
Mechanical Properties of Cubene Crystals

 , , , and
, , , and 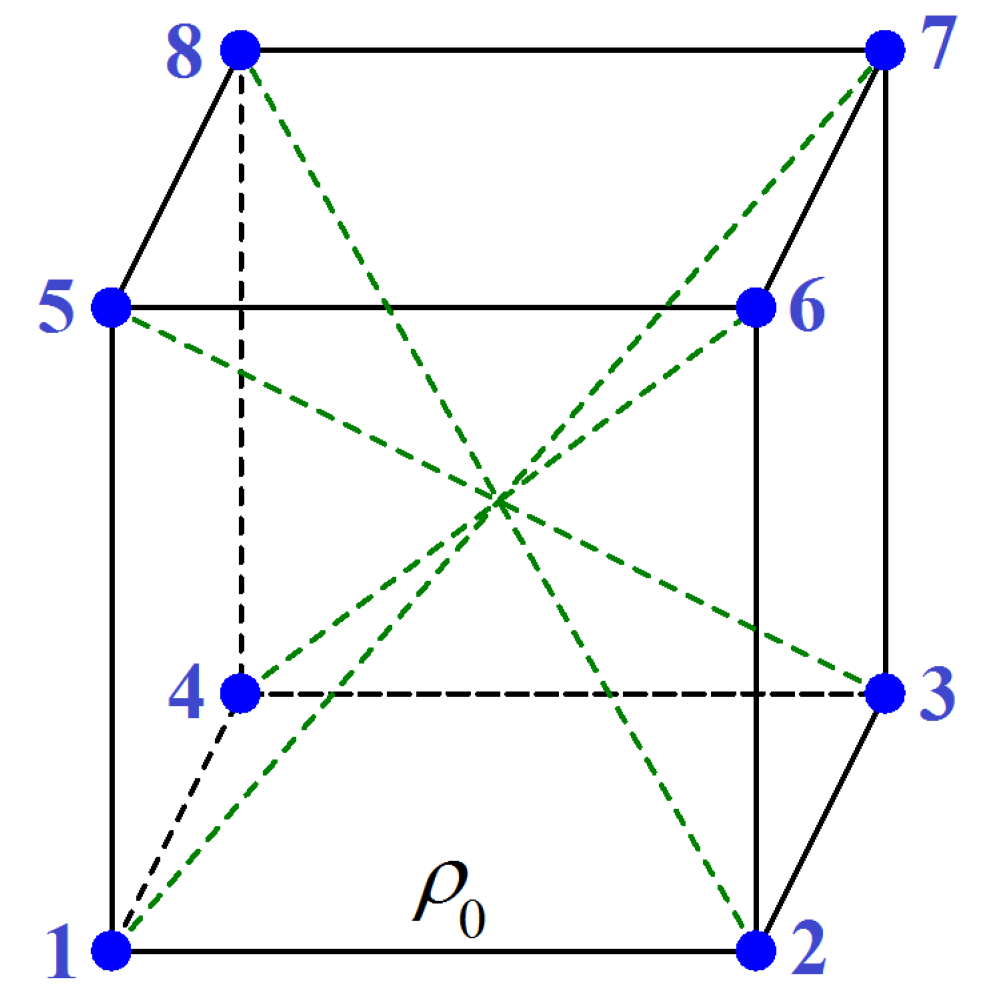{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Two Equilibrium Phases
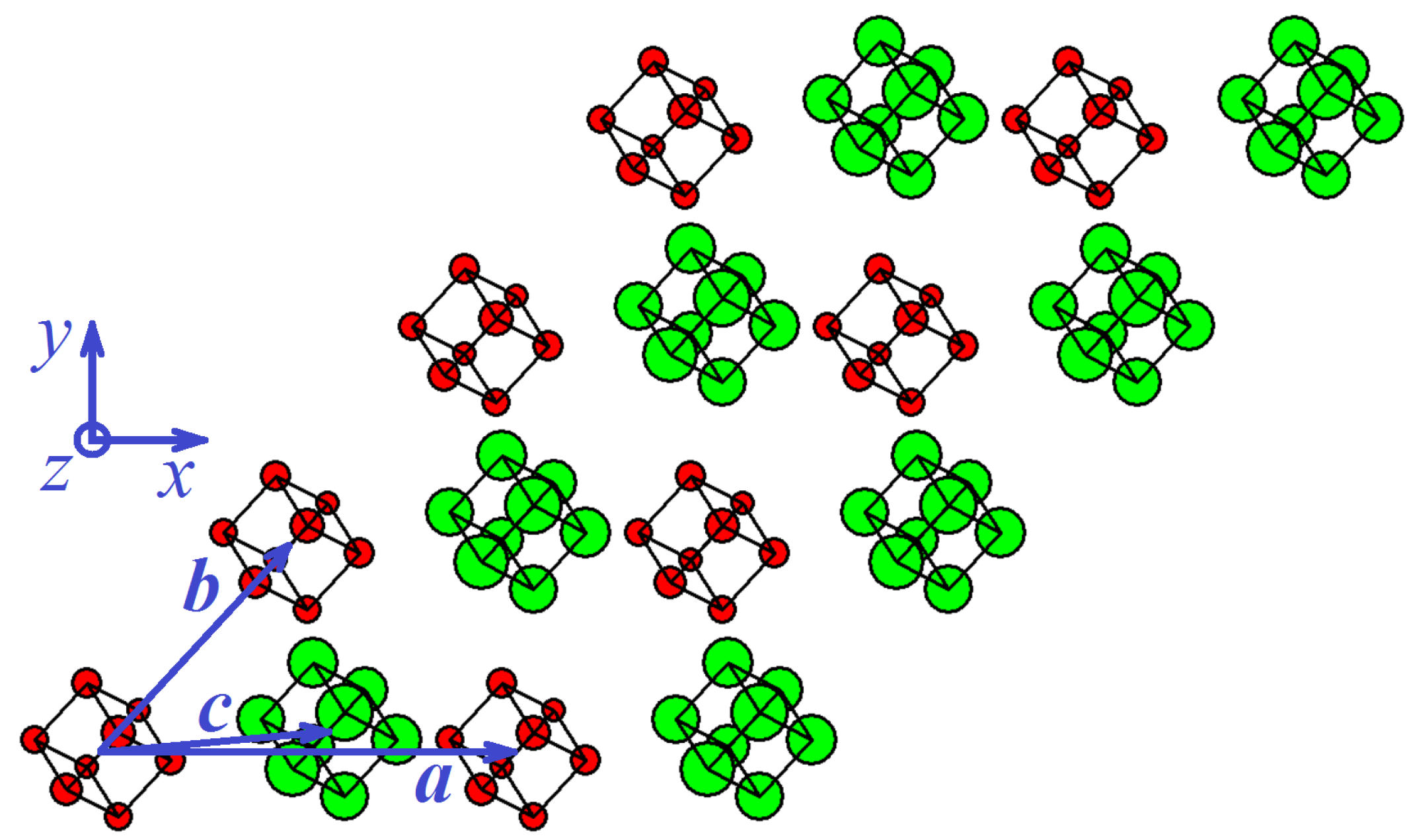
3.1.1. Metastable Cubic Phase
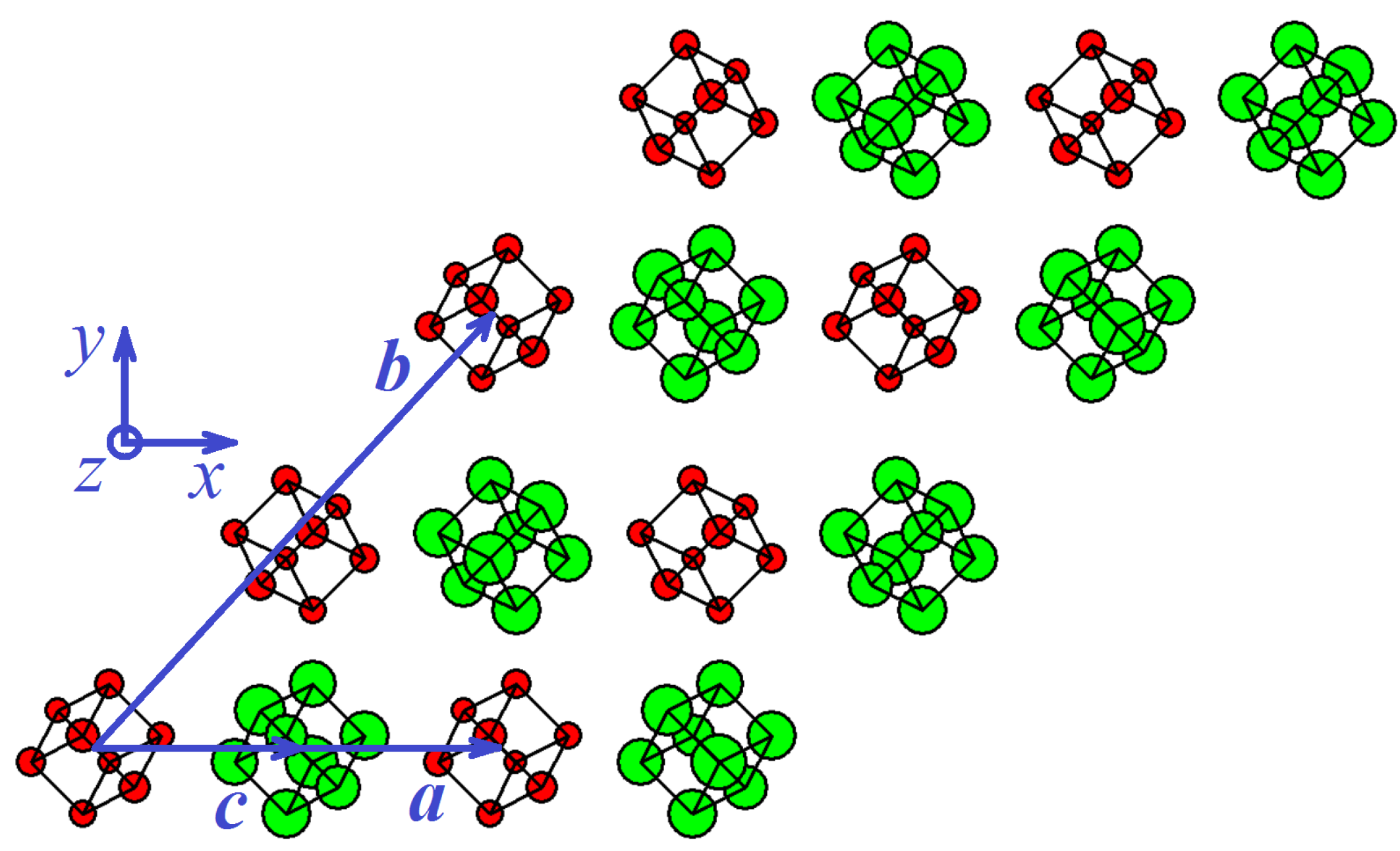
3.1.2. Stable Triclinic Phase
3.2. Energy of Cubic and Triclinic Cubene Crystals
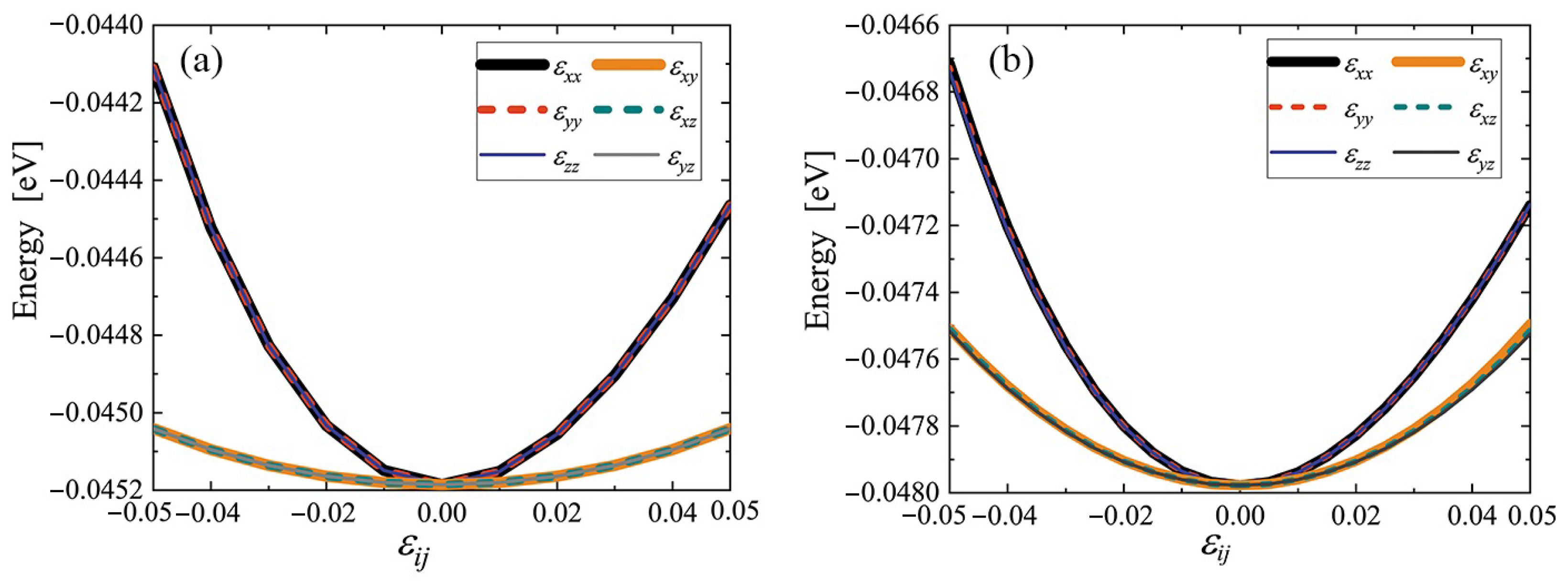
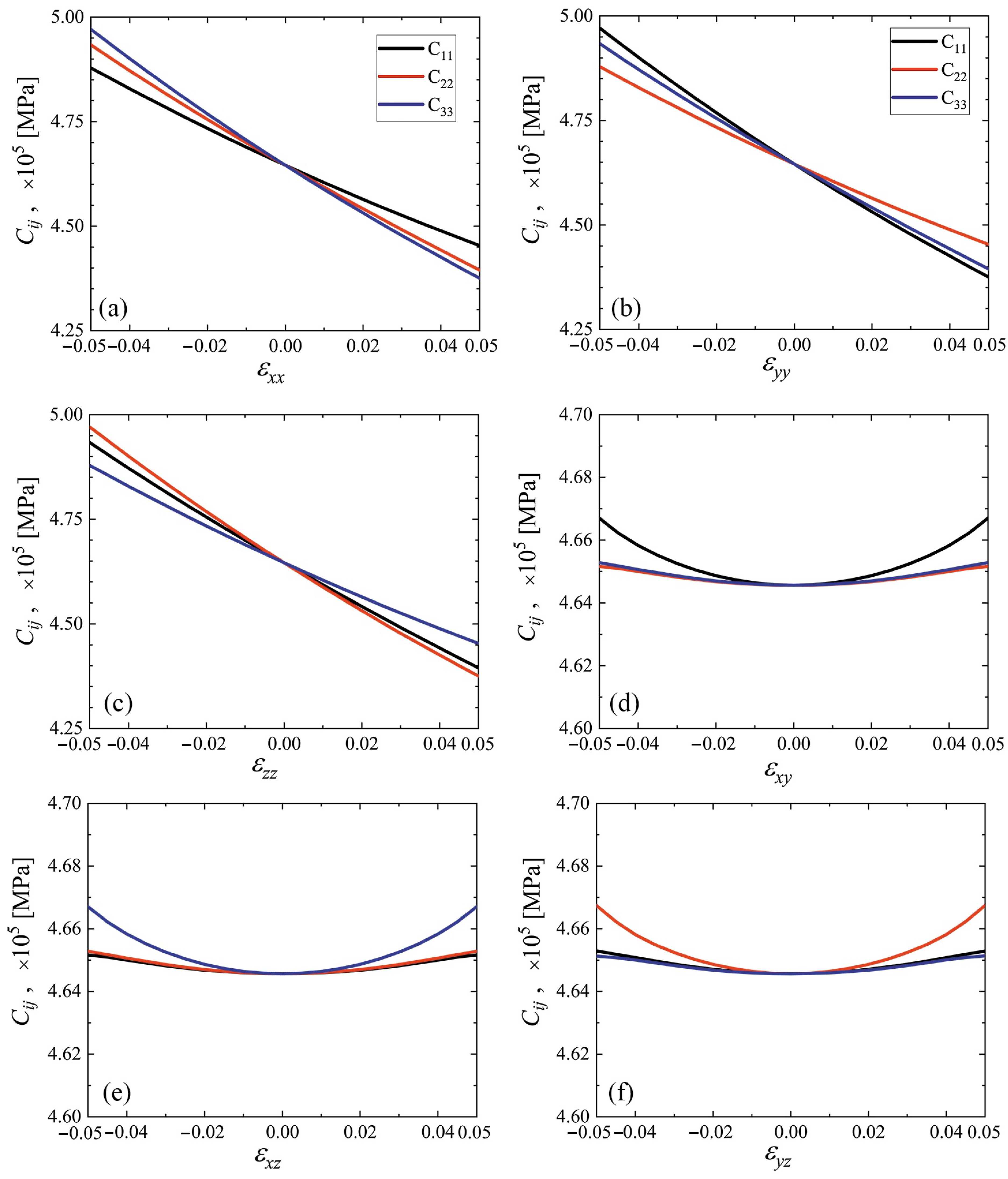
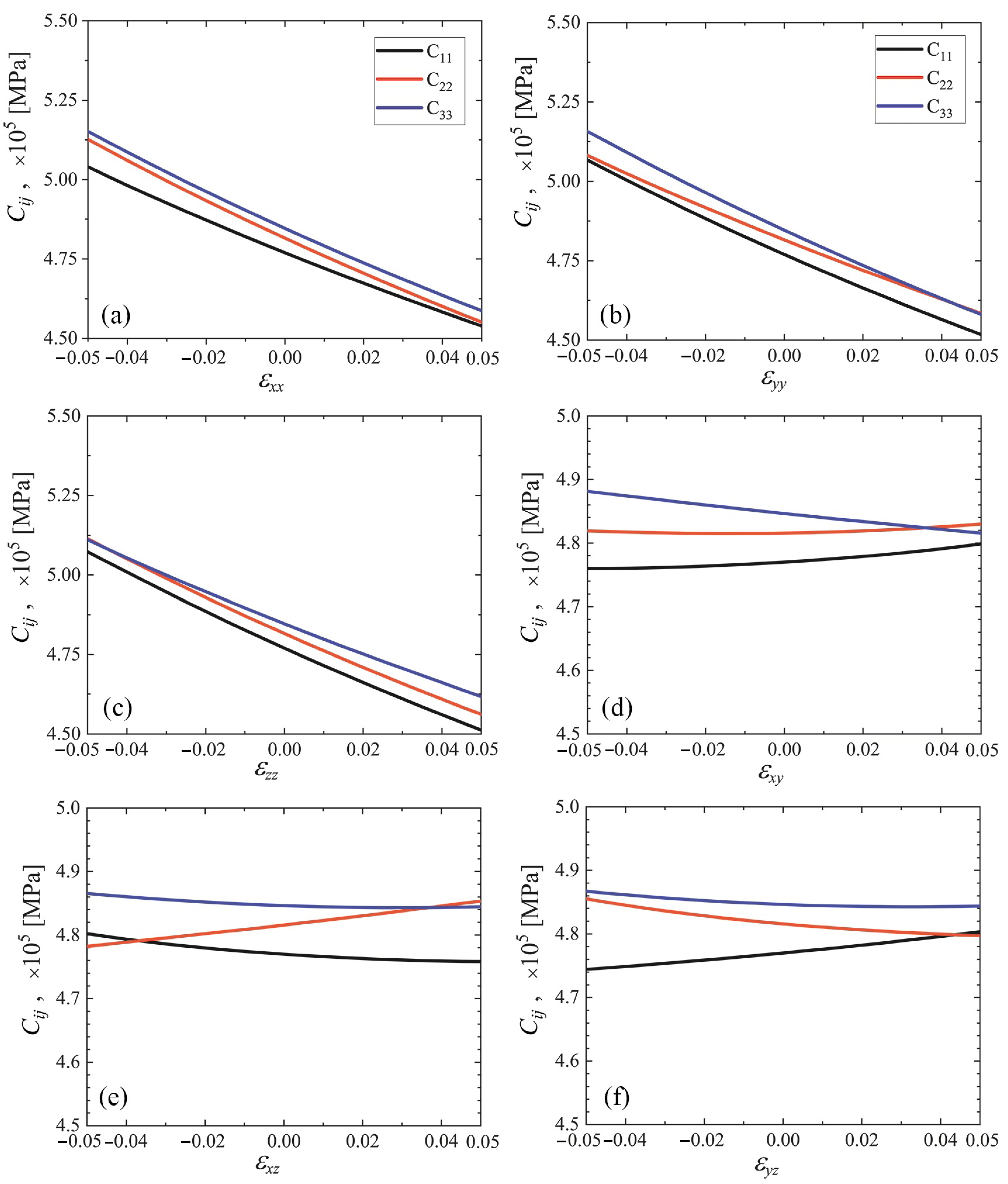
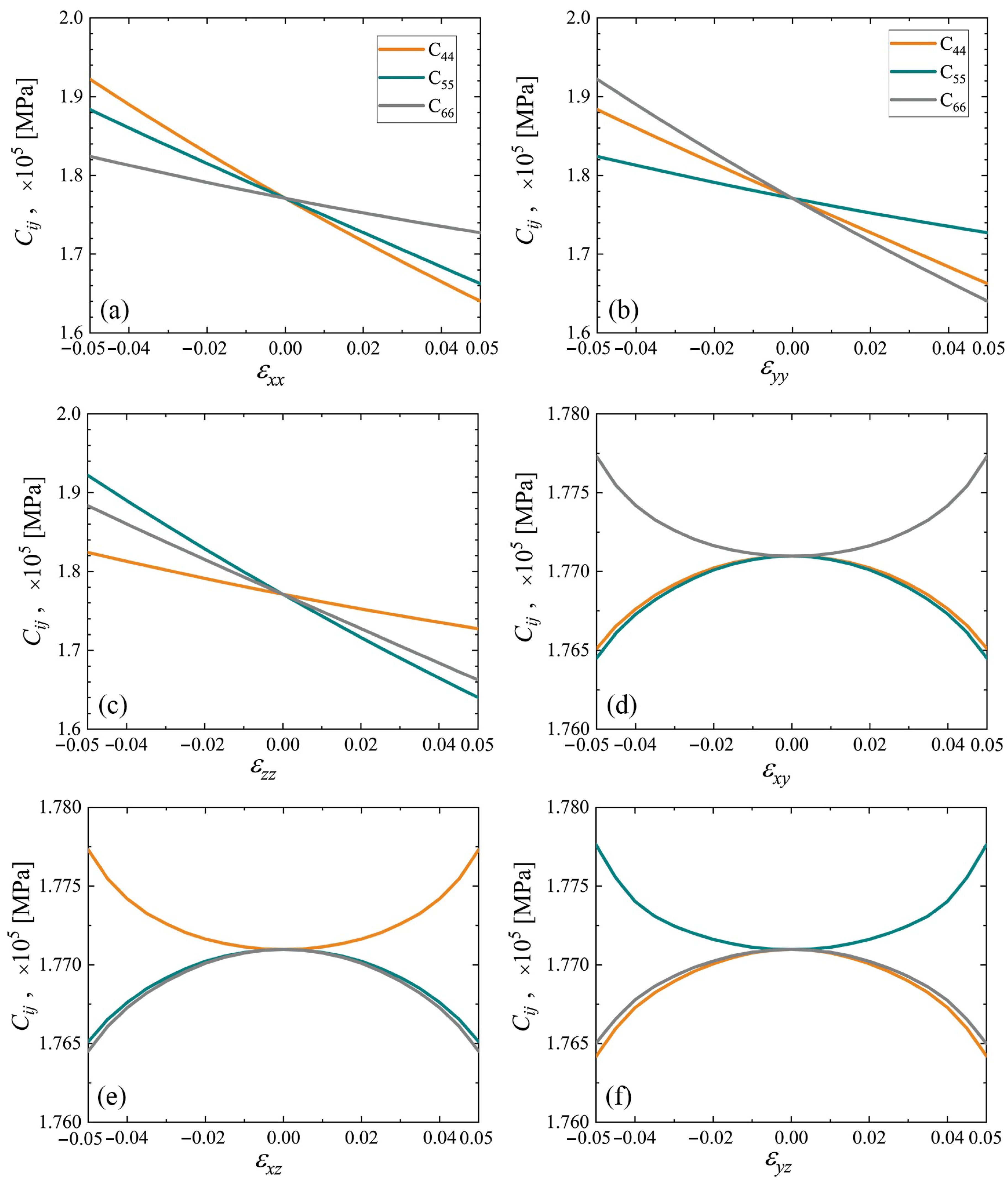
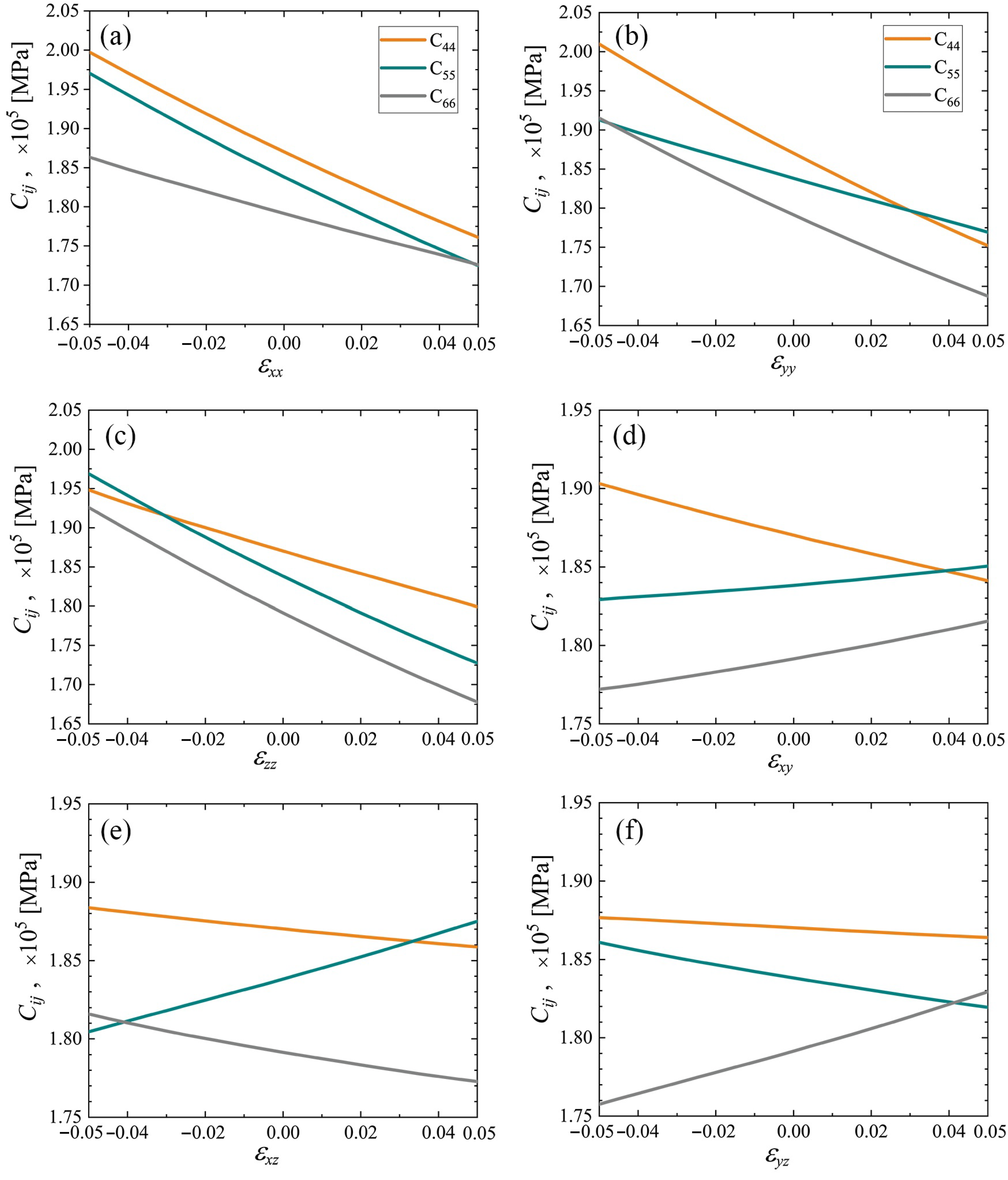
3.3. Elastic Constants for the Cubene Crystals
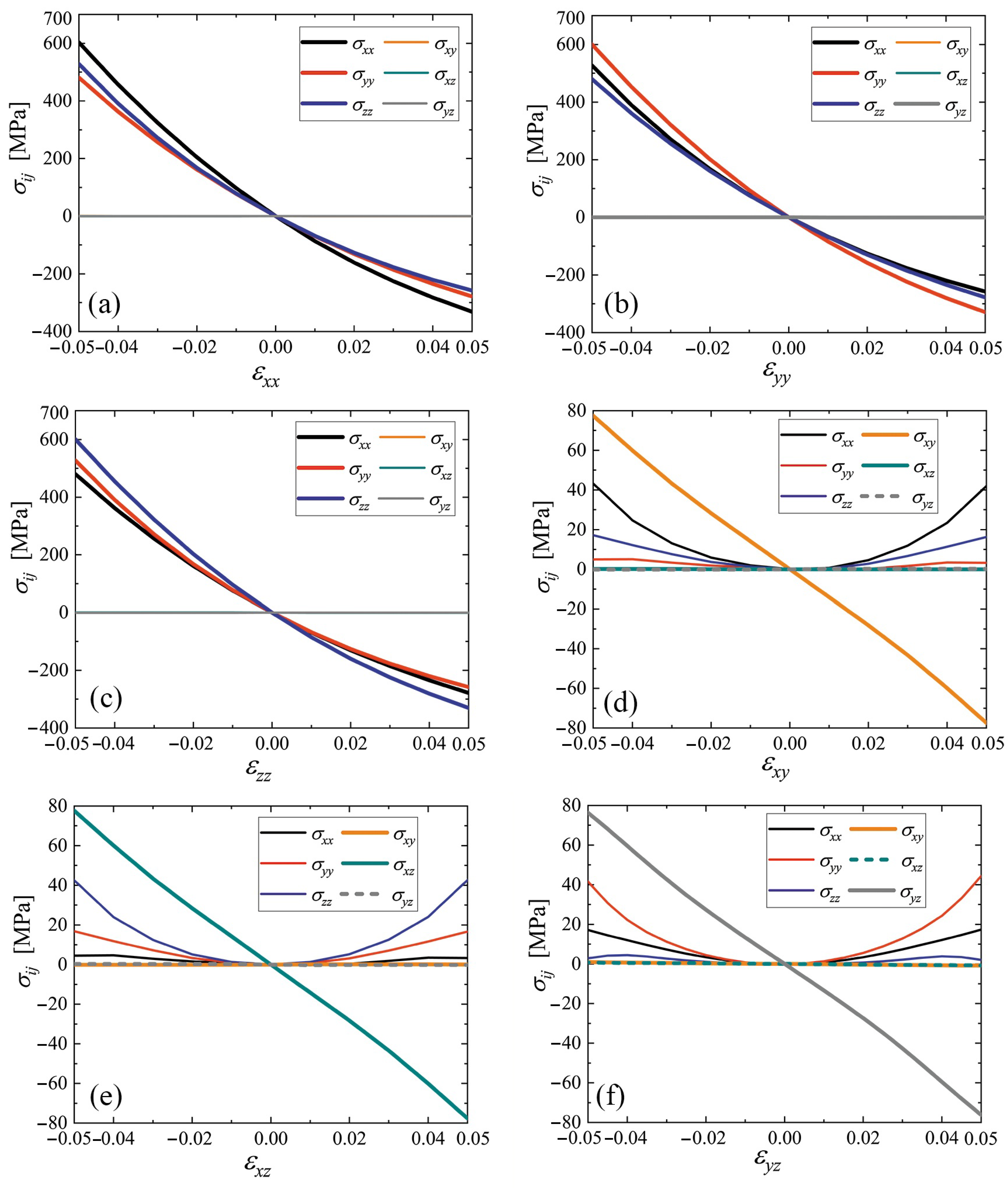
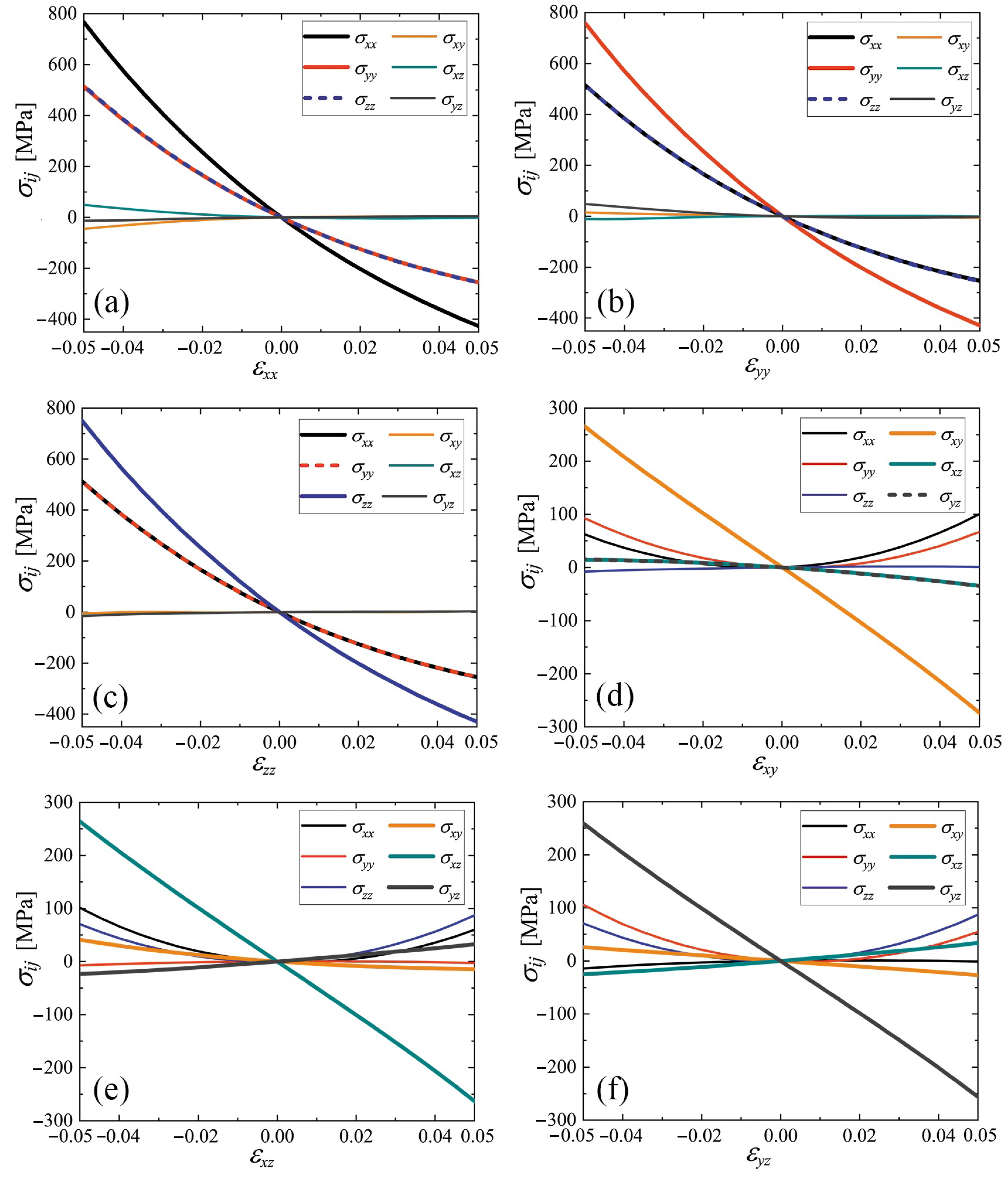
3.4. Stress–Strain Curves
4. Discussion
5. Conclusions and Future Work
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kroto, H.W.; Heath, J.R.; Obrien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Kawada, H.; Fujii, Y.; Nakao, H.; Murakami, Y.; Watanuki, T.; Suematsu, H.; Kikuchi, K.; Achiba, Y.; Ikemoto, I. Structural aspects of C82 and C76 crystals studied by x-ray diffraction. Phys. Rev. B 1995, 51, 8723–8730. [Google Scholar] [CrossRef] [PubMed]
- Bubenchikov, M.; Mamontov, D.; Azheev, A.; Azheev, S. Study of the influence of the Dzhanibekov effect on the dynamic state of fullerene C100. J. Phys. Conf. Ser. 2022, 2211, 012006. [Google Scholar] [CrossRef]
- Visser, S.; Filatov, M.; Schreiner, P.; Shaik, S. A REKS assessment of the face-diagonal bond in 1,3-didehydrocubane and a comparison with benzyne biradicals. Eur. J. Org. Chem. 2003, 2003, 4199–4204. [Google Scholar] [CrossRef]
- Eaton, P.E.; Cole, T.W., Jr. The cubane system. J. Am. Chem. Soc. 1964, 86, 962–964. [Google Scholar] [CrossRef]
- Maslov, M.M.; Lobanov, D.A.; Podlivaev, A.I.; Openov, L.A. Thermal stability of cubane C8H8. Phys. Solid State 2009, 51, 645–648. [Google Scholar] [CrossRef]
