

Tuning the Electronic and Charge Transport Properties of Schiff Base Compounds by Electron Donor and/or Acceptor Groups

Abstract
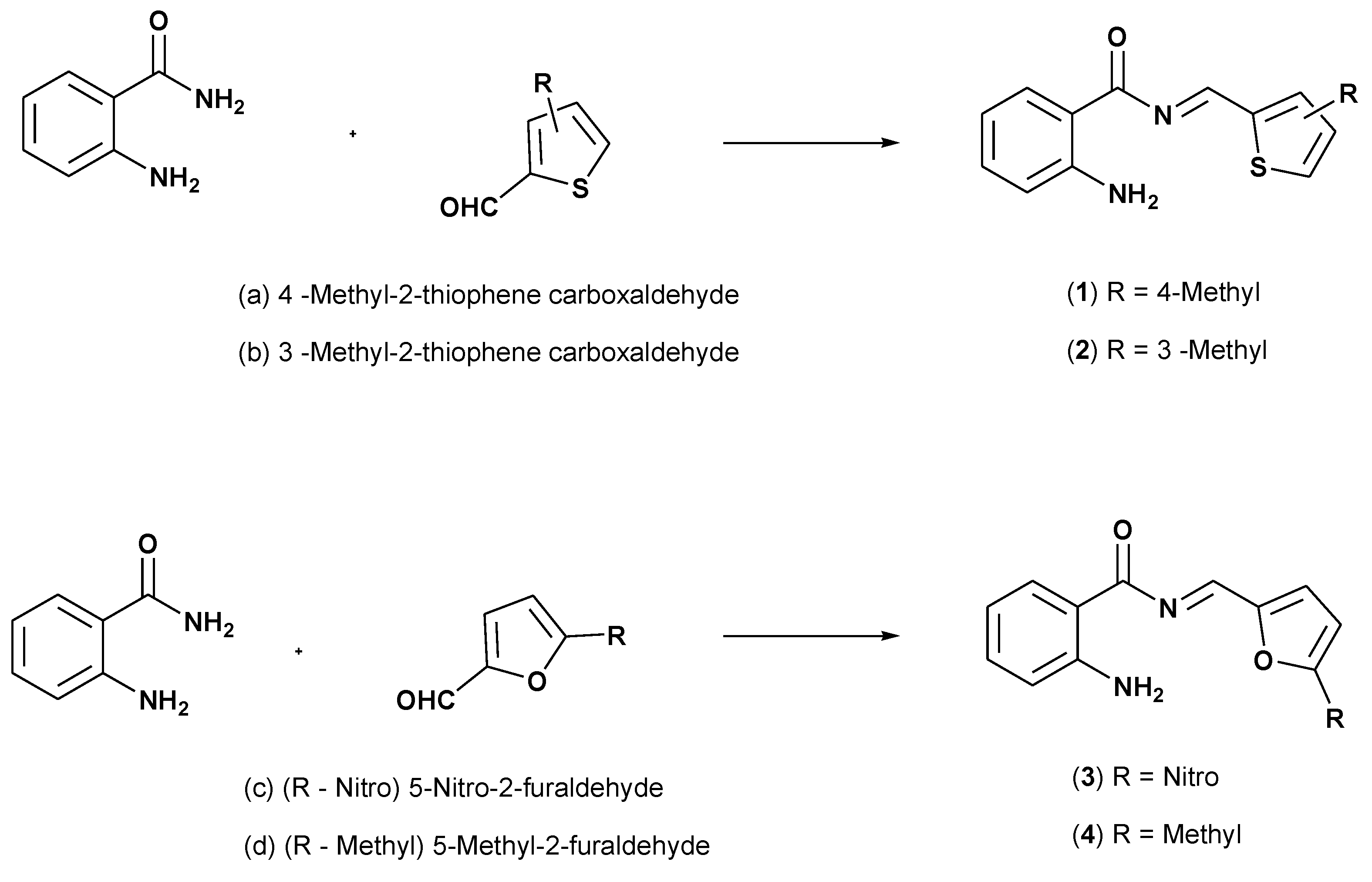
:1. Introduction
2. Computational Details
3. Results and Discussion
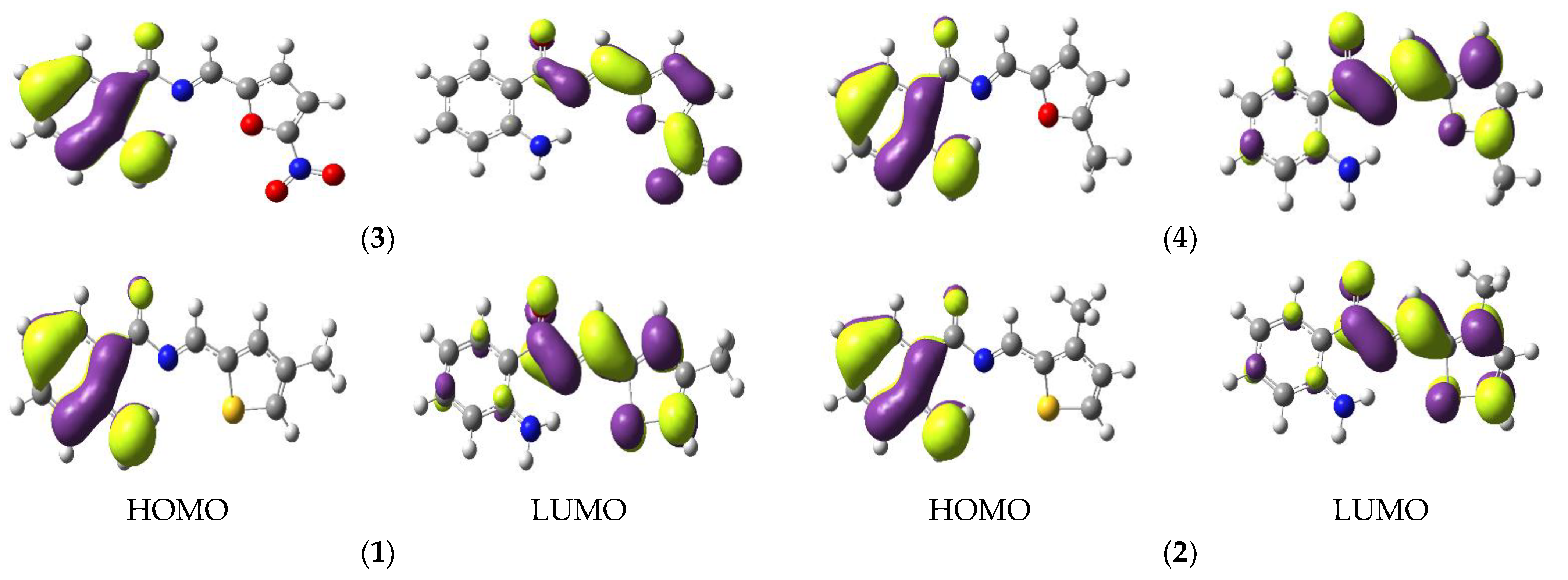
3.1. Electronic Properties
3.2. Absorption Spectra
 to * for the 3 (2.07 eV), while the other Schiff based derivatives showed larger band gaps as established from the optical property values and the occurrence of UV values (see Table 3). These results clearly show that the interaction between the donor and the acceptor, either in an alternating manner or in a separate block in the molecule, performs a significant role in controlling the planarity and the photophysical properties.
