
Advances and Challenges of Self-Healing Elastomers: A Mini Review
Abstract
:1. Introduction
2. Healing Process of Intrinsic Self-Healing Polymers
3. Recent Advances of Self-Healing Elastomer with Fast Healing Speed
3.1. Thermally Triggered Fast Self-Healing Elastomers
3.2. Light Triggered Fast Self-Healing Elastomers
3.3. Mechanical Force Enhanced Fast Self-Healing Glassy Polymers
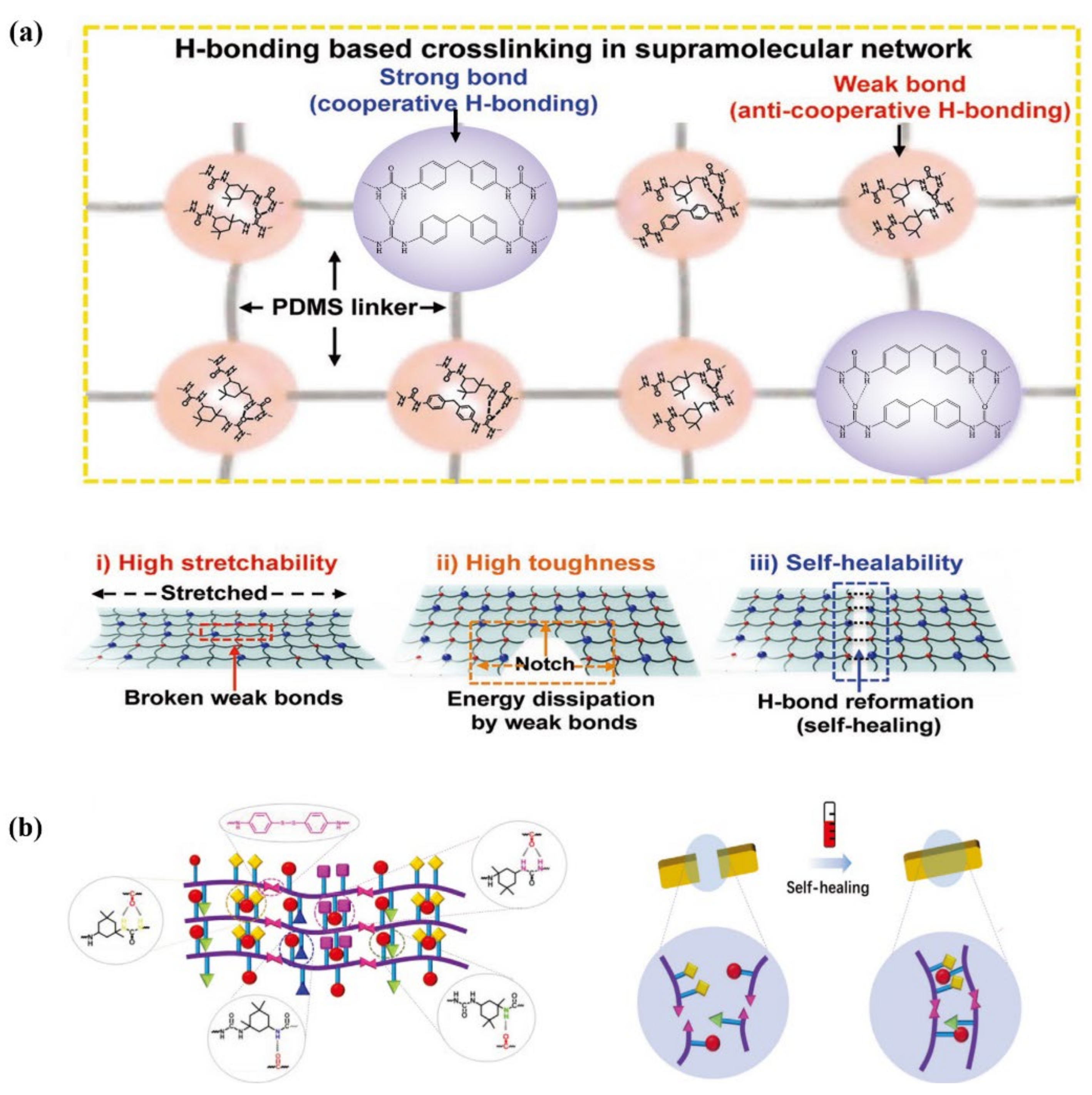
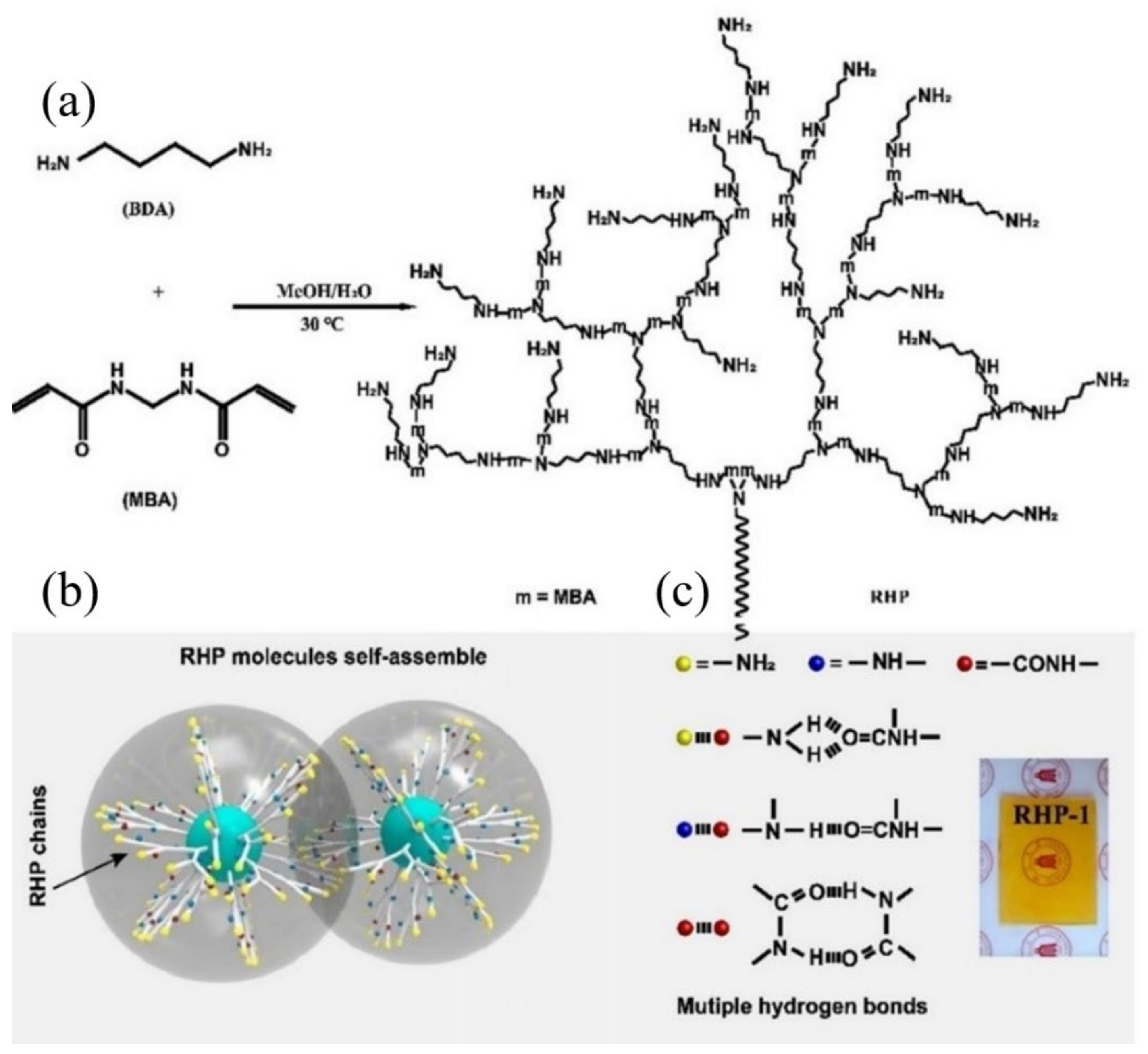
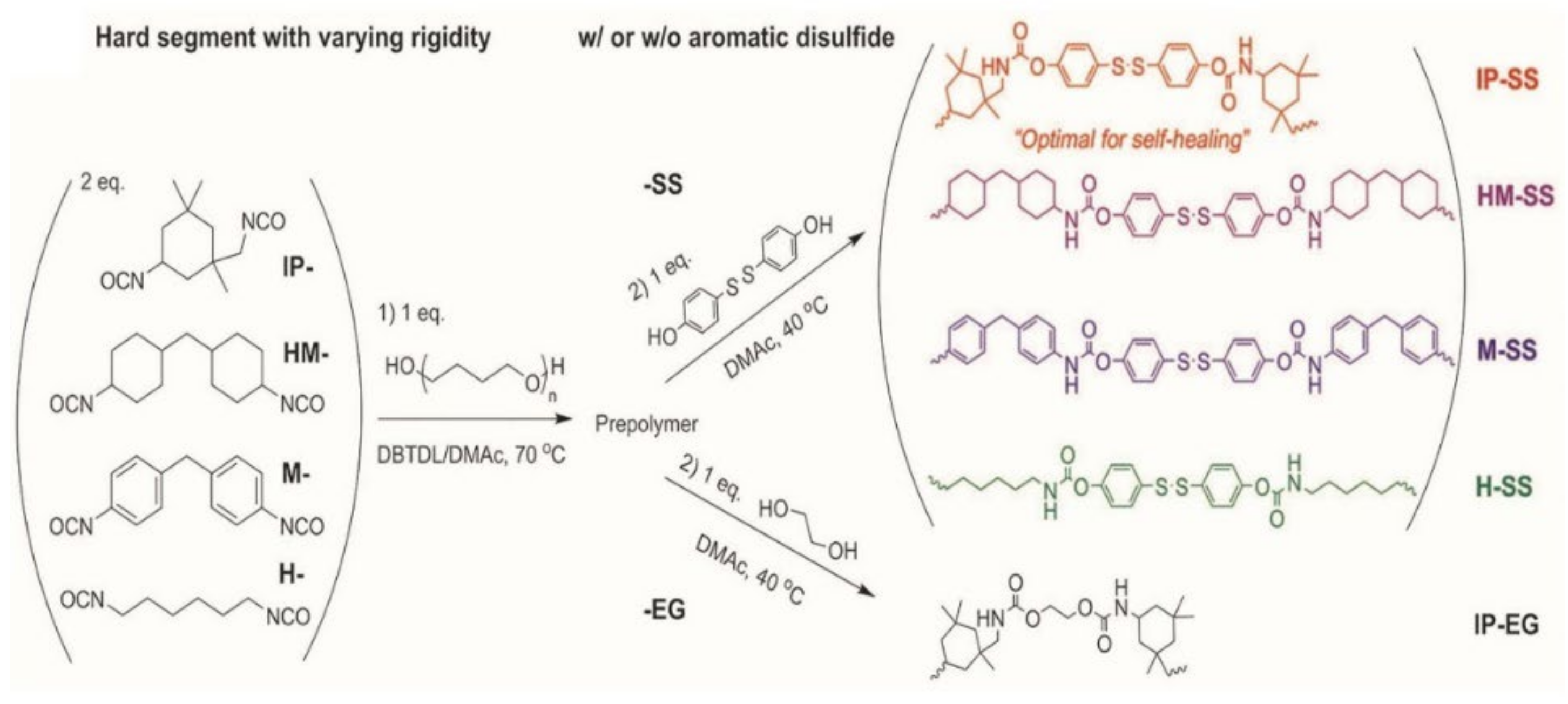
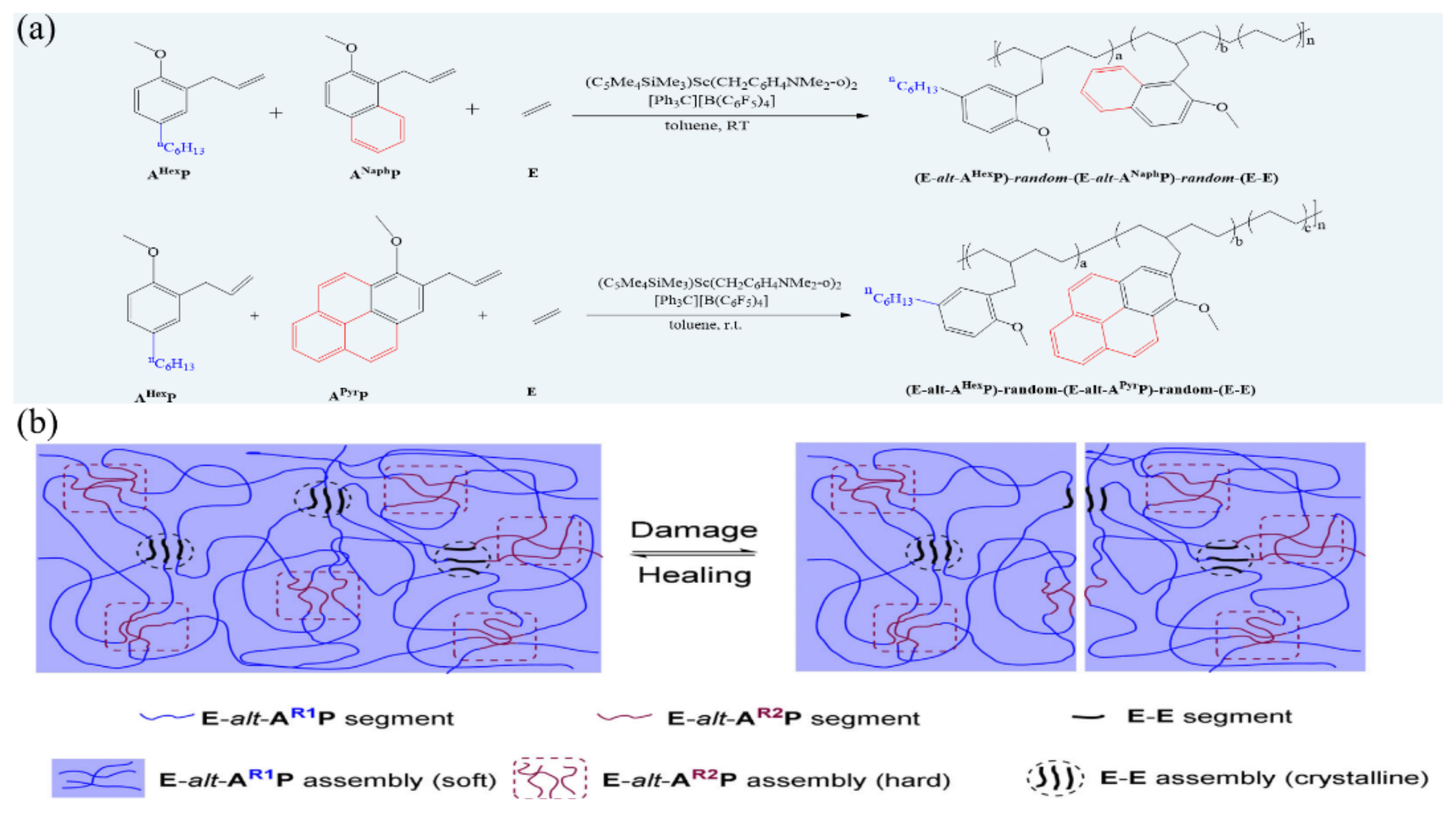
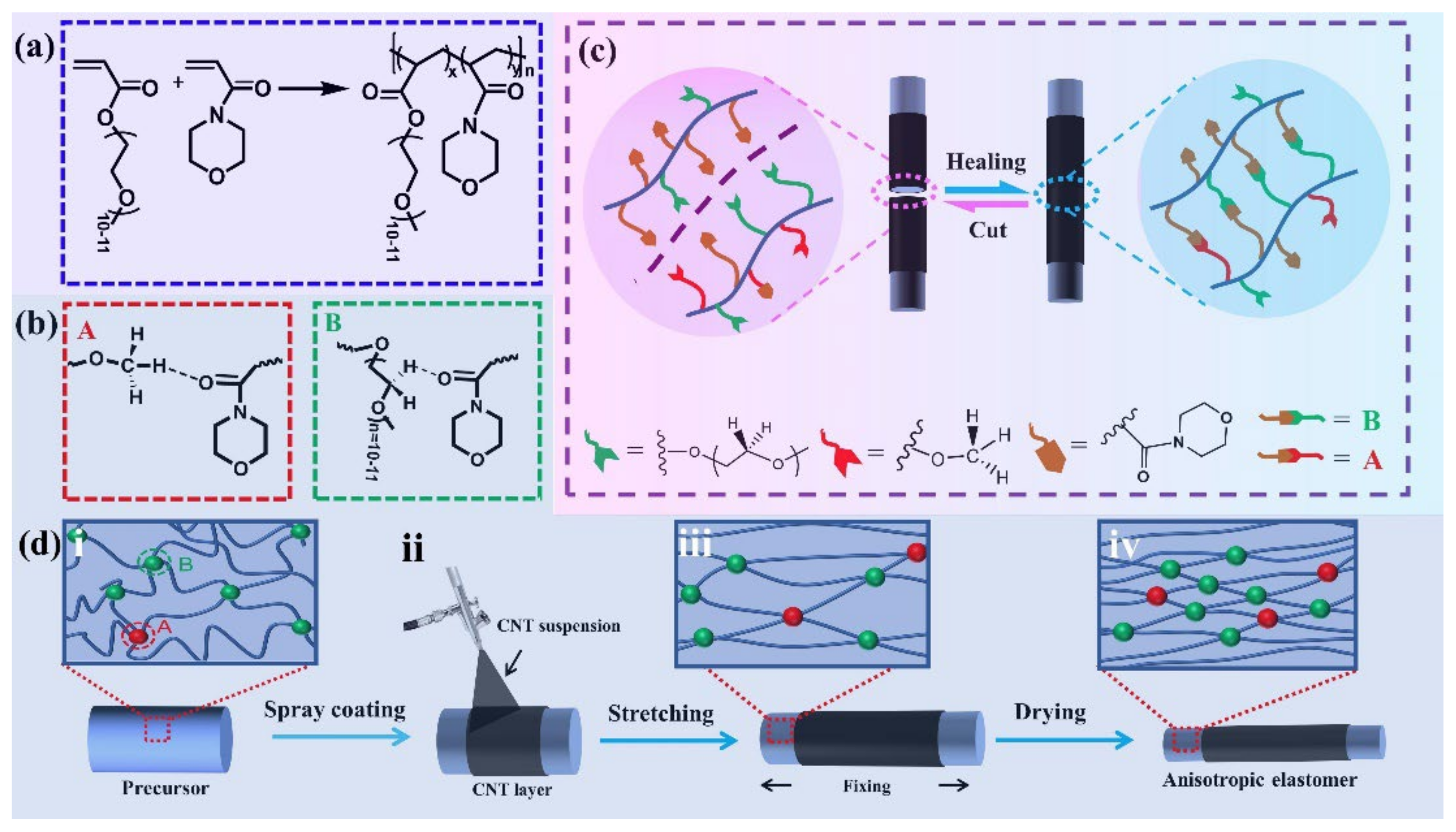
3.4. Room-Temperature Fast Self-Healing Elastomers
4. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, X.C.; Huang, W.P.; Ren, K.F.; Ji, J. Self-healing label materials based on photo-cross-linkable polymeric films with dynamic surface structures. ACS Nano 2018, 12, 8686–8696. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Mei, J.F.; Jia, X.Y.; Li, C.H.; You, X.Z.; Bao, Z.N. A stiff and healable polymer based on dynamic-covalent boroxine bonds. Adv. Mater. 2016, 28, 8277–8282. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ding, X.C.; Urban, W.M. Chemical and physical aspects of self-healing materials. Prog. Polym. Sci. 2015, 49–50, 34–59. [Google Scholar] [CrossRef]
- Li, M.X.; Rong, M.Z.; Zhang, M.Q. Reversible mechanochemistry enabled autonomous sustaining of robustness of polymers—An example of next generation self-healing strategy. Chin. J. Polym. Sci. 2021, 39, 545–553. [Google Scholar] [CrossRef]
- Wu, D.Y.; Meure, S.; Solomon, D. Self-healing polymeric materials: A review of recent developments. Prog. Polym. Sci. 2008, 33, 479–522. [Google Scholar] [CrossRef]
- Wei, Z.; Yang, J.H.; Zhou, J.X.; Xu, F.; Zrı’nyi, M.; Dussault, P.H.; Osadag, Y.; Chen, Y.M. Self-healing gels based on constitutional dynamic chemistry and their potential applications. Chem. Soc. Rev. 2014, 43, 8114–8131. [Google Scholar] [CrossRef]
- Roy, N.; Bruchmannb, B.; Lehn, J.M. Dynamers: Dynamic polymers as self-healing materials. Chem. Soc. Rev. 2015, 44, 3786–3807. [Google Scholar] [CrossRef]
- Murphy, E.B.; Wudl, F. The world of smart healable materials. Prog. Polym. Sci. 2010, 35, 223–251. [Google Scholar] [CrossRef]
- Hou, R.; Li, G.Q.; Zhang, Y.; Li, M.J.; Zhou, G.M.; Chai, X.M. Self-healing polymers materials based on dynamic supramolecular motifs. Prog. Chem. 2019, 31, 690–698. [Google Scholar]
- Espinosaa, L.M.D.; Fiorea, G.L.; Wedera, C.; Fosterb, E.J.; Simon, Y.C. Healable supramolecular polymer solids. Prog. Polym. Sci. 2015, 49–50, 60–78. [Google Scholar] [CrossRef]
- Burattini, S.; Greenland, B.W.; Chappell, D.; Colquhoun, H.M.; Hayes, W. Healable polymeric materials: A tutorial review. Chem. Soc. Rev. 2010, 39, 1973–1985. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Urban, M.W. Self-healing polymeric materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef]
- Zhang, L.Z.; You, Z.W. Dynamic oxime-urethane bonds, a versatile unit of high performance self-healing polymers for diverse applications. Chin. J. Polym. Sci. 2021, 39, 1281–1291. [Google Scholar] [CrossRef]
- White, S.R.; Sottos, N.R.; Geubelle, P.H.; Moore, J.S.; Kessler, M.R.; Sriram, S.R.; Brown, E.N.; Viswanathan, S. Autonomic healing of polymer composites. Nature 2001, 409, 794–797. [Google Scholar] [CrossRef]
- Chen, X.X.; Dam, M.A.; Ono, K.J.; Mal, A.; Shen, H.B.; Nutt, S.R.; Sheran, K.; Wudl, F. A thermally re-mendable cross-linked polymeric material. Science 2002, 295, 1698–1702. [Google Scholar] [CrossRef]
- Canadell, J.; Goossens, H.; Klumperman, B. Self-Healing Materials Based on Disulfide Links. Macromolecules 2011, 44, 2536–2541. [Google Scholar] [CrossRef]