- Sharapa, D.; Hirsch, A.; Meyer, B.; Clark, T. Cubic C8: An observable allotrope of carbon? ChemPhysChem 2015, 16, 2165. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, L.S.; Moreira, E.; Lopes, A.R.; Fonseca, A.L.A.; Azevedo, D.L. Cubane and cubanoid: Structural, optoelectronic and thermodynamic properties from DFT and TD-DFT method. J. Mol. Graph. Model. 2021, 103, 107820. [Google Scholar] [CrossRef]
- Chaban, V.V.; Prezhdo, O.V. Energy storage in cubane derivatives and their real-time decomposition: Computational molecular dynamics and thermodynamics. ACS Energy Lett. 2016, 1, 189–194. [Google Scholar] [CrossRef]
- Houston, S.D.; Fahrenhorst-Jones, T.; Xing, H.; Chalmers, B.A.; Sykes, M.L.; Stok, J.E.; Soto, C.F.; Burns, J.M.; Bernhardt, P.V.; De Voss, J.J.; et al. The cubane paradigm in bioactive molecule discovery: Further scope, limitations and the cyclooctatetraene complement. Org. Biomol. Chem. 2019, 17, 6790–6798. [Google Scholar] [CrossRef]
- Gopalsamy, K.; Subramanian, V. Hydrogen storage capacity of alkali and alkaline earth metal ions doped carbon based materials: A DFT study. Int. J. Hydrogen Energy 2014, 39, 2549–2559. [Google Scholar] [CrossRef]
- Yildirim, T.; Gülseren, O.; Ciraci, S. Exohydrogenated single-wall carbon nanotubes. Phys. Rev. B 2001, 64, 075404. [Google Scholar] [CrossRef] [Green Version]
- Macreadie, L.K.; Babarao, R.; Setter, C.J.; Lee, S.J.; Qazvini, O.T.; Seeber, A.J.; Tsanaktsidis, J.; Telfer, S.G.; Batten, S.R.; Hill, M.R. Enhancing multicomponent metal-organic frameworks for low pressure liquid organic hydrogen carrier separations. Angew. Chem. Int. Edit. 2020, 59, 6090–6098. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Valencia, F.; Romero, A.H.; Kiwi, M.; Ramírez, R.; Toro-Labbé, A. Cubane oligomers: A density functional theory study. J. Mol. Struc. THEOCHEM 2006, 769, 183–187. [Google Scholar] [CrossRef]
