
Quantum-Chemical Consideration of Al2M2 Tetranuclear Metal Clusters (M–3d-Element): Molecular/Electronic Structures and Thermodynamics



Abstract
:1. Introduction
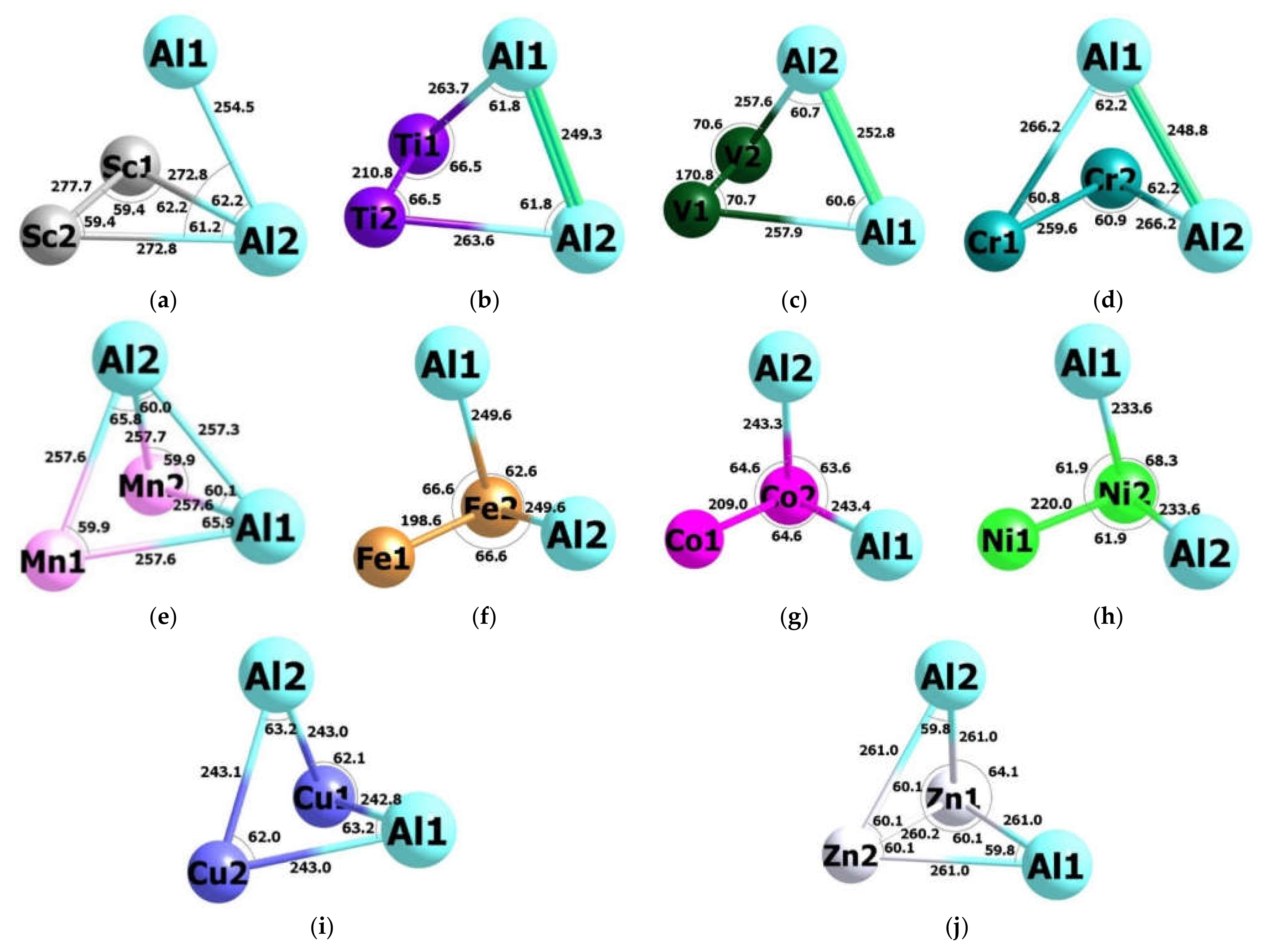
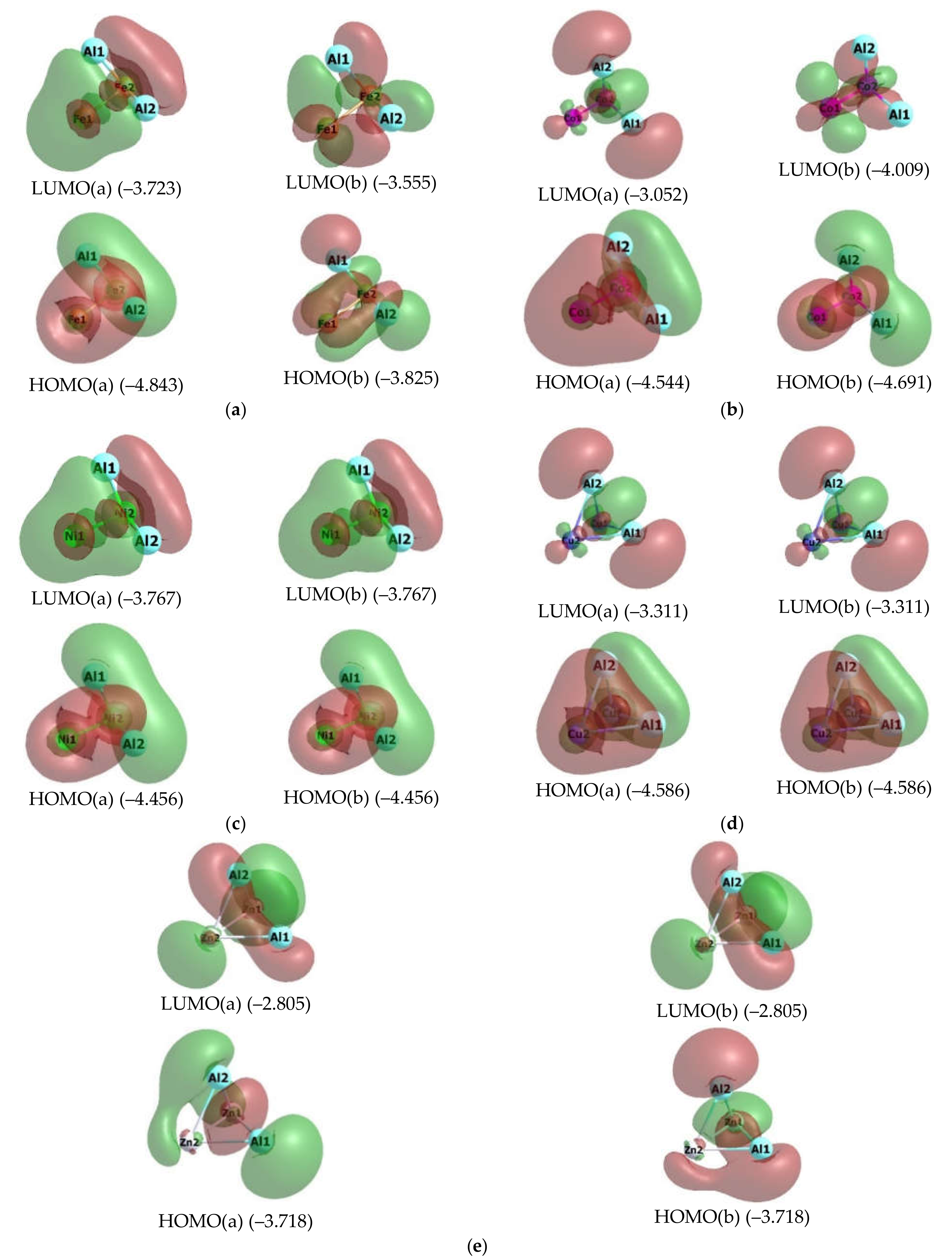
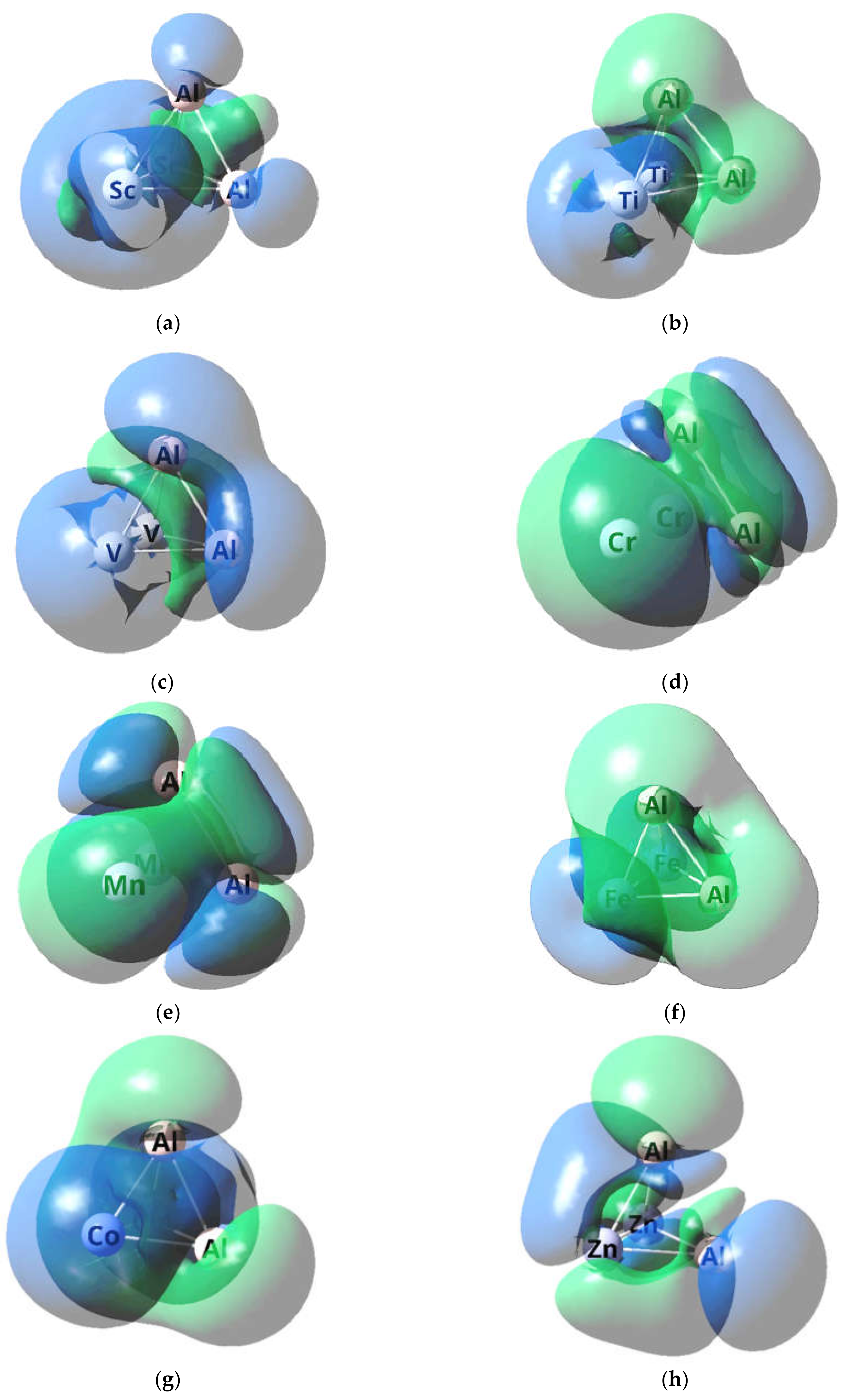
2. Results
3. Discussion
4. Calculation Method
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maroun, F.; Ozanam, F.; Magnussen, O.M.; Behm, R.J. The role of atomic ensembles in the reactivity of bimetallic electrocatalysts. Science 2001, 293, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, W. Clusters as new materials. Surf. Sci. 2002, 500, 242–270. [Google Scholar] [CrossRef]
- Yang, J.X.; Guo, J.J.; Die, D. Ab initio study of AunIr (n=1–8) clusters. Comput. Theor. Chem. 2011, 963, 435–438. [Google Scholar] [CrossRef]
- Singh, N.B.; Sarkar, U. A density functional study of chemical, magnetic and thermodynamic properties of small palladium clusters. Mol. Simul. 2014, 40, 1255–1264. [Google Scholar] [CrossRef]
- Bouderbala, W.; Boudjahem, A.G.; Soltani, A. Geometries, stabilities, electronic and magnetic properties of small PdnIr (n = 1–8) clusters from first principles calculations. Mol. Phys. 2014, 112, 1789–1798. [Google Scholar] [CrossRef]
- Ling, W.; Dong, D.; Shi-Jian, W.; Zheng-Quan, Z. Geometrical, electronic, and magnetic properties of CunFe (n = 1–12) clusters: A density functional study. J. Phys. Chem. Solids 2015, 76, 10–16. [Google Scholar] [CrossRef]
- Kilimis, D.A.; Papageorgiou, D.G. Density functional study of small bimetallic Ag–Pd clusters. J. Mol. Struct. 2010, 939, 112–117. [Google Scholar] [CrossRef]