- Yang, X.; Liu, J.Z.; Fan, D.Y.; Cao, J.; Huang, X.; Zheng, Z.; Zhang, X.X. Scalable manufacturing of real-time self-healing strain sensors based on brominated natural rubber. Chem. Eng. J. 2020, 389, 124448. [Google Scholar] [CrossRef]
- Cao, J.; Lu, C.H.; Zhuang, J.; Liu, M.X.; Zhang, X.X.; Yu, Y.M.; Tao, Q.C. Multiple hydrogen bonding enables the self-healing of sensors for human-machine interactions. Angew. Chem. Int. Ed. Engl. 2017, 56, 8795–8800. [Google Scholar] [CrossRef]
- Liu, X.H.; Su, G.H.; Guo, Q.Q.; Lu, C.H.; Zhou, T.; Zhou, C.L.; Zhang, X.X. Hierarchically structured self-healing sensors with tunable positive/negative piezoresistivity. Adv. Funct. Mater. 2018, 28, 1706658. [Google Scholar] [CrossRef]
- Yin, Q.Y.; Dai, C.H.; Chen, H.; Gou, K.; Guan, H.Z.; Wang, P.H.; Jiang, J.T.; Weng, G.S. Tough double metal-ion cross-linked elastomers with temperature-adaptable self-healing and luminescence properties. Chin. J. Polym. Sci. 2021, 39, 554–565. [Google Scholar] [CrossRef]
- Döhler, D.; Kang, J.; Cooper, C.B.; Tok, J.B.H.; Rupp, H.; Binder, W.H.; Bao, Z.N. Tuning the self-healing response of poly (dimethylsiloxane)-based elastomers. ACS Appl. Polym. Mater. 2020, 2, 4127–4139. [Google Scholar] [CrossRef]
- Chen, S.B.; Mahmood, N.; Beiner, M.; Binder, W.H. Self-healing materials from V-and H-Shaped supramolecular architectures. Angew. Chem. Int. Ed. 2015, 127, 10326–10330. [Google Scholar] [CrossRef]
- Bueche, F.; Cashin, W.M.; Debye, P. The measurement of self-diffusion in solid polymers. J. Chem. Phys. 1952, 20, 1956–1958. [Google Scholar] [CrossRef]
- Zhai, L.; Narkarb, A.; Ahn, K. Self-healing polymers with nanomaterials and nanostructures. Nano Today 2020, 30, 100826. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Qi, T.; Li, G.L. Towards dynamic but supertough healable polymers through biomimetic hierarchical hydrogen-bonding interactions. Angew. Chem. Int. Edit. 2018, 57, 13838–13842. [Google Scholar] [CrossRef]
- Feng, X.Q.; Zhang, G.Z.; Xu, B.; Jiang, H.Y.; Baia, Q.; Li, H.J. Self-healing elastomer assembly towards three-dimensional shape memory devices. Rsc Adv. 2015, 5, 70000–70004. [Google Scholar] [CrossRef]
- Feng, X.Q.; Zhang, G.Z.; Bai, Q.M.; Jiang, H.Y.; Bo, X.; Li, H.J. High strength Self-healing magnetic elastomers with shape memory effect. Macromol. Mater. Eng. 2016, 301, 125–132. [Google Scholar] [CrossRef]
- Fan, C.J.; Huang, Z.C.; Li, B.; Xiao, W.X.; Zheng, E.; Yang, K.K.; Wang, Y.Z. A robust self-healing polyurethane elastomer: From H-bonds and stacking interactions to well-defined microphase morphology. Sci. China. Mater. 2019, 62, 1188–1198. [Google Scholar] [CrossRef]
- Li, Y.H.; Li, W.J.; Sun, A.L.; Jing, M.F.; Liu, X.J.; Wei, L.H.; Wu, K.; Fu, Q. A self-reinforcing and self-healing elastomer with high strength, unprecedented toughness and room-temperature reparability. Mater. Horiz. 2021, 8, 267–275. [Google Scholar] [CrossRef]
- Chen, C.X.; Chen, S.; Guo, Z.H.; Hu, W.R.; Chen, Z.P.; Wang, J.W.; Hu, J.S.; Guo, J.; Yang, L.Q. Highly efficient self-healing materials with excellent shape memory and unprecedented mechanical properties. J. Mater. Chem. A 2020, 8, 16203–16211. [Google Scholar] [CrossRef]
- Wang, D.; Wang, Z.F.; Ren, S.Y.; Xu, J.H.; Wang, C.; Hu, P.; Fu, J.J. Molecular engineering of a colorless, extremely tough, superiorly self-recoverable, and healable poly(urethane-urea) elastomer for impact-resistant applications. Mater. Horiz. 2021, 8, 2238–2250. [Google Scholar] [CrossRef]
- Eom, Y.; Kim, S.M.; Lee, M.; Jeon, H.; Park, J.; Lee, E.S.; Hwang, S.Y.; Park, J.; Oh, D.X. Mechano-responsive hydrogen-bonding array of thermoplastic polyurethane elastomer captures both strength and self-healing. Nat. Commun. 2021, 12, 621. [Google Scholar] [CrossRef]