to * for the 3 (2.07 eV), while the other Schiff based derivatives showed larger band gaps as established from the optical property values and the occurrence of UV values (see Table 3). These results clearly show that the interaction between the donor and the acceptor, either in an alternating manner or in a separate block in the molecule, performs a significant role in controlling the planarity and the photophysical properties.3.3. Charge Transport Properties
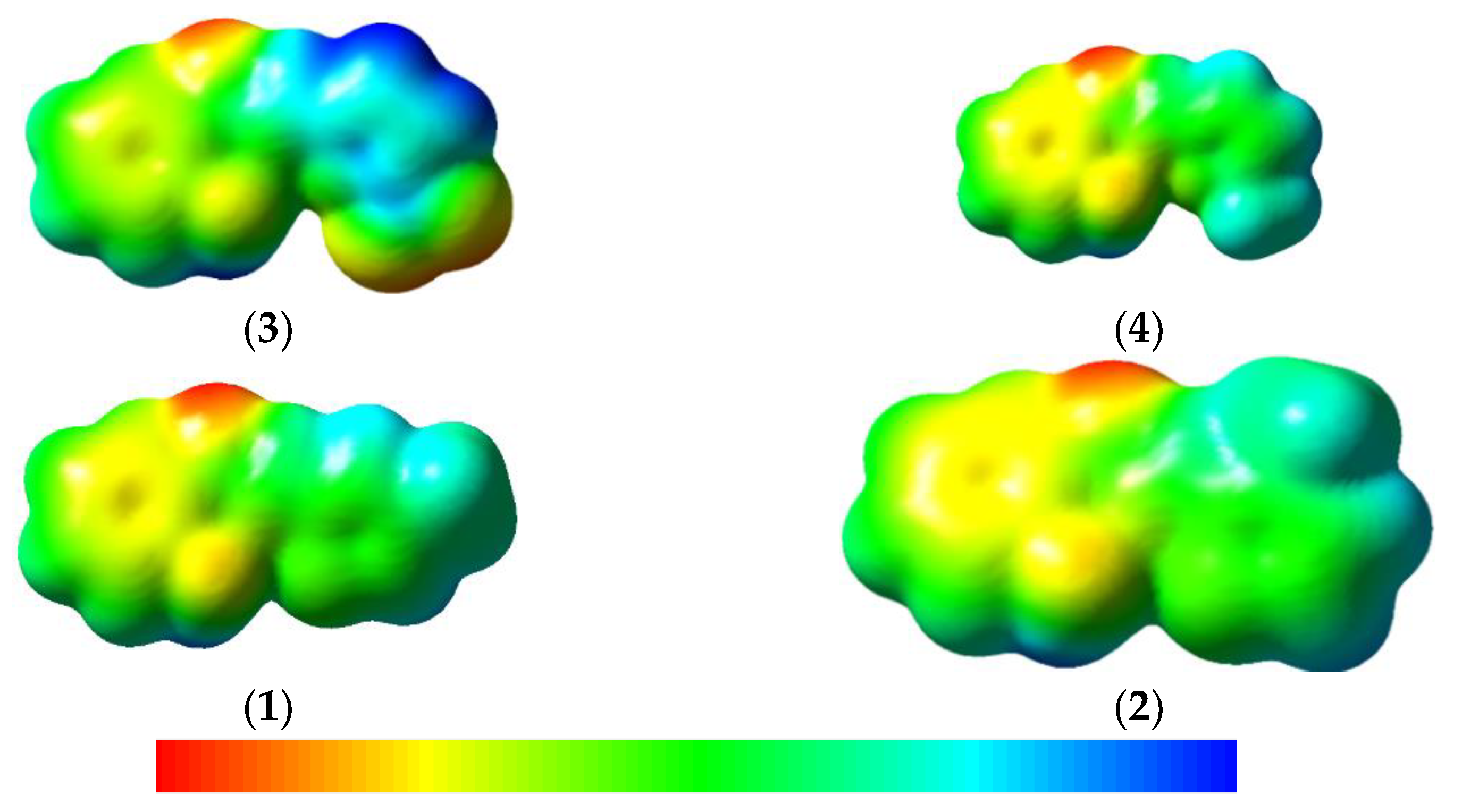
-conjugated organic materials with an electronic charge motion are carried out by a hopping mechanism. The internal reorganization energy (), because of its structural variation from neutral to ionic states (cation and/or anion), is a vital parameter for the charge transfer in organic electronic materials, as it is one of the most important factors that influence the rate of the charge of hopping and considering the mobility of field-effect transistors. In order to have a high degree of mobility of materials, the needs to be reduced. The is a significant parameter for the estimation of the rate of charge transfer. Previously, it was exposed that the DFT can be a trustworthy way to imitate the experimental data [41,42,43,44]. The and external polarization () are two components of total reorganization energy [45]. Here, was projected for the hole () and the electron . The was estimated by Equations (1) and (2) [46]:3.4. Molecular Electrostatic Potential
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reimers, J.; Picconnatto, C.; Ellenbogen, J. (Eds.) Molecular Electronics III (Annals of the New York Academy of Sciences); 2003. Available online: https://www.nyas.org/annals/molecular-electronics-iii/ (accessed on 11 December 2021).