- Zhao, S.; Ren, Y.; Wang, J.; Yin, W. Density functional study of NO binding on small AgnPdm (n + m ≤ 5) clusters. Comput. Theor. Chem. 2011, 964, 298–303. [Google Scholar] [CrossRef]
- Al-Odail, F.; Mazher, J.; Abuelela, A.M. A density functional theory study of structural, electronic and magnetic properties of small PdnAg (n = 1–8) clusters. Comput. Theor. Chem. 2017, 1125, 103–111. [Google Scholar] [CrossRef]
- Ma, L.; Wang, J.; Hao, Y.; Wang, G. Density functional theory study of FePdn (n = 2–14) clusters and interactions with small molecules. Comput. Mater. Sci. 2013, 68, 166–173. [Google Scholar] [CrossRef]
- Chaves, A.S.; Rondina, G.G.; Piotrowski, M.J.; Da Silva, J.L.F. Structural formation of binary PtCu clusters: A density functional theory investigation. Comput. Mater. Sci. 2015, 98, 278–286. [Google Scholar] [CrossRef]
- Dong, D.; Xiao-Yu, K.; Jian-Jun, G.; Ben-Xia, Z. First-principle study of AunFe (n = 1–7) clusters. J. Mol. Struct. 2009, 902, 54–58. [Google Scholar]
- Liu, X.; Tian, D.; Meng, C. DFT study on stability and H2 adsorption activity of bimetallic Au79−nPdn (n = 1–55) clusters. Chem. Phys. 2013, 415, 179–185. [Google Scholar] [CrossRef]
- Hong, L.; Wang, H.; Cheng, J.; Huang, X.; Sai, L.; Zhao, J. Atomic structures and electronic properties of small Au–Ag binary clusters: Effects of size and composition. Comput. Theor. Chem. 2012, 993, 36–44. [Google Scholar] [CrossRef]