- An, N.; Wang, X.H.; Li, Y.X.; Zhang, L.; Lu, Z.Y.; Sun, J.Q. Healable and mechanically super-Strong polymeric composites derived from hydrogen-bonded polymeric complexes. Adv. Mater. 2019, 31, 1904882. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Huang, X.; Zhang, X.X. Ultrarobust, tough and highly stretchable self-healing materials based on cartilage-inspired noncovalent assembly nanostructure. Nat. Commun. 2021, 12, 1291. [Google Scholar] [CrossRef]
- Weng, W.; Yang, J.J.; Zhang, Y.; Li, Y.; Yang, S.Y.; Zhu, L.P.; Zhu, M.F. A route toward smart system integration: From fiber design to device construction. Adv. Mater. 2020, 32, 1902301. [Google Scholar] [CrossRef]
- Shi, Q.W.; Sun, J.Q.; Hou, C.Y.; Li, Y.G.; Zhang, Q.H.; Wang, H.Z. Advanced functional fiber and smart textile. Adv. Fiber Mater. 2019, 1, 3–31. [Google Scholar] [CrossRef]
- Xu, K. Manipulating interphases in batteries. Natl. Sci. Rev. 2017, 4, 19–20. [Google Scholar] [CrossRef]
- Liao, S.; Lian, X.; Wang, Y. Self-healing ionic liquid-based electronics and beyond. Chin. J. Polym. Sci. 2021, 39, 1235–1245. [Google Scholar] [CrossRef]
- Liu, W.X.; Zhang, C.; Zhang, H.; Zhao, N.; Yu, Z.X.; Xu, J. Oxime-based and catalyst-free dynamic covalent polyurethanes. J. Am. Chem. Soc. 2017, 139, 8678–8684. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, Z.; Wu, X.; Guan, Q.; Chen, S.; Sun, L.; Guo, Y.; Wang, S.; Song, J.; Jeffries, E.M.; et al. A highly efficient self-healing elastomer with unprecedented mechanical properties. Adv. Mater. 2019, 31, 1901402. [Google Scholar] [CrossRef]
- Chen, L.; Sun, T.L.; Cui, K.P.; King, D.R.; Kurokawa, T.; Saruwatari, Y.; Gong, J.P. Facile synthesis of novel elastomers with tunable dynamics for toughness, self-healing and adhesion. J. Mater. Chem. A. 2019, 7, 17334–17344. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Z.H.; Zhang, X.H.; Liu, Y.J.; Wu, S.W.; Guo, B.C. Covalently cross-linked elastomers with self-healing and malleable abilities enabled by boronic ester bonds. ACS. Appl. Mater. Interfaces. 2018, 10, 24224–24231. [Google Scholar] [CrossRef] [PubMed]
- Yue, D.W.; Wang, H.Q.; Tao, H.Q.; Zheng, P.; Li, C.H.; Zuo, J.L. A fast and room-temperature self-healing thermal conductive polymer composite. Chin. J. Polym. Sci. 2021, 39, 1328–1336. [Google Scholar] [CrossRef]
- Wool, R.P.; O’Connor, K.M. A theory crack healing in polymers. J. Appl. Phys. 1981, 52, 5953–5963. [Google Scholar] [CrossRef]
- Kim, Y.H.; Wool, R.P. A theory of healing at a polymer-polymer inter face. Macromolecules 1983, 16, 1115–1120. [Google Scholar] [CrossRef]
- Wool, R.P. Self-healing materials: A review. Soft. Matter. 2008, 4, 400–418. [Google Scholar] [CrossRef]
- Mordvinkin, A.; Döhler, D.; Binder, W.H.; Colby, R.H.; Saalwächter, K. Rheology, Sticky Chain, and Sticker Dynamics of Supramolecular Elastomers Based on Cluster-Forming Telechelic Linear and Star Polymers. Macromolecules 2021, 54, 5065–5076. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Russell, T.P.; Deline, V.R.; Dozier, W.D.; Felcher, G.P.; Agrawal, G.; Wool, R.P.; Mays, J.W. Direct observation of reputation at polymer interfaces. Nature 1993, 365, 235–237. [Google Scholar] [CrossRef]
- Aguirresarobe, R.H.; Nevejans, S.; Reck, B.; Irusta, L.; Sardon, H.; Asua, J.M.; Ballard, N. Healable and self-healing polyurethanes using dynamic chemistry. Prog. Polym. Sci. 2021, 114, 101362. [Google Scholar] [CrossRef]
- Cordier, P.; Tournilhac, F.; Soulie-Ziakovic, C.; Leibler, L. Self-healing and thermoreversible rubber from supramolecular assembly. Nature 2008, 451, 977–980. [Google Scholar] [CrossRef]