- Pichierri, F. Cubane oligomers: Substituent effects in cubane and hypercubane: A DFT and QTAIM study. Theor. Chem. Acc. 2017, 136, 114. [Google Scholar] [CrossRef]
- Chaglayan, B.; Huran, A.W.; Ben Amor, N.; Brumas, V.; Evangelisti, S.; Leininger, T. Cubane oligomers: Spherical aromaticity and electron delocalization in C8 and B4N4 cubic systems. Theor. Chem. Acc. 2018, 138, 5. [Google Scholar] [CrossRef]
- Katin, K.P.; Maslov, M.M. Chemical functionalization effects on cubane-based chain electronic transport. Adv. Cond. Mat. Phys. 2015, 2015, 754873. [Google Scholar] [CrossRef] [Green Version]
- Katin, K.P.; Maslov, M.M. Toward CL-20 crystalline covalent solids: On the dependence of energy and electronic properties on the effective size of CL-20 chains. J. Phys. Chem. Solids 2017, 108, 82–87. [Google Scholar] [CrossRef] [Green Version]
- Khorobrykh, F.; Kulnitskiy, B.; Churkin, V.; Skryleva, E.; Parkhomenko, Y.; Zholudev, S.; Blank, V.; Popov, M. The effect of C60 fullerene polymerization processes on the mechanical properties of clusters forming ultrahard structures of 3D C60 polymers. Diam. Relat. Mater. 2022, 124, 108911. [Google Scholar] [CrossRef]
- Savin, A.V.; Kivshar, Y.S. Anomalous thermal relaxation in carbon nanoclusters. Appl. Phys. Lett. 2011, 98, 193106. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Gao, Q.; Gao, Y.; Luo, Y.; Guo, J.; Sun, Q.; Liang, E. Uniaxial negative thermal expansion behavior of β-CuSCN. Appl. Phys. Lett. 2021, 118, 222105. [Google Scholar] [CrossRef]
- Koocher, N.Z.; Huang, L.-F.; Rondinelli, J.M. Negative thermal expansion in the Ruddlesden-Popper calcium titanates. Phys. Rev. Mater. 2021, 5, 053601. [Google Scholar] [CrossRef]
- D’Ambrumenil, S.; Zbiri, M.; Chippindale, A.M.; Hibble, S.J.; Marelli, E.; Hannon, A.C. Lattice dynamics and negative thermal expansion in the framework compound ZnNi(CN)4 with two-dimensional and three-dimensional local environments. Phys. Rev. B 2019, 99, 024309. [Google Scholar] [CrossRef] [Green Version]