- Wen, J.Q.; Xia, T.; Zhou, H.; Wang, J.F. A density functional theory study of small bimetallic PdnAl (n = 1–8) clusters. J. Phys. Chem. Solids 2014, 75, 528–534. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structures of Aluminum–Iron Clusters AlFe3, Al2Fe3, and Al2Fe4 according to Quantum-Chemical DFT Calculations. Russ. J. Inorg. Chem. 2017, 62, 336–343. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular Structures of Tetranuclear (Al, Fe) Metal Clusters. Glass Phys. Chem. 2018, 44, 339–345. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structures of Al2Cr3 and Al2Mo3 Metal Clusters according to Density Functional Theory Calculations. Russ. J. Inorg. Chem. 2018, 63, 786–799. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. DFT calculation of molecular structures of Al2Fe3 and Al2Cu3 heterobinuclear clusters. Struct. Chem. 2018, 29, 1543–1549. [Google Scholar] [CrossRef]
- Chachkov, D.V.; Mikhailov, O.V. DFT Quantum Chemical Calculation of the Molecular Structures of the Metal Clusters Al2Cu3 and Al2Ag3. Russ. J. Inorg. Chem. 2019, 64, 79–87. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Quantum-chemical calculation of molecular structures of Al2Mn3 and Al2Zn3 clusters by using DFT method. Struct. Chem. 2019, 30, 1289–1299. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structure models of Al2Ti3 and Al2V3 clusters according to DFT quantum-chemical calculations. Eur. Chem. Bull. 2019, 9, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, O.V.; Chachkov, D.V. Molecular structures of five atomic aluminum-titanium and aluminum-vanadium metal clusters: Theoretical consideration. Tsvetnye Met. 2020, 49–56. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Models of Molecular Structures of Hexa-Nuclear AlnFem Metal Clusters (n + m = 6): DFT Quantum-Chemical Design. Materials 2021, 14, 597. [Google Scholar] [CrossRef]