- Chen, Y.L.; Kushner, A.M.; Williams, G.A.; Guan, Z.B. Multiphase design of autonomic self-healing thermoplastic elastomers. Nat. Chem. 2012, 4, 467–472. [Google Scholar] [CrossRef]
- Zhang, R.C.; Yan, T.Z.; Lechner, B.D.; Schröter, K.; Liang, Y.; Li, B.H.; Furtado, F.; Sun, P.C.; Saalwächter, K. Heterogeneity, segmental and hydrogen bond dynamics, and aging of supramolecular self-healing rubber. Macromolecules 2013, 46, 1841–1850. [Google Scholar] [CrossRef]
- Wang, D.; Xu, J.H.; Chen, J.Y.; Hu, P.; Wang, Y.; Jiang, W.; Fu, J.J. Transparent, mechanically strong, extremely tough, self-recoverable, healable supramolecular elastomers facilely fabricated via dynamic hard domains design for multifunctional Applications. Adv. Funct. Mater. 2019, 30, 1907109. [Google Scholar] [CrossRef]
- Lai, J.C.; Li, L.; Wang, D.P.; Zhang, M.H.; Mo, S.R.; Wang, X.; Zeng, K.Y.; Li, C.H.; Jiang, Q.; You, X.Z.; et al. A rigid and healable polymer cross-linked by weak but abundant Zn(II)-carboxylate interactions. Nat. Commun. 2018, 9, 2725. [Google Scholar] [CrossRef]
- Cao, Y.; Morrissey, T.G.; Acome, E.; Allec, S.I.; Wong, B.M.; Keplinger, C.; Wang, C. A transparent, self-healing, highly stretchable ionic conductor. Adv. Mater. 2017, 29, 1605099. [Google Scholar] [CrossRef]
- Rao, Y.L.; Chortos, A.; Pfattner, R.; Lissel, F.; Chiu, Y.C.; Feig, V.; Xu, J.; Kurosawa, T.; Gu, X.D.; Wang, C.; et al. Stretchable self-healing polymeric dielectrics cross-linked through metal-ligand coordination. J. Am. Chem. Soc. 2016, 138, 6020–6027. [Google Scholar] [CrossRef]
- Grande, A.M.; Garcia, S.J.; Van der Zwaag, S. On the interfacial healing of a supramolecular elastomer. Polymer 2015, 56, 435–442. [Google Scholar] [CrossRef]
- Santana, M.H.; Huete, M.; Lameda, P.; Araujo, J.; Verdejo, R.; López-Manchado, M.A. Design of a new generation of sustainable SBR compounds with good trade-off between mechanical properties and self-healing ability. Eur. Polym. J. 2018, 106, 273–283. [Google Scholar] [CrossRef]
- AbdolahZadeh, M.; Esteves, A.C.C.; van der Zwaag, S.; Garcia, S.J. Healable dual organic–inorganic crosslinked Sol–Gel based polymers: Crosslinking density and tetrasulfide content effect. J. Polym. Sci. Part A Polym. Chem. 2014, 52, 1953–1961. [Google Scholar] [CrossRef]
- Hernández, M.; Grande, A.M.; van der Zwaag, S.; Garcia, S.J. Monitoring network and interfacial healing processes by broadband dielectric spectroscopy: A case study on natural rubber. ACS Appl. Mater. Interfaces 2016, 8, 10647–10656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.P.; Rong, M.Z.; Zhang, M.Q. Self-healing, reshaping, and recycling of vulcanized chloroprene rubber: A case study of multitask cyclic utilization of cross-linked polymer. ACS Sustain. Chem. Eng. 2016, 4, 2715–2724. [Google Scholar] [CrossRef]
- Hernández, M.; Grande, A.M.; Dierkes, W.; Bijleveld, J.; Van Der Zwaag, S.; García, S.J. Turning vulcanized natural rubber into a self-healing polymer: Effect of the disulfide/polysulfide ratio. ACS Sustain. Chem. Eng. 2016, 4, 5776–5784. [Google Scholar] [CrossRef]
- Yu, H.T.; Feng, Y.Y.; Gao, L.; Chen, C.; Zhang, Z.X.; Feng, W. Self-healing high strength and thermal conductivity of 3D graphene/PDMS composites by the optimization of multiple molecular interactions. Macromolecules 2020, 53, 7161–7170. [Google Scholar] [CrossRef]
- De Alwis Watuthanthrige, N.; Ahammed, B.; Dolan, M.T.; Fang, Q.H.; Wu, J.; Sparks, J.L.; Zanjani, M.B.; Konkolewicz, D.; Ye, Z.J. Accelerating dynamic exchange and self-healing using mechanical forces in crosslinked polymers. Mater. Horiz. 2020, 7, 1581–1587. [Google Scholar] [CrossRef]
- Wang, S.Y.; Urban, M.W. Self-healing polymers. Nat. Rev. Mater. 2020, 5, 562–583. [Google Scholar] [CrossRef]