- Campbell, B.; Howard, C.J.; Averett, T.B.; Whittle, T.A.; Schmid, S.; Machlus, S.; Yost, C.; Stokes, H.T. An algebraic approach to cooperative rotations in networks of interconnected rigid units. Acta Crystallogr. A 2018, 74, 408. [Google Scholar] [CrossRef]
- Dove, M.T.; Du, J.; Wei, Z.; Keen, D.A.; Tucker, M.G.; Phillips, A.E. Quantitative understanding of negative thermal expansion in scandium trifluoride from neutron total scattering measurements. Phys. Rev. B 2020, 102, 094105. [Google Scholar] [CrossRef]
- Pavlov, I.S.; Erofeev, V.I.; Muravieva, A.V.; Vasiliev, A.A. On estimation of the rotational wave velocity in a simple cubic lattice of a fullerite crystal. Bull. Russ. Acad. Sci. Phys. 2021, 85, 686–690. [Google Scholar] [CrossRef]
- Savin, A.V.; Kivshar, Y.S. Nonlinear breatherlike localized modes in C60 nanocrystals. Phys. Rev. B 2012, 85, 125427. [Google Scholar] [CrossRef] [Green Version]
- Bubenchikov, A.M.; Bubenchikov, M.A.; Lun-Fu, A.V.; Ovchinnikov, V.A. Gyroscopic effects in fullerite crystal upon deformation. Eur. Phys. J. Plus 2021, 136, 388. [Google Scholar] [CrossRef]
- Bubenchikov, A.M.; Bubenchikov, M.A.; Lun-Fu, A.V.; Ovchinnikov, V.A. Rotational dynamics of fullerenes in the molecular crystal of fullerite. Phys. Status Solidi A 2021, 218, 2000174. [Google Scholar] [CrossRef]
- Bubenchikov, A.; Bubenchikov, M.; Mamontov, D. The dynamic state of a pseudo-crystalline structure of B42 molecules. Crystals 2020, 10, 510. [Google Scholar] [CrossRef]
- Savin, A.V.; Dmitriev, S.V. The frequency spectrum of rotobreathers with many degrees of freedom. Europhys. Lett. 2022, 137, 36005. [Google Scholar] [CrossRef]
- Dmitriev, S.V.; Sunagatova, I.R.; Ilgamov, M.A.; Pavlov, I.S. Natural frequencies of bending vibrations of carbon nanotubes. Tech. Phys. 2022, 67, 7–13. [Google Scholar] [CrossRef]