- Tour, J. Molecular Electronics; World Scientific: Singapore, 2003. [Google Scholar]
- Cunimberti, G.; Fagas, G.; Richter, K. (Eds.) Introducing Molecular Electronics; Lecture Notes in Physics; Springer: Berlin/Heidelberg, Germany, 2005. [Google Scholar]
- Kaltenbrunner, M.; White, M.S.; Głowacki, E.D.; Sekitani, T.; Someya, T.; Sarıçiftçi, N.S.; Bauer, S. Ultrathin and lightweight organic solar cells with high flexibility. Nat. Commun. 2012, 3, 770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irfan, A.; Al-Sehemi, A.G.; Al-Assiri, M.S. Push–pull effect on the electronic, optical and charge transport properties of the benzo[2,3-b]thiophene derivatives as efficient multifunctional materials. Comput. Theor. Chem. 2014, 1031, 76–82. [Google Scholar] [CrossRef]
- Szlachcic, P.; Danel, K.S.; Gryl, M.; Stadnicka, K.; Usatenko, Z.; Nosidlak, N.; Lewińska, G.; Sanetra, J.; Kuźnik, W. Organic light emitting diodes (OLED) based on helical structures containing 7-membered fused rings. Dyes Pigm. 2015, 114, 184–195. [Google Scholar] [CrossRef]
- Marks, T.J.; Hersam, M.C. Materials science: Semiconductors grown large and thin. Nature 2015, 520, 631–632. [Google Scholar] [CrossRef]
- Beaujuge, P.M.; Fréchet, J.M.J. Molecular design and ordering effects in π-functional materials for transistor and solar cell applications. J. Am. Chem. Soc. 2011, 133, 20009–20029. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, L.; Wang, C.; Yang, F.; Ren, X.; Zhang, X.; Dong, H.; Hu, W. Organic crystalline materials in flexible electronics. Chem. Soc. Rev. 2019, 48, 1492–1530. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Dong, H.; Jiang, L.; Hu, W. Organic semiconductor crystals. Chem. Soc. Rev. 2018, 47, 422–500. [Google Scholar] [CrossRef]
- Surendar, P.; Pooventhiran, T.; Rajam, S.; Rao, D.J.; Manigandan, N.; Irfan, A.; Thomas, R. Organic quasi-liquid Schiff bases from biomolecules: Synthesis, structure and quantum mechanical studies. Biointerface Res. Appl. Chem. 2023, 13, 107. [Google Scholar] [CrossRef]
- Zhao, K.; Yu, F.; Liu, W.; Huang, Y.; Said, A.A.; Li, Y.; Zhang, Q. Unexpected synthesis, properties, and nonvolatile memory device application of imidazole-fused azaacenes. J. Org. Chem. 2020, 85, 101–107. [Google Scholar] [CrossRef]
- Yi, W.; Zhao, S.; Sun, H.; Kan, Y.; Shi, J.; Wan, S.; Li, C.; Wang, H. Isomers of organic semiconductors based on dithienothiophenes: The effect of sulphur atoms positions on the intermolecular interactions and field-effect performances. J. Mater. Chem. C 2015, 3, 10856–10861. [Google Scholar] [CrossRef]
- Pajak, A.K.; Kotowicz, S.; Gnida, P.; Małecki, J.G.; Ciemiega, A.; Tuczak, A.; Jung, J.; Schab-Balcerzak, E. Synthesis and characterization of new conjugated azomethines end-capped with amino-thiophene-3,4-dicarboxylic acid diethyl ester. Int. J. Mol. Sci. 2022, 23, 8160. [Google Scholar] [CrossRef]