- Zedler, L.; Hager, M.D.; Schubert, U.S.; Harrington, M.J.; Schmitt, M.; Popp, J.; Dietzek, B. Monitoring the chemistry of self-healing by vibrational spectroscopy—Current state and perspectives. Mater. Today 2014, 17, 57–69. [Google Scholar] [CrossRef]
- Van der Kooij, H.M.; Susa, A.; Garcia, S.J.; Van der Zwaag, S.; Sprakel, J. Imaging the molecular motions of autonomous repair in a self-healing polymer. Adv. Mater. 2017, 29, 1701017. [Google Scholar] [CrossRef]
- Neumann, L.N.; Oveisi, E.; Petzold, A.; Style, R.W.; Thurn-Albrecht, T.; Weder, C.; Schrettl, S. Dynamics and healing behavior of metallosupramolecular polymers. Sci. Adv. 2021, 7, 4154. [Google Scholar] [CrossRef]
- Li, Z.J.; Zhong, J.; Liu, M.C.; Rong, J.C.; Yang, K.; Zhou, J.Y.; Shen, L.; Gao, F.; He, H.F. Investigation on self-healing property of epoxy resins based on disulfide dynamic links. Chin. J. Polym. Sci. 2020, 38, 932–940. [Google Scholar] [CrossRef]
- Wang, B.S.; Zhai, W.Z.; Fan, J.B.; Xu, J.; Zhao, W.P.; Feng, X.Q. An interfacially polymerized self-healing organo/hydro copolymer with shape memory. Nanoscale 2019, 11, 6846–6851. [Google Scholar] [CrossRef]
- Zhuo, S.Y.; Liu, Y.X.; Zhou, L.L.; Feng, X.Q. Enhanced dual-responsive shape memory nanocomposites with rapid and efficient self-healing capability. J. Mater. Sci. 2018, 53, 13936–13948. [Google Scholar] [CrossRef]
- Wu, H.H.; Xie, H.P.; Tian, X.X.; Sun, Y.L.; Shi, B.R.; Zhou, Y.; Sheng, D.K.; Liu, X.D.; Yang, Y.M. Hard, tough and fast self-healing thermoplastic polyurethane. Prog. Org. Coat. 2021, 159, 106409. [Google Scholar] [CrossRef]
- Kang, J.; Son, D.; Wang, G.N.; Liu, Y.X.; Lopez, J.; Kim, Y.; Oh, J.Y.; Katsumata, T.; Mun, J.; Lee, Y.; et al. Tough and water-insensitive self-healing elastomer for robust electronic skin. Adv. Mater. 2018, 30, 1706846. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Y.J.; Zhao, X.J.; Yang, X.; Yu, R.; Zhang, Y.; Huang, W. A high strength but fast fracture-self-healing thermoplastic elastomer. Macromol. Rapid. Commun. 2021, 42, 2100135. [Google Scholar] [CrossRef]
- Yuan, D.; Bonab, V.S.; Patel, A.; Manas-Zloczower, I. Self-healing epoxy coatings with enhanced properties and facile processability. Polymer 2018, 147, 196–201. [Google Scholar] [CrossRef]
- Wang, Z.Y.; Yang, H.T.; Fairbanks, B.D.; Liang, H.B.; Ke, J.J.; Zhu, C.F. Fast self-healing engineered by UV-curable polyurethane contained Diels-Alder structure. Prog. Org. Coat. 2019, 131, 131–136. [Google Scholar] [CrossRef]
- Wang, X.Y.; Zhao, M.Y.; Zhang, L.; Li, K.; Wang, D.; Zhang, L.; Zhang, A.M.; Xu, Y. Liquid metal bionic instant self-healing flexible electronics with full recyclability and high reliability. Chem. Eng. J. 2022, 431, 133965. [Google Scholar] [CrossRef]
- Yang, Y.; Davydovich, D.; Hornat, C.C.; Liu, X.L.; Urban, M.W. Leaf-inspired self-healing polymers. Chem 2018, 4, 1928–1936. [Google Scholar] [CrossRef]
- Yang, S.W.; Wang, S.; Du, X.S.; Cheng, X.; Wang, H.B.; Du, Z.L. Mechanically and thermo-driven self-healing polyurethane elastomeric composites using inorganic–organic hybrid material as crosslinker. Polym. Chem. 2020, 11, 1161–1170. [Google Scholar] [CrossRef]
- Lai, Y.; Kuang, X.; Zhu, P.; Huang, M.M.; Dong, X.; Wang, D.J. Colorless, transparent, robust, and fast scratch-self-healing elastomers via a phase-locked dynamic bonds design. Adv. Mater. 2018, 30, 1802556. [Google Scholar] [CrossRef]
- Zhang, L.D.; Qiu, T.; Zhu, Z.Q.; Guo, L.H.; Li, X.Y. Self-healing polycaprolactone networks through thermo-induced reversible disulfide bond formation. Macromol. Rapid. Commun. 2018, 39, 1800121. [Google Scholar] [CrossRef]
