rehman, G.; Ali, L.; Shafiq, M.; Iqbal, R.; Ahmad, R.; Khan, T.; Jalali-Asadabadi, S.; Maqbool, M.; Ahmad, I. Structural, electronic and optical properties of CsPbX3 (X=Cl, Br, I) for energy storage and hybrid solar cell applications. J. Alloys Compd. 2017, 705, 828–839. [Google Scholar] [CrossRef]
- Ghaithan, H.M.; Alahmed, Z.A.; Qaid, S.M.H.; Hezam, M.; Aldwayyan, A.S. Density functional study of cubic, tetragonal, and orthorhombic CsPbBr3 Perovskite. ACS Omega 2020, 5, 7468–7480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, C.A.; Abia, C.; Alvarez-Galván, M.C.; Hong, B.-K.; Martínez-Huerta, M.V.; Serrano-Sánchez, F.; Carrascoso, F.; Castellanos-Gómez, A.; Fernández-Díaz, M.T.; Alonso, J.A. Crystal Structure Features of CsPbBr3 Perovskite Prepared by Mechanochemical Synthesis. ACS Omega 2020, 5, 5931–5938. [Google Scholar] [CrossRef] [Green Version]
- Singh, H.; Singh, A.; Indu, B.D. The Born-Mayer-Huggins potential in high temperature superconductors. Mod. Phys. Lett. B 2016, 30, 1650283. [Google Scholar] [CrossRef]
- Almishal, S.S.I.; Rashwan, O. New accurate molecular dynamics potential function to model the phase transformation ofcesium lead triiodide perovskite. RSC Adv. 2020, 10, 44503–44511. [Google Scholar] [CrossRef]
- Balestra, S.R.; Vicent-Luna, J.M.; Calero, S.; Tao, S.; Anta, J.A. Efficient modelling of ion structure and dynamics in inorganic metal halide perovskites. J. Mater. Chem. A 2020, 8, 11824–11836. [Google Scholar] [CrossRef]
- Haeger, T.; Wilmes, M.; Heiderhoff, R.; Riedl, T. Simultaneous Mapping of Thermal Conductivity, Thermal Diffusivity, and Volumetric Heat Capacity of Halide Perovskite Thin Films: A Novel Nanoscopic Thermal Measurement Technique. J. Phys. Chem. Lett. 2019, 10, 3019–3023. [Google Scholar] [CrossRef]
- Lee, W.; Li, H.; Wong, A.; Zhang, D.; Lai, M.; Yu, Y.; Kong, Q.; Lin, E.; Urban, J.J.; Grossman, J.C.; et al. Ultralow thermal conductivity in all-inorganic halide perovskites. Proc. Natl. Acad. Sci. USA 2017, 114, 8693–8697. [Google Scholar] [CrossRef] [Green Version]
- Brown, I.D. Bond valences—A simple structural model for inorganic chemistry. Chem. Soc. Rev. 1978, 7, 359–376. [Google Scholar] [CrossRef]
- Brown, I.D. Recent Developments in the Methods and Applications of the Bond Valence Model. Chem. Rev. 2009, 109, 6858–6919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, I.D. What is the best way to determine bond-valence parameters? IUCrJ 2017, 4, 514–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anbalagan, K.; Thomas, T. Size-dependent disproportionation (in~ 2–20 nm regime) and hybrid Bond Valence derived interatomic potentials for BaTaO2N. Appl. Nanosci. 2018, 8, 1379–1388. [Google Scholar] [CrossRef]
- Liu, S.; Grinberg, I.; Takenaka, H. Reinterpretation of the bond-valence model with bond-order formalism: An improved bond-valence-based interatomic potential for PbTiO3. Phys. Rev. B 2016, 94, 134308. [Google Scholar] [CrossRef] [Green Version]
- Shin, Y.-H.; Cooper, V.R.; Grinberg, I.; Rappe, A.M. Development of a bond-valence molecular-dynamics model for complex oxides. Phys. Rev. B 2005, 71, 054104. [Google Scholar] [CrossRef] [Green Version]
- Atourki, L.; Vega, E.; Mollar, M.; Marí, B.; Kirou, H.; Bouabid, K.; Ihlal, A. Impact of iodide substitution on the physical propertiesand stability of cesium lead halide perovskite thin films CsPbBr3-xIx (0 ≤ x ≤ 1). J. Alloys Compd. 2017, 702, 404–409. [Google Scholar] [CrossRef]