- Grigoras, M.; Catanescu, C.O. Imine oligomers and polymer. J. Macromol. Sci. Part C 2004, 44, 131–173. [Google Scholar] [CrossRef]
- Isık, D.; Santato, C.; Barik, S.; Skene, W.G. Charge-carrier transport in thin films of π-conjugated thiopheno-azomethines. Org. Electron. 2012, 13, 3022–3031. [Google Scholar] [CrossRef]
- Sicard, L.; Navarathne, D.; Skalski, T.; Skene, W.G. On-substrate preparation of an electroactive conjugated polyazomethine from solution-processable monomers and its application in electrochromic devices. Adv. Funct. Mater. 2013, 23, 3549–3559. [Google Scholar] [CrossRef]
- Jeevadason, A.W.; Murugavel, K.K.; Neelakantan, M.A. Review on Schiff bases and their metal complexes as organic photovoltaic materials. Renew. Sustain. Energy Rev. 2014, 36, 220–227. [Google Scholar] [CrossRef]
- García-López, M.C.; Muñoz-Flores, B.M.; Jiménez-Pérez, V.M.; Moggio, I.; Arias, E.; Chan-Navarro, R.; Santillan, R. Santillan, Synthesis and photophysical characterization of organotin compounds derived from Schiff bases for organic light emitting diodes. Dyes Pigm. 2014, 106, 188–196. [Google Scholar] [CrossRef]
- Ivanova, B.B.; Spiteller, M. Optical and nonlinear optical properties of new Schiff’s bases: Experimental versus theoretical study of inclusion interactions. J. Incl. Phenom. Macrocycl. Chem. 2013, 75, 211–221. [Google Scholar] [CrossRef]
- Sıdır, Y.G.; Arslan, C.; Berber, H.; Sıdır, I. The electronic structure, solvatochromism, and electric dipole moments of new schiff base derivatives using absorbance and fluorescence spectra. Struc. Chem. 2019, 30, 835–851. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Q.; Ren, H.; Yan, H.; Zhang, J.; Zhang, H.; Gu, J. Calculation of band gap in long alkyl-substituted heterocyclic-thiophene-conjugated polymers with electron donor–acceptor fragment. Sol. Energy Mater. Sol. Cells 2008, 92, 581–587. [Google Scholar] [CrossRef]
- Hassanali, A.A.; Li, T.; Zhong, D.; Singer, S.J. A molecular dynamics study of lys-trp-lys: Structure and dynamics in solution following photoexcitation. J. Phy. Chem. B 2006, 110, 10497–10508. [Google Scholar] [CrossRef] [PubMed]
- Irfan, A.; Imran, M.; Thomas, R.; Basra, M.A.R.; Ullah, S.; Al-Sehemi, A.G.; Assiri, M.A. Exploring the effect of oligothiophene and acene cores on the optoelectronic properties and enhancing p- and n-type ability of semiconductor materials. J. Sulfur Chem. 2021, 42, 180–192. [Google Scholar] [CrossRef]
- Irfan, A. Push-pull effect on the charge transport characteristics in v-shaped organic semiconductor materials. Bull. Mater. Sci. 2021, 44, 43. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Promising architectures modifying the d-π-a architecture of 2,3-dipentyldithieno[3,2-f:2′,3′-h]quinoxaline-based dye as efficient sensitizers in dye-sensitized solar cells: A DFT study. Mater. Sci. Semiconduc. Process. 2020, 120, 105260. [Google Scholar] [CrossRef]