- Mikhailov, O.V.; Chachkov, D.V. Quantum-Chemical Design of Molecular Structures of Tetra-, Penta- and Hexanuclear Metal Clusters Containing Aluminum and 3d-Element Atoms. Materials 2020, 13, 1852. [Google Scholar] [CrossRef] [Green Version]
- Mikhailov, O.V.; Chachkov, D.V. Quantum Chemical Calculation of Molecular Structures of Al2Fe2 and Al2FeCo Tetranuclear Metalloclusters. Glass Phys. Chem. 2017, 43, 597–604. [Google Scholar] [CrossRef]
- Schaefer, A.; Horn, H.; Ahlrichs, R. Fully optimized contracted Gaussian basis sets for atoms Li to Kr. J. Chem. Phys. 1992, 97, 2571–2577. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Hoe, W.M.; Cohen, A.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1997, 78, 1396–1396. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, H.; Duelund, L.; Winkler, H.; Toftlund, H.; Trautwein, A.X. Free Energy of Spin-Crossover Complexes Calculated with Density Functional Methods. Inorg. Chem. 2001, 40, 2201–2203. [Google Scholar] [CrossRef]
- Swart, M.; Groenhof, A.R.; Ehlers, A.W.; Lammertsma, K. Validation of Exchange−Correlation Functionals for Spin States of Iron Complexes. J. Phys. Chem. A 2004, 108, 5479–5483. [Google Scholar] [CrossRef]
- Swart, M.; Ehlers, A.W.; Lammertsma, K. Performance of the OPBE exchange-correlation functional. Mol. Phys. 2004, 102, 2467–2474. [Google Scholar] [CrossRef]