- Pena-Francesch, A.; Jung, H.H.; Demirel, M.C.; Sitti, M. Biosynthetic self-healing materials for soft machines. Nat. Mater. 2020, 19, 1230–1235. [Google Scholar] [CrossRef]
- Solouki Bonab, V.; Karimkhani, V.; Manas-Zloczower, I. Ultra-fast microwave assisted self-healing of covalent adaptive polyurethane networks with carbon nanotubes. Macromol. Mater. Eng. 2019, 304, 1800405. [Google Scholar] [CrossRef]
- Chen, Q.M.; Yu, X.W.; Pei, Z.Q.; Yang, Y.; Wei, Y.; Ji, Y. Multi-stimuli responsive and multi-functional oligoaniline-modified vitrimers. Chem. Sci. 2017, 8, 724–733. [Google Scholar] [CrossRef]
- Li, Z.; Yao, Y.G.; Lin, Z.Y.; Moon, K.S.; Lin, W.; Wong, C.P. Ultrafast, dry microwave synthesis of graphene sheets. J. Mater. Chem. 2010, 20, 4781. [Google Scholar] [CrossRef]
- Huang, L.; Yi, N.B.; Wu, Y.P.; Zhang, Y.; Zhang, Q.; Huang, Y.; Ma, Y.F.; Chen, Y.S. Multichannel and repeatable self-healing of mechanical enhanced graphene-thermoplastic polyurethane composites. Adv. Mater. 2013, 25, 2224–2228. [Google Scholar] [CrossRef]
- Yang, Y.; Pei, Z.Q.; Zhang, X.Q.; Tao, L.; Wei, Y.; Ji, Y. Carbon nanotube-vitrimer composite for facile and efficient photo-welding of epoxy. Chem. Sci. 2014, 5, 3486–3492. [Google Scholar] [CrossRef]
- Yan, J.; Li, M.F.; Wang, Z.W.; Chen, C.; Ma, C.Q.; Yang, G. Highly tough, multi-stimuli-responsive, and fast self-healing supramolecular networks toward strain sensor application. Chem. Eng. J. 2020, 389, 123468. [Google Scholar] [CrossRef]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-healing of covalently cross-linked polymers by reshuffling thiuram disulfide moieties in air under visible light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Ji, S.B.; Cao, W.; Yu, Y.; Xu, H.P. Visible-light-induced self-healing diselenide-containing polyurethane elastomer. Adv. Mater. 2015, 27, 7740–7745. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xia, J.H.; Ji, S.B.; Fan, Z.Y.; Xu, H.P. Visible-light-induced metathesis reaction between diselenide and ditelluride. Chem. Commun. 2019, 55, 2813–2816. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.H.; Jin, Y.; Shi, L.J.; Du, W.N.; Zhou, R.; Lai, S.Q.; Shen, Y.C.; Li, Y.P. Achieving fast self-healing and reprocessing of supertough water-dispersed “Living” supramolecular polymers containing dynamic ditelluride bonds under visible light. ACS. Appl. Mater. Interfaces 2020, 12, 6383–6395. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.J.; Zhang, Y.F.; Montazerian, M.; Gulbiten, O.; Mauro, J.C.; Zanotto, E.D.; Yue, Y.Z. Understanding glass through differential scanning calorimetry. Chem. Rev. 2019, 119, 7848–7939. [Google Scholar] [CrossRef]
- Yan, P.Y.; Zhao, W.; Fu, X.W.; Liu, Z.M.; Kong, W.B.; Zhou, C.L.; Lei, J.X. Multifunctional polyurethane-vitrimers completely based on transcarbamoylation of carbamates: Thermally-induced dual-shape memory effect and self-welding. RSC Adv. 2017, 7, 26858–26866. [Google Scholar] [CrossRef]
- Wang, H.; Liu, H.C.; Cao, Z.X.; Li, W.H.; Huang, X.; Zhu, Y.; Ling, F.W.; Xu, H.; Wu, Q.; Peng, Y.; et al. Room-temperature autonomous self-healing glassy polymers with hyperbranched structure. Proc. Natl. Acad. Sci. USA 2020, 117, 11299–11305. [Google Scholar] [CrossRef]
- Yanagisawa, Y.; Nan, Y.L.; Okuro, K.; Aida, T. Mechanically robust, readily repairable polymers via tailored noncovalent cross-linking. Science 2018, 359, 72–76. [Google Scholar] [CrossRef]
- Xu, J.H.; Chen, J.Y.; Zhang, Y.N.; Liu, T.; Fu, J.J. A fast room-temperature self-healing glassy polyurethane. Angew. Chem. Int. Ed. Engl. 2021, 60, 7947–7955. [Google Scholar] [CrossRef]
- Guo, H.S.; Han, Y.; Zhao, W.Q.; Yang, J.; Zhang, L. Universally autonomous self-healing elastomer with high stretchability. Nat. Commun. 2020, 11, 2037. [Google Scholar] [CrossRef]