- Kumar, M.U.; Jeyakumari, A.P.; Suresh, M. Senthilkumar Chandran, G Vinitha. Mater. Res. Express 2019, 6, 075102. [Google Scholar]
- Ermis, E. Synthesis, spectroscopic characterization and DFT calculations of novel Schiff base containing thiophene ring. J. Mol. Struc. 2018, 1156, 91–104. [Google Scholar] [CrossRef]
- Kohn, W.; Becke, A.D.; Parr, R.G. Density functional theory of electronic structure. J. Phys. Chem. 1996, 100, 12974–12980. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D. Density-functional thermochemistry. Iii. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Miehlich, B.; Savin, A.; Stoll, H.; Preuss, H. Results obtained with the correlation energy density functionals of becke and lee, yang and parr. Chem. Phys. Lett. 1989, 157, 200–206. [Google Scholar] [CrossRef]
- Tang, S.-S.; Liu, J.-B.; Chen, G.; Jin, R.-F. Theoretical study on electronic and charge transfer properties of oligo[8]thiophene and its circular, hooped, and helical derivatives. Chin. J. Struc. Chem. 2014, 33, 104–114. [Google Scholar]
- Li, P.; Bu, Y.; Ai, H. Theoretical determinations of ionization potential and electron affinity of glycinamide using density functional theory. J. Phys. Chem. A 2004, 108, 1200–1207. [Google Scholar] [CrossRef]
- Furche, F.; Ahlrichs, R. Adiabatic time-dependent density functional methods for excited state properties. J. Chem. Phys. 2002, 117, 7433–7447. [Google Scholar] [CrossRef]
- Frisch, M.J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.01; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Takano, Y.; Houk, K.N. Benchmarking the conductor-like polarizable continuum model (cpcm) for aqueous solvation free energies of neutral and ionic organic molecules. J. Chem. Theo. Comp. 2005, 1, 70–77. [Google Scholar] [CrossRef]
- Some data from Handbook of Chemistry and Physics. Available online: http://www2.chemistry.msu.edu/faculty/harrison/cem483/work_functions.pdf (accessed on 10 January 2022).
- Sonar, P.; Singh, S.P.; Leclère, P.; Surin, M.; Lazzaroni, R.; Lin, T.T.; Dodabalapur, A.; Sellinger, A. Synthesis, characterization and comparative study of thiophene–benzothiadiazole based donor–acceptor–donor (D-A-D) materials. J. Mater. Chem. 2009, 19, 3228–3237. [Google Scholar] [CrossRef]
- Nikolka, M.; Nasrallah, I.; Rose, B.; Ravva, M.K.; Broch, K.; Sadhanala, A.; Harkin, D.; Charmet, J.; Hurhangee, M.; Brown, A.; et al. High operational and environmental stability of high-mobility conjugated polymer field-effect transistors through the use of molecular additives. Nature Mater. 2017, 16, 356–362. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, D.M.; Simenon, M.M.J.; Brown, A.R.; Einerhand, R.E.F. Stability of n-type doped conducting polymers and consequences for polymeric microelectronic devices. Synth. Met. 1997, 87, 53–59. [Google Scholar] [CrossRef]