- Moller, C.K. The structure of perovskite-like caesium plumbotrihalides. Mater. Fys. Medd. Dan. Vid. Selsk. 1959, 32, 1–27. [Google Scholar]
- Giannozzi, P.; Baroni, S.; Bonini, N.; Calandra, M.; Car, R.; Cavazzoni, C.; Ceresoli, D.; Chiarotti, G.L.; Cococcioni, M.; Dabo, I.; et al. QUANTUM ESPRESSO: A modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter 2009, 21, 395502. [Google Scholar] [CrossRef]
- Michael, T.H.; Yong, Z.; Edward, J.M. PyLAT: Python LAMMPS Analysis Tools. J. Chem. Inf. Model. 2019, 4, 1301–1305. [Google Scholar]
- Nagaoka, Y.; Hills-Kimball, K.; Tan, R.; Li, R.; Wang, Z.; Chen, O. Nanocube Superlattices of Cesium Lead Bromide Perovskites and Pressure-Induced Phase Transformations at Atomic and Mesoscale Levels. Adv. Mater. 2017, 29, 1606666. [Google Scholar] [CrossRef]
- Zheng, C.; Rubel, O. Ionization Energy as a Stability Criterion for Halide Perovskites. J. Phys. Chem. C 2017, 121, 11977–11984. [Google Scholar] [CrossRef] [Green Version]
- Golesorkhtabar, R.; Pavone, P.; Spitaler, J.; Puschnig, P.; Draxl, C. ElaStic: A tool for calculating second-order elastic constants from first principles. Comput. Phys. Commun. 2013, 184, 1861–1873. [Google Scholar] [CrossRef]
- Azzouz, L.; Halit, M.; Sidoumou, M.; Charifi, Z.; Allal, A.; Bouchenafa, M.; Baaziz, H. Electronic structure, elastic and optical properties of AEuS2 (A = Na, K, Rb, and Cs) ternary sulfides: First-principles study. Phys. Status Solidi. 2019, 256, 1900136. [Google Scholar] [CrossRef]
- Tan, J.; Li, Y.; Ji, G. Elastic constants and bulk modulus of semiconductors: Performance of plane-wave pseudopotential and local-density-approximation density functional theory. Comput. Mater. Sci. 2012, 58, 243–247. [Google Scholar] [CrossRef]
- Rakita, Y.; Cohen, S.R.; Kedem, N.K.; Hodes, G.; Cahen, D. Mechanical properties of APbX3 (A = Cs or CH3NH3; X = I or Br) perovskite single crystals. MRS Commun. 2015, 5, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Maphoto, R.I.; Ngoepe, P.; Masedi, M.; Morukuladi, M.; Malatji, K.T. First-Principle Study of CsPbBr3 and CsPbI3 Perovskite Solar Cells. ECS J. Solid State Sci. Technol. 2022, 11, 035012. [Google Scholar] [CrossRef]
- Jinnouchi, R.; Lahnsteiner, J.; Karsai, F.; Kresse, G.; Bokdam, M. Phase Transitions of Hybrid Perovskites Simulated by Machine-Learning Force Fields Trained on the Fly with Bayesian Inference. Phys. Rev. Lett. 2019, 122, 225701. [Google Scholar] [CrossRef] [Green Version]
- Haider, N.; Hossain, F. CSV2RDF: Generating RDF data from CSV file using semantic web technologies. J. Theor. Inf. Technol. 2018, 96, 6889–6902. [Google Scholar]
- Stoumpos, C.C.; Malliakas, C.D.; Peters, J.A.; Liu, Z.; Sebastian, M.; Im, J.; Chasapis, T.C.; Wibowo, A.C.; Chung, D.Y.; Freeman, A.J.; et al. Crystal Growth of the Perovskite Semiconductor CsPbBr3: A New Material for High-Energy Radiation Detection. Cryst. Growth Des. 2013, 13, 2722–2727. [Google Scholar] [CrossRef]
- Chang, X.; Li, W.; Zhu, L.; Liu, H.; Geng, H.; Xiang, S.; Liu, J.; Chen, H. Carbon-Based CsPbBr3 Perovskite Solar Cells: All-Ambient Processes and High Thermal Stability. ACS Appl. Mater. Interfaces 2016, 8, 33649–33655. [Google Scholar] [CrossRef]
- Pisoni, A.; Jacimovic, J.; Barisic, O.S.; Spina, M.; Gaál, R.; Forró, L.; Horváth, E. Ultra-low thermal conductivity in organic-inorganic hybrid. J. Phys. Chem. Lett. 2014, 5, 2488–2492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalsky, A.; Wang, L.; Marek, G.T.; Burda, C.; Dyck, J.S. Thermal conductivity of CH3NH3PbI3 and CsPbI3: Measuring the effect of the methylammonium ion on phonon scattering. J. Phys.Chem. C 2017, 121, 3228–3233. [Google Scholar] [CrossRef]