- Hentschel, J.; Kushner, A.M.; Ziller, J.; Guan, Z.B. Self-healing supramolecular block copolymers. Angew. Chem. Int. Ed. Engl. 2012, 51, 10561–10565. [Google Scholar] [CrossRef]
- Xu, J.H.; Chen, P.; Wu, J.W.; Hu, P.; Fu, Y.S.; Jiang, W.; Fu, J.J. Notch-insensitive, ultrastretchable, efficient self-healing supramolecular polymers constructed from multiphase active hydrogen bonds for electronic applications. Chem. Mater. 2019, 31, 7951–7961. [Google Scholar] [CrossRef]
- Wang, D.; Liu, D.Y.; Xu, J.H.; Fu, J.J.; Wu, K. Highly thermoconductive yet ultraflexible polymer composites with superior mechanical properties and autonomous self-healing functionality via a binary filler strategy. Mater. Horiz. 2022, 9, 640–652. [Google Scholar] [CrossRef]
- Zhao, W.P.; Huang, B.; Zhu, L.; Feng, X.Q.; Xu, J.; Zhang, H.; Yan, S.K. Printable hydrogels based on starch and natural rubber latex with high toughness and self-healing capability. Int. J. Biol. Macromol. 2022, 218, 580–587. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior toughness and fast self-healing at room temperature engineered by transparent elastomers. Adv. Mater. 2018, 30, 1705145. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.H.; Yan, Y.Q.; Liu, S.; Li, Z.Q.; Wang, Y.G.; Li, H.R. Highly stretchable and fast self-healing luminescent materials. ACS. Appl. Mater. Interfaces 2020, 12, 13239–13247. [Google Scholar] [CrossRef]
- Guo, W.J.; Wang, X.H.; Lu, X.Y.; Li, X.; Li, Y.; Sun, J.Q. Plant oil and amino acid-derived elastomers with rapid room temperature self-healing ability. J. Mater. Chem. A 2019, 7, 21927–21933. [Google Scholar] [CrossRef]
- Zhou, J.H.; Yang, Y.L.; Qin, R.; Xu, M.; Sheng, Y.M.; Lu, X. Robust poly(urethane-amide) protective film with fast self-healing at room temperature. ACS Appl. Polym. Mater. 2019, 2, 285–294. [Google Scholar] [CrossRef]
- He, C.L.; Liang, F.C.; Veeramuthu, L.; Cho, C.J.; Benas, J.S.; Tzeng, Y.R.; Tseng, Y.L.; Chen, W.C.; Rwei, A.; Kuo, C.C. Super tough and spontaneous water-assisted autonomous self-healing elastomer for underwater wearable electronics. Adv. Sci. 2021, 8, 2102275. [Google Scholar] [CrossRef]
- Autumn, K.; Sitti, M.; Liang, Y.C.; Peattie, A.; Hansen, W.; Sponberg, S.; Kenny, T.; Fearing, R.; Israelachvili, J.; Robert, F. Evidence for van der Waals adhesion in gecko setae. Proc. Natl. Acad. Sci. USA 2002, 99, 12252–12256. [Google Scholar] [CrossRef]
- Urban, M.W.; Davydovich, D.; Yang, Y.; Demir, T.; Zhang, Y.Z.; Casabianca, L. Key-and-lock commodity self-healing copolymers. Science 2018, 362, 220–225. [Google Scholar] [CrossRef]
- Wang, H.B.; Yang, Y.; Nishiura, M.; Higaki, Y.; Takahara, A.; Hou, Z.M. Synthesis of self-healing polymers by scandium-catalyzed copolymerization of ethylene and anisylpropylenes. J. Am. Chem. Soc. 2019, 141, 3249–3257. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, H.B.; Huang, L.; Nishiura, M.; Higaki, Y.; Hou, Z.M. Terpolymerization of ethylene and two different methoxyaryl-substituted propylenes by scandium catalyst makes tough and fast self-healing elastomers. Angew. Chem. Int. Ed. Engl. 2021, 60, 26192–26198. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Liu, J.F.; Thundat, T.; Zeng, H.B. Polypyrrole-doped conductive supramolecular elastomer with stretchability, rapid self-healing, and adhesive property for flexible electronic sensors. ACS. Appl. Mater. Interfaces 2019, 11, 18720–18729. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.Q.; Yue, D.W.; Li, C.H. A fast self-healing magnetic nanocomposite for magnetic actuators. Macromol. Mater. Eng. 2021, 307, 2100649. [Google Scholar] [CrossRef]