- Abbaszadeh, D.; Kunz, A.; Kotadiya, N.B.; Mondal, A.; Andrienko, D.; Michels, J.J.; Wetzelaer, G.-J.A.H.; Blom, P.W.M. Electron trapping in conjugated polymers. Chem. Mater. 2019, 31, 6380–6386. [Google Scholar] [CrossRef] [Green Version]
- Usta, H.; Risko, C.; Wang, Z.; Huang, H.; Deliomeroglu, M.K.; Zhukhovitskiy, A.; Facchetti, A.; Marks, T.J. Design, synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory. J. Am. Chem. Soc. 2009, 131, 5586–5608. [Google Scholar] [CrossRef] [PubMed]
- Felscia, U.R.; Rajkumar, B.J.M.; Mary, M.B. Charge transport properties of pyrene and its derivatives: Optoelectronic and nonlinear optical applications. J. Mater. Sci. 2018, 53, 15213–15225. [Google Scholar] [CrossRef]
- Yang, G.; Su, Z.; Qin, C. Theoretical study on the second-order nonlinear optical properties of asymmetric spirosilabifluorene derivatives. J. Phy. Chem. A 2006, 110, 4817–4821. [Google Scholar] [CrossRef]
- Wazzan, N.; El-Shishtawy, R.M.; Irfan, A. DFT and TD-DFT calculations of the electronic structures and photophysical properties of newly designed pyrene-core arylamine derivatives as hole-transporting materials for perovskite solar cells. Theor. Chem. Acc. 2017, 137, 9. [Google Scholar] [CrossRef]
- Wazzan, N.; Irfan, A. Theoretical study of triphenylamine-based organic dyes with mono-, di-, and tri-anchoring groups for dye-sensitized solar cells. Org. Electron. 2018, 63, 328–342. [Google Scholar] [CrossRef]
- Brédas, J.-L.; Beljonne, D.; Coropceanu, V.; Cornil, J. Charge-transfer and energy-transfer processes in π-conjugated oligomers and polymers: A molecular picture. Chem. Rev. 2004, 104, 4971–5004. [Google Scholar] [CrossRef] [PubMed]
- Chai, W.; Jin, R. Theoretical investigations into optical and charge transfer properties of donor-acceptor 1,8-naphthalimide derivatives as possible organic light-emitting materials. J. Mol. Struct. 2016, 1103, 177–182. [Google Scholar] [CrossRef]
- Gruhn, N.E.; da Silva Filho, D.A.; Bill, T.G.; Malagoli, M.; Coropceanu, V.; Kahn, A.; Brédas, J.-L. The vibrational reorganization energy in pentacene: Molecular influences on charge transport. J. Am. Chem. Soc. 2002, 124, 7918–7919. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Comp. | EHOMO | ELUMO | Eg |
|---|---|---|---|
| 1 | −5.65 | −2.68 | 2.97 |
| 2 | −5.62 | −2.64 | 2.98 |
| 3 | −5.88 | −3.81 | 2.07 |
| 4 | −5.55 | −2.55 | 3.00 |
| Comp. | HIE (Ag) | EIE (Ag) | HIE (Al) | EIE (Al) |
|---|---|---|---|---|
| 1 | 0.91 | 2.06 | 1.57 | 1.40 |
| 2 | 0.88 | 2.02 | 1.54 | 1.44 |
| 3 | 1.14 | 0,93 | 1.80 | 0.27 |
| 4 | 0.81 | 2.19 | 1.47 | 1.53 |
| Comp. | λabs | f | Tran | %Con | λabs | f | Tran | %Con |