- Swart, M. Metal–ligand bonding in metallocenes: Differentiation between spin state, electrostatic and covalent bonding. Inorg. Chim. Acta 2007, 360, 179–189. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Foster, J.P.; Weinhold, F. Natural hybrid orbitals. J. Am. Chem. Soc. 1980, 102, 7211–7218. [Google Scholar] [CrossRef]
- Glendening, E.D.; Reed, A.E.; Carpenter, J.E.; Weinhold, F. NBO 3.0. QCPE Bull. 1990, 10, 58. [Google Scholar]
- Ochterski, J.W. Thermochemistry in Gaussian; Gaussian, Inc.: Wallingford, CT, USA, 2000. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn |
|---|---|---|---|---|---|---|---|---|---|---|
| N | 7 | 13 | 14 | 9 | 20 | 22 | 21 | 6 | 12 | 8 |
| Al2Sc2 (3-I) | Al2Ti2 (3-I) | Al2V2 (3-I) | Al2Cr2 (1-I)* | Al2Mn2 (1-I)* | |
|---|---|---|---|---|---|
| All distances between metal atoms, pm | |||||
| Al1Al2 | 254.5 | 249.3 | 252.8 | 248.8 | 257.3 |
| Al1M1 | 272.8 | 263.7 | 257.9 | 266.2 | 257.6 |
| Al1M2 | 272.8 | 263.8 | 257.9 | 266.3 | 257.6 |
| Al2M1 | 272.8 | 263.7 | 257.6 | 266.4 | 257.6 |
| Al2M2 | 272.8 | 263.6 | 257.6 | 266.2 | 257.7 |
| M1M2 | 277.7 | 210.3 | 170.8 | 259.6 | 280.0 |
| All plane angles between metal atoms, deg | |||||
| Al1Al2M1 | 62.2 | 61.8 | 60.7 | 62.1 | 60.0 |
| Al1Al2M2 | 62.2 | 61.8 | 60.7 | 62.2 | 60.0 |
| Al2Al1M1 | 62.2 | 61.8 | 60.6 | 62.2 | 60.0 |
| Al2Al1M2 | 62.2 | 61.7 | 60.6 | 62.1 | 60.0 |
| Al1M1Al2 | 55.6 | 56.4 | 58.7 | 55.7 | 59.9 |
| Al1M1M2 | 59.4 | 66.5 | 70.7 | 60.8 | 57.1 |
| M1Al1M2 | 61.2 | 47.1 | 38.7 | 58.3 | 65.8 |
| Al1M2Al2 | 55.6 | 56.4 | 58.7 | 55.7 | 59.9 |
| Al1M2M1 | 59.4 | 66.4 | 70.7 | 60.8 | 57.1 |