- Savin, A.V.; Korznikova, E.A.; Dmitriev, S.V. Plane vibrational modes and localized nonlinear excitations in carbon nanotube bundle. J. Sound Vib. 2022, 520, 116627. [Google Scholar] [CrossRef]
- Savin, A.V.; Sunagatova, I.R.; Dmitriev, S.V. Rotobreathers in a chain of coupled elastic rotators. Phys. Rev. E 2021, 104, 034207. [Google Scholar] [CrossRef]
- Dmitriev, S.; Semenov, A.; Savin, A.; Ilgamov, M.; Bachurin, D.V. Rotobreather in a carbon nanotube bundle. J. Micromechanics Mol. Phys. 2020, 2020, 2050010. [Google Scholar] [CrossRef]
- Kuzkin, V.A.; Krivtsov, A.M. Description for mechanical properties of graphene using particles with rotational degrees of freedom. Dokl. Phys. 2011, 56, 527–530. [Google Scholar] [CrossRef]
- Berinskii, I.; Kuzkin, V.A. Equilibration of energies in a two-dimensional harmonic graphene lattice. Philos. T. R. Soc. A 2020, 378, 20190114. [Google Scholar] [CrossRef]
- Dudek, K.K.; Attard, D.; Caruana-Gauci, R.; Wojciechowski, K.W.; Grima, J.N. Unimode metamaterials exhibiting negative linear compressibility and negative thermal expansion. Smart Mater. Struct. 2016, 25, 025009. [Google Scholar] [CrossRef]
- Dudek, K.K.; Gatt, R.; Mizzi, L.; Dudek, M.R.; Attard, D.; Evans, K.E.; Grima, J.N. On the dynamics and control of mechanical properties of hierarchical rotating rigid unit auxetics. Sci. Rep. 2017, 7, 46529. [Google Scholar] [CrossRef] [Green Version]
- Gatt, R.; Mizzi, L.; Azzopardi, J.I.; Azzopardi, K.M.; Attard, D.; Casha, A.; Briffa, J.; Grima, J.N. Hierarchical auxetic mechanical metamaterials. Sci. Rep. 2015, 5, 8395. [Google Scholar] [CrossRef] [Green Version]
- Vasiliev, A.A.; Dmitriev, S.V.; Ishibashi, Y.; Shigenari, T. Elastic properties of a two-dimensional model of crystals containing particles with rotational degrees of freedom. Phys. Rev. B 2002, 65, 8395. [Google Scholar] [CrossRef] [Green Version]