|---|---|---|---|---|---|---|---|---|
| Gas Phase | In Ethanol | |||||||
| 1 | 512 | 0.0432 | H → L | 71% | 477 | 0.0721 | H → L | 71% |
| 316 | 0.2682 | H → L+1 | 34% | 334 | 0.4736 | H → L+1 | 12% | |
| 2 | 510 | 0.0439 | H → L | 71% | 474 | 0.0740 | H → L | 70% |
| 312 | 0.2970 | H → L+1 | 47% | 325 | 0.6117 | H → L+1 | 60% | |
| 3 | 745 | 0.0259 | H → L | 71% | 680 | 0.0343 | H → L | 70% |
| 410 | 0.0518 | H → L+1 | 16% | 334 | 0.5260 | H → L+2 | 10% | |
| 4 | 502 | 0.0507 | H → L | 70% | 468 | 0.0860 | H → L | 70% |
| 308 | 0.4157 | H → L+1 | 37% | 330 | 0.7663 | H → L+1 | 12% | |
| 230 | 0.0632 | H → L+2 | 28% | 240 | 0.0918 | H → L+1 | 2% | |
| Comp. | λabs | f | Tran | %Con | λabs | f | Tran | %Con |
| In Acetone | In DMF | |||||||
| 1 | 478 | 0.0717 | H → L | 70% | 477 | 0.0747 | H → L | 70% |
| 334 | 0.4744 | H → L+1 | 12% | 335 | 0.4839 | H → L+1 | 12% | |
| 2 | 474 | 0.0736 | H → L | 70% | 474 | 0.0769 | H → L | 70% |
| 325 | 0.6109 | H → L+1 | 60% | 326 | 0.6271 | H → L+1 | 60% | |
| 3 | 682 | 0.0342 | H → L | 71% | 682 | 0.0342 | H → L | 71% |
| 334 | 0.4860 | H → L+2 | 9% | 335 | 0.5838 | H → L+2 | 9% | |
| 4 | 468 | 0.0855 | H → L | 70% | 468 | 0.0893 | H → L | 70% |
| 330 | 0.7661 | H → L+1 | 13% | 332 | 0.7773 | H → L+1 | 12% | |
| 240 | 0.0918 | H → L+1 | 2% | 240 | 0.0921 | H → L+2 | 3% | |
| Comp. | λabs | f | Tran | %Con | ||||
| In DMSO | ||||||||
| 1 | 477 | 0.0745 | H → L | 70% | ||||
| 335 | 0.4808 | H → L+1 | 12% | |||||
| 2 | 473 | 0.0766 | H → L | 70% | ||||
| 326 | 0.6244 | H → L+1 | 60% | |||||
| 3 | 678 | 0.0352 | H → L | 71% | ||||
| 335 | 0.5819 | H → L+2 | 9% | |||||
| 4 | 467 | 0.0891 | H → L | 70% | ||||
| 331 | 0.7750 | H → L+1 | 12% | |||||
| 240 | 0.0926 | H → L+2 | 3% | |||||
| Comp. | IPa | EAa | IPv | EAv | |||
|---|---|---|---|---|---|---|---|
| 1 | 7.07 | 1.47 | 7.37 | 1.19 | 0.533 | 0.527 | 0.006 |
| 2 | 7.04 | 1.48 | 7.35 | 1.17 | 0.542 | 0.541 | 0.001 |
| 3 | 7.34 | 2.55 | 7.61 | 2.22 | 0.479 | 0.630 | 0.151 |
| 4 | 6.98 | 1.28 | 7.26 | 1.03 | 0.522 | 0.477 | 0.045 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irfan, A.; Al-Sehemi, A.G.; Kalam, A. Tuning the Electronic and Charge Transport Properties of Schiff Base Compounds by Electron Donor and/or Acceptor Groups. Materials 2022, 15, 8590. https://doi.org/10.3390/ma15238590
Irfan A, Al-Sehemi AG, Kalam A. Tuning the Electronic and Charge Transport Properties of Schiff Base Compounds by Electron Donor and/or Acceptor Groups. Materials. 2022; 15(23):8590. https://doi.org/10.3390/ma15238590
Chicago/Turabian StyleIrfan, Ahmad, Abdullah G. Al-Sehemi, and Abul Kalam. 2022. "Tuning the Electronic and Charge Transport Properties of Schiff Base Compounds by Electron Donor and/or Acceptor Groups" Materials 15, no. 23: 8590. https://doi.org/10.3390/ma15238590