| Al2M1M2 | 59.4 | 66.4 | 70.7 | 60.8 | 57.1 |
| M1Al2M2 | 61.2 | 47.1 | 38.7 | 58.3 | 65.8 |
| Al2M2M1 | 59.4 | 66.5 | 70.6 | 60.9 | 57.1 |
| Selected torsion angles between metal atoms, deg | |||||
| Al1Al2M1M2 | −71.8 | −76.5 | −78.6 | 72.9 | −68.1 |
| Al1Al2M2M1 | 71.8 | 76.4 | 78.6 | −72.8 | 68.1 |
| Al2Al1M1M2 | 71.8 | 76.4 | 78.6 | −72.8 | 68.1 |
| Al2Al1M2M1 | −71.8 | −76.5 | −78.6 | 72.9 | −68.0 |
| M1Al1Al2M2 | −70.3 | −54.0 | −44.7 | 66.9 | −77.7 |
| M1Al2Al1M2 | 70.3 | 54.0 | 44.7 | −66.9 | 77.7 |
| Al1M1M2Al2 | −65.6 | −62.1 | −62.6 | 64.7 | −73.0 |
| Al1M2M1Al2 | 65.6 | 62.1 | 62.6 | −64.7 | 73.0 |
| Al1M1Al2M2 | 71.8 | 76.5 | 78.6 | −72.9 | 68.1 |
| Al1M2Al2M1 | −71.8 | −76.4 | −78.6 | 72.8 | −68.1 |
| Al2M1Al1M2 | −71.8 | −76.4 | −78.6 | 72.9 | −68.1 |
| Al2M2Al1M1 | 71.8 | 76.5 | 78.6 | −729.8 | 68.1 |
| Al2Fe2 (5-I) | Al2Co2 (5-I) | Al2Ni2 (1-I) | Al2Cu2 (1-I) | Al2Zn2 (1-I)* | |
|---|---|---|---|---|---|
| All distances between metal atoms, pm | |||||
| Al1Al2 | 259.3 | 256.6 | 262.2 | 250.5 | 276.9 |
| Al1M1 | 249.8 | 243.5 | 233.6 | 242.8 | 261.0 |
| Al1M2 | 249.6 | 243.4 | 233.6 | 243.0 | 261.0 |
| Al2M1 | 249.8 | 243.3 | 233.6 | 243.0 | 261.0 |
| Al2M2 | 249.6 | 243.3 | 233.6 | 243.1 | 261.0 |
| M1M2 | 198.6 | 209.0 | 220.0 | 254.7 | 260.2 |
| All plane angles between metal atoms, deg | |||||
| Al1Al2M1 | 58.7 | 58.2 | 55.9 | 58.9 | 58.0 |
| Al1Al2M2 | 58.7 | 58.2 | 55.9 | 59.0 | 58.0 |
| Al2Al1M1 | 58.7 | 58.2 | 55.9 | 59.0 | 58.0 |
| Al2Al1M2 | 58.7 | 58.2 | 55.9 | 59.0 | 58.0 |
| Al1M1Al2 | 62.5 | 63.6 | 68.3 | 62.1 | 64.1 |
| Al1M1M2 | 66.5 | 64.6 | 61.9 | 58.4 | 60.1 |