- Vasiliev, A.A.; Pavlov, I.S. Auxetic properties of chiral hexagonal Cosserat lattices composed of finite-sized particles. Phys. Status Solidi B 2020, 257, 1900389. [Google Scholar] [CrossRef]
- Jamari, J.; Ammarullah, M.I.; Santoso, G.; Sugiharto, S.; Supriyono, T.; Prakoso, A.T.; Basri, H.; van der Heide, E. Computational Contact Pressure Prediction of CoCrMo, SS 316L and Ti6Al4V Femoral Head against UHMWPE Acetabular Cup under Gait Cycle. J. Funct. Biomater. 2022, 13, 64. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, J.; Yan, Z.; Feng, M.; Yu, Z.; Wang, L. Pressure-induced dimerization of C60 at room temperature as revealed by an in situ spectroscopy study using an infrared laser. Crystals 2020, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Meletov, K.P.; Christofilos, D.; Kourouklis, G.A.; Ves, S. Pressure induced phase transitions in C60 single crystals. Chem. Phys. Lett. 1995, 236, 265–270. [Google Scholar] [CrossRef]
- Quo, Y.; Karasawa, N.; Goddard, W.A., III. Prediction of fullerene packing in C60 and C70 crystals. Nature 1991, 351, 464–467. [Google Scholar] [CrossRef]
- Bortel, G.; Kováts, E.; Földes, D.; Jakab, E.; Durkó, G.; Pekker, S. Recognition-control and host-guest interactions in high-symmetry cocrystals of fullerenes with cubane and mesitylene. Cryst. Growth Des. 2020, 20, 4169–4175. [Google Scholar] [CrossRef]
- Savin, A.V.; Kivshar, Y.S.; Hu, B. Suppression of thermal conductivity in graphene nanoribbons with rough edges. Phys. Rev. B 2010, 82, 195422. [Google Scholar] [CrossRef] [Green Version]
- Savin, A.V.; Korznikova, E.A.; Dmitriev, S.V. Dynamics of surface graphene ripplocations on a flat graphite substrate. Phys. Rev. B 2019, 99, 235411. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458–9471. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783–802. [Google Scholar] [CrossRef]