- Tadano, T.; Saidi, W.A. First-Principles Phonon Quasiparticle Theory Applied to a Strongly Anharmonic Halide Perovskite. Phys. Rev. Lett. 2022, 129, 185901. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Tadano, T.; Iikubo, S. Effect of Halogen Ions on the Low Thermal Conductivity of Cesium Halide Perovskite. J. Phys. Chem. C 2021, 125, 91–97. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cs | Pb | Br | |||||
|---|---|---|---|---|---|---|---|
| Cs | 2.304 | 2.585 | 2.539 | 1.448 | 2.094 | 1.00 | 0.151 |
| Pb | - | 2.390 | 2.495 | 1.497 | 1.932 | 2.00 | 0.080 |
| Br | - | - | 2.455 | 1.714 | 1.909 | −1.00 | 0.396 |
| Structure | Lattice Parameters | Bulk Modulus | Shear Modulus | Young’s Modulus | Pugh Ratio | ||
|---|---|---|---|---|---|---|---|
| a (Å) | b(Å) | c (Å) | B (GPa) | G (GPa) | E (GPa) | B/G | |
| Orthorhombic | |||||||
| Experimental | a 8.250 | a 11.700 | a 8.212 | d 15.50 | d 5.93 | d 15.80 | d 2.63 |
| LDA | b 8.561 | b 12.190 | b 8.51 | e 18.45 | e 7.90 | e 20.74 | e 2.34 |
| DFT | 8.404 | 11.971 | 8.355 | 18.29 | 7.14 | 18.95 | 2.56 |
| BV model | 8.292 | 11.600 | 8.291 | 14.95 | 6.90 | 17.94 | 2.17 |
| Error | 0.51% | 0.85% | 0.96% | 3.55% | 16.36% | 13.54% | 17.49% |
| Tetragonal | |||||||
| Experimental | c 5.864 | c 5.864 | c 5.811 | - | - | - | - |
| LDA | b 5.772 | b 5.772 | b 5.828 | - | - | - | - |
| DFT | 5.991 | 5.991 | 5.952 | 21.56 | 7.92 | 21.17 | 2.72 |
| BV model | 5.776 | 5.771 | 5.804 | 18.16 | 7.43 | 19.61 | 2.44 |
| Error | 1.50% | 1.59% | 0.12% | - | - | - | - |
| Cubic | |||||||
| Experimental | c 5.874 | c 5.874 | c 5.874 | - | - | - | - |
| LDA | b 5.772 | b 5.772 | b 5.772 | f 19.43 | f 8.31 | f 21.69 | f 2.34 |
| DFT | 5.878 | 5.878 | 5.878 | 18.37 | 6.33 | 17.03 | 2.90 |
| BV model | 5.802 | 5.802 | 5.802 | 18.13 | 6.91 | 18.27 | 2.62 |
| Error | 1.22% | 1.22% | 1.22% | - | - | - | - |
| Structure | Experiment | DFT | BV Model/Error |
|---|---|---|---|
| Orthorhombic | |||
| ac plane | 2.931 | 2.906 | 2.945/0.48% |
| b axis | 2.977 | 2.943 | 3.006/0.82% |
| Tetragonal | |||
| ac plane | 2.821 | 2.910 | 2.851/1.06% |
| b axis | 3.310 | 3.350 | 3.25/1.81% |
| Cubic | |||
| Average | 2.952 | 2.935 | 2.955/0.10% |
| 300 K | 380 K | 600 K | ||
|---|---|---|---|---|
| CsPbBr3 | Experimental | 0.42 | - | - |
| BV model | 0.497 | 0.412 | 0.395 | |
| DFT | ~0.80 | ~0.76 | ~0.70 | |
| CsPbI3 | Experimental | 0.45 | - | - |
| DFT | ~0.38 | ~0.37 | ~0.35 | |
| CsSnI3 | Experimental | 0.38 | - | - |
| DFT | ~0.63 | ~0.55 | ~0.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Q.; Gu, S.; Gou, X. The Highly Accurate Interatomic Potential of CsPbBr3 Perovskite with Temperature Dependence on the Structure and Thermal Properties. Materials 2023, 16, 2043. https://doi.org/10.3390/ma16052043
You Q, Gu S, Gou X. The Highly Accurate Interatomic Potential of CsPbBr3 Perovskite with Temperature Dependence on the Structure and Thermal Properties. Materials. 2023; 16(5):2043. https://doi.org/10.3390/ma16052043
Chicago/Turabian StyleYou, Qianyu, Shun Gu, and Xiaofan Gou. 2023. "The Highly Accurate Interatomic Potential of CsPbBr3 Perovskite with Temperature Dependence on the Structure and Thermal Properties" Materials 16, no. 5: 2043. https://doi.org/10.3390/ma16052043