| M1Al1M2 | 46.9 | 50.8 | 56.2 | 63.2 | 59.8 |
| Al1M2Al2 | 62.5 | 63.6 | 68.3 | 62.0 | 64.1 |
| Al1M2M1 | 66.6 | 64.6 | 61.9 | 58.3 | 60.1 |
| Al2M1M2 | 66.5 | 64.6 | 61.9 | 58.4 | 60.1 |
| M1Al2M2 | 46.9 | 50.9 | 56.2 | 63.2 | 59.8 |
| Al2M2M1 | 66.6 | 64.6 | 61.9 | 58.4 | 60.1 |
| Selected torsion angles between metal atoms, deg | |||||
| Al1Al2M1M2 | 74.7 | −72.8 | 68.8 | 68.3 | 68.9 |
| Al1Al2M2M1 | −74.8 | 72.9 | −68.8 | −68.2 | −68.9 |
| Al2Al1M1M2 | −74.7 | 72.8 | −68.8 | −68.3 | −68.9 |
| Al2Al1M2M1 | 74.8 | −72.8 | 68.8 | 68.3 | 68.9 |
| M1Al1Al2M2 | 55.5 | −60.7 | 69.4 | 75.4 | 72.1 |
| M1Al2Al1M2 | −55.5 | 60.7 | −69.4 | −75.4 | −72.1 |
| Al1M1M2Al2 | 68.9 | −71.4 | 79.0 | 74.5 | 75.5 |
| Al1M2M1Al2 | −68.9 | 71.4 | −79.0 | −74.5 | −75.5 |
| Al1M1Al2M2 | −74.7 | 72.8 | −68.8 | −68.3 | −68.9 |
| Al1M2Al2M1 | 74.8 | −72.9 | 68.8 | 68.2 | 68.9 |
| Al2M1Al1M2 | 74.7 | −72.8 | 68.8 | 68.3 | 68.9 |
| Al2M2Al1M1 | −74.8 | 72.8 | −68.8 | −68.3 | −68.9 |
| Metal Cluster | ΔfH0(298 K) kJ/mol | Sf0(298 K) J/mol K | ΔfG0(298 K), kJ/mol |
|---|---|---|---|
| Al2Sc2 (3-I) | 677.9 | 387.8 | 599.9 |
| Al2Ti2 (3-I) | 833.4 | 375.9 | 756.5 |
| Al2V2 (3-I) | 521.0 | 378.5 | 439.2 |
| Al2Cr2 (1-I)* | 923.0 | 361.6 | 848.1 |
| Al2Mn2 (1-I)* | 469.8 | 362.2 | 397.7 |
| Al2Fe2 (5-I) | 691.7 | 387.4 | 609.4 |
| Al2Co2 (5-I) | 701.8 | 380.5 | 623.3 |
| Al2Ni2 (1-I) | 669.8 | 375.6 | 592.5 |
| Al2Cu2 (1-I) | 662.7 | 358.9 | 592.5 |
| Al2Zn2 (1-I)* | 604.7 | 372.9 | 535.4 |
| Charges on the Al and M Atoms in the Al2M2 Metal Cluster, ē | Charges on the Al and M Atoms in the Al2M2 Metal Cluster, ē | ||||||