- Marks, N.A. Generalizing the environment-dependent interaction potential for carbon. Phys. Rev. B 2000, 63, 035401. [Google Scholar] [CrossRef]
- Tersoff, J. Empirical interatomic potential for carbon, with applications to amorphous carbon. Phys. Rev. Lett. 1988, 61, 2879–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Los, J.H.; Fasolino, A. Intrinsic long-range bond-order potential for carbon: Performance in Monte Carlo simulations of graphitization. Phys. Rev. B 2003, 68, 24107. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, S.G.; van Duin, A.C.T.; Ganesh, P. Development of a ReaxFF potential for carbon condensed phases and its application to the thermal fragmentation of a large fullerene. J. Phys. Chem. 2015, 119, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef] [Green Version]
- Rowe, P.; Deringer, V.L.; Gasparotto, P.; Csányi, G.; Michaelides, A. An accurate and transferable machine learning potential for carbon. J. Chem. Phys. 2020, 153, 10897690. [Google Scholar] [CrossRef]
- Babicheva, R.I.; Semenov, A.S.; Shcherbinin, S.A.; Korznikova, E.A.; Kudreyko, A.A.; Vivegananthan, P.; Zhou, K.; Dmitriev, S.V. Effect of the stiffness of interparticle bonds on properties of delocalized nonlinear vibrational modes in an fcc lattice. Phys. Rev. E 2022, 105, 064204. [Google Scholar] [CrossRef]
- Goldstein, R.V.; Gorodtsov, V.A.; Lisovenko, D.S. Classification of cubic auxetics. Phys. Status Solidi B 2013, 250, 2038–2043. [Google Scholar] [CrossRef]
- Epishin, A.I.; Lisovenko, D.S. Extreme values of the Poisson’s ratio of cubic crystals. Tech. Phys. 2016, 61, 1516–1524. [Google Scholar] [CrossRef]
- Goldstein, R.V.; Gorodtsov, V.A.; Lisovenko, D.S. Cubic auxetics. Dokl. Phys. 2011, 56, 399–402. [Google Scholar] [CrossRef]
- Lisovenko, D.S.; Baimova, J.A.; Rysaeva, L.K.; Gorodtsov, V.A.; Rudskoy, A.I.; Dmitriev, S.V. Equilibrium diamond-like carbon nanostructures with cubic anisotropy: Elastic properties. Phys. Status Solidi B 2016, 253, 1295–1302. [Google Scholar] [CrossRef]