- Wang, C.; Liu, N.; Allen, R.; Tok, J.B.; Wu, Y.P.; Zhang, F.; Chen, Y.S.; Bao, Z.N. A rapid and efficient self-healing thermo-reversible elastomer crosslinked with graphene oxide. Adv. Mater. 2013, 25, 5785–5790. [Google Scholar] [CrossRef]
- Tee, B.C.; Wang, C.; Allen, R.; Bao, Z.N. An electrically and mechanically self-healing composite with pressure- and flexion-sensitive properties for electronic skin applications. Nat. Nanotechnol. 2012, 7, 825–832. [Google Scholar] [CrossRef]
- Tang, M.; Li, Z.L.; Wang, K.Q.; Jiang, Y.Z.; Tian, M.; Qin, Y.J.; Gong, Y.; Li, Z.; Wu, L.M. Ultrafast self-healing and self-adhesive polysiloxane towards reconfigurable on-skin electronics. J. Mater. Chem. A 2022, 10, 1750–1759. [Google Scholar] [CrossRef]
- Yoon, J.H.; Kim, S.M.; Eom, Y.; Koo, J.M.; Cho, H.W.; Lee, T.J.; Lee, K.G.; Park, H.J.; Kim, Y.K.; Yoo, H.J.; et al. Extremely fast self-healable bio-based supramolecular polymer for wearable real-time sweat-monitoring sensor. ACS. Appl. Mater. Interfaces 2019, 11, 46165–46175. [Google Scholar] [CrossRef]
- Zhao, W.P.; Liu, Y.Y.; Zhao, C.; Shi, X.Y.; Feng, X.Q.; Xu, J.; Wang, S.G.; Wu, Y.M.; Yan, S.K. A fast self-healable and stretchable conductor based on hierarchical wrinkled structure for flexible electronics. Compos. Sci. Technol. 2021, 211, 108834. [Google Scholar] [CrossRef]
- Xu, J.; Zhang, Z.X.; Nie, Y.J.; Liu, Y.X.; Fan, Y.; Zhao, W.P.; Feng, X.Q. Dual physical cross-linked self-healing elastomer for the triple shape memory. J. Mater. Sci. 2022, 57, 11430–11442. [Google Scholar] [CrossRef]
- Zhao, W.P.; Liu, Y.Y.; Zhang, Z.X.; Feng, X.Q.; Xu, H.; Xu, J.; Hu, J.; Wang, S.G.; Wu, Y.M.; Yan, S.K. High-strength, fast self-healing, aging-insensitive elastomers with shape memory effect. ACS. Appl. Mater. Interfaces 2020, 12, 35445–35452. [Google Scholar] [CrossRef]
- Zhao, W.P.; Zhang, Z.X.; Hu, J.; Feng, X.Q.; Xu, J.; Wu, Y.M.; Yan, S.K. Robust and ultra-fast self-healing elastomer with hierarchically anisotropic structures and used for wearable sensors. Chem. Eng. J. 2022, 446, 137305. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dynamic Bonding | Pressure | Temperature (°C) | Time (h) | Ref. |
|---|---|---|---|---|
| Disulfide bond | 10 bar | 70 | 7 | [59] |
| Disulfide bond | 30 kPa | 70–90 | 1 | [60] |
| Disulfide bond | 1 bar | 70 | 7 | [61] |
| Disulfide bond | 0.01 MPa | 120 | 5 | [62] |
| Disulfide bond | 1 bar | 70 | 7 | [63] |
| Boron-based bond | Slight force | 40 | 6 | [64] |
| Matrix | Healing Mechanism | T (°C) | t | Ref. |
|---|---|---|---|---|
| poxy/TPU | Hydrogen bond | 80 | 10 min | [76] |
| PLA/PEG/HPDMS | Diels-Alder reaction | 120 | 100 s | [77] |
| TPUs | Disulfide bond | 50 | 1 h | [73] |
| Polyacrylate composite | Hydrogen/Disulfide bonds | 60 | 5 min | [78] |
| PCL-PU | Shape memory effects | 65 | 2 h | [79] |
| PU | Diels-Alder reaction | 130 | 2 h | [80] |
| TPU | Disulfide bond | 70 | 60 s | [81] |
| Polyacrylate composite | Coordination bond | 60-80 | 10 min | [20] |
| PCL | Disulfide bond | 60 | 1 h | [82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Zhu, L.; Nie, Y.; Li, Y.; Wei, S.; Chen, X.; Zhao, W.; Yan, S. Advances and Challenges of Self-Healing Elastomers: A Mini Review. Materials 2022, 15, 5993. https://doi.org/10.3390/ma15175993
Xu J, Zhu L, Nie Y, Li Y, Wei S, Chen X, Zhao W, Yan S. Advances and Challenges of Self-Healing Elastomers: A Mini Review. Materials. 2022; 15(17):5993. https://doi.org/10.3390/ma15175993
Chicago/Turabian StyleXu, Jun, Lei Zhu, Yongjia Nie, Yuan Li, Shicheng Wei, Xu Chen, Wenpeng Zhao, and Shouke Yan. 2022. "Advances and Challenges of Self-Healing Elastomers: A Mini Review" Materials 15, no. 17: 5993. https://doi.org/10.3390/ma15175993