- Zarochentsev, E.V.; Troitskaya, E.P.; Chabanenko, V.V. Elastic constants of noble-gas crystals under pressure and the Cauchy relations. Phys. Solid State 2004, 46, 249. [Google Scholar] [CrossRef]
- Hoen, S.; Chopra, N.G.; Xiang, X.-D.; Mostovoy, R.; Hou, J.; Vareka, W.A.; Zettl, A. Elastic properties of a van der Waals solid: C60. Phys. Rev. B 1992, 46, 12737. [Google Scholar] [CrossRef]
- Funamori, Y.; Suzuki, R.; Wakahara, T.; Ohmura, T.; Nakagawa, E.; Tachibana, M. Large elastic deformation of C60 nanowhiskers. Carbon 2020, 169, 65–72. [Google Scholar] [CrossRef]
- Asaka, K.; Kato, R.; Miyazawa, K.; Kizuka, T. Buckling of C60 whiskers. Appl. Phys. Lett. 2006, 89, 071912. [Google Scholar] [CrossRef] [Green Version]
- Shepelev, I.A.; Bachurin, D.V.; Korznikova, E.A.; Dmitriev, S.V. Highly efficient energy and mass transfer in bcc metals by supersonic 2-crowdions. J. Nucl. Mater. 2022, 568, 153841. [Google Scholar] [CrossRef]
- Prokhorov, V.M.; Blank, V.D.; Dubitsky, G.A.; Berezina, S.; Levin, V.M. Elastic and microstructural properties of C60- and C70-based polymerized fullerites exposed to high pressure (15 GPa) and elevated temperatures. J. Phys. Condens. Mat. 2002, 14, 11305–11310. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galiakhmetova, L.K.; Pavlov, I.S.; Bayazitov, A.M.; Kosarev, I.V.; Dmitriev, S.V. Mechanical Properties of Cubene Crystals. Materials 2022, 15, 4871. https://doi.org/10.3390/ma15144871
Galiakhmetova LK, Pavlov IS, Bayazitov AM, Kosarev IV, Dmitriev SV. Mechanical Properties of Cubene Crystals. Materials. 2022; 15(14):4871. https://doi.org/10.3390/ma15144871
Chicago/Turabian StyleGaliakhmetova, Leysan Kh., Igor S. Pavlov, Ayrat M. Bayazitov, Igor V. Kosarev, and Sergey V. Dmitriev. 2022. "Mechanical Properties of Cubene Crystals" Materials 15, no. 14: 4871. https://doi.org/10.3390/ma15144871