|---|---|---|---|---|---|---|---|
| Al2Sc2 (3-I) (<S**2> = 2.0534) | Al2Fe2 (5-I) (<S**2> = 6.7424) | ||||||
| Al1 | Al2 | Sc1 | Sc2 | Al1 | Al2 | Fe1 | Fe2 |
| −0.0493 | −0.0494 | +0.0492 | +0.0495 | +0.2022 | +0.2020 | −0.1996 | −0.2048 |
| Al2Ti2 (3-I) (<S**2> = 2.2578) | Al2Co2 (5-I) (<S**2> = 6.2633) | ||||||
| Al1 | Al2 | Ti1 | Ti2 | Al1 | Al2 | Co1 | Co2 |
| +0.1420 | +0.1416 | −0.1416 | −0.1420 | +0.2443 | +0.2439 | −0.2436 | −0.2446 |
| Al2V2 (3-I) (<S**2> = 2.0204) | Al2Ni2 (1-I) (<S**2> = 0.0000) | ||||||
| Al1 | Al2 | V1 | V2 | Al1 | Al2 | Ni1 | Ni2 |
| +0.1607 | +0.1599 | −0.1606 | −0.1600 | +0.3525 | +0.3525 | −0.3525 | −0.3525 |
| Al2Cr2 (1-I)* (<S**2> = 4.6791) | Al2Cu2 (1-I) (<S**2> = 0.0000) | ||||||
| Al1 | Al2 | Cr1 | Cr2 | Al1 | Al2 | Cu1 | Cu2 |
| −0.0315 | −0.0317 | +0.0312 | +0.0320 | +0.1116 | +0.1127 | −0.1128 | −0.1115 |
| Al2Mn2 (1-I)* (<S**2> = 4.7528) | Al2Zn2 (1-I)* (<S**2> = 0.4335) | ||||||
| Al1 | Al2 | Mn1 | Mn2 | Al1 | Al2 | Zn1 | Zn2 |
| −0.1351 | −0.1344 | +0.1347 | +0.1348 | −0.2163 | +0.1179 | +0.0492 | +0.0492 |
| Metal Cluster | ΔrH0(298 K), kJ | ΔrS0(298 K), J/K | Metal Cluster | ΔrH0(298 K), kJ | ΔrS0(298 K), J/K |
|---|---|---|---|---|---|
| Al2Sc2 (3-I) | −738.5 | −290.4 | Al2Fe2 (5-I) | −799.5 | −302.3 |
| Al2Ti2 (3-I) | −772.0 | −313.4 | Al2Co2 (5-I) | −805.7 | −307.2 |
| Al2V2 (3-I) | −1167.9 | −315.2 | Al2Ni2 (1-I) | −847.6 | −317.5 |
| Al2Cr2 (1-I) | −528.3 | −315.7 | Al2Cu2 (1-I) | −672.1 | −302.5 |
| Al2Mn2(1-I) | −749.6 | −313.9 | Al2Zn2 (1-I) | −315.0 | −278.2 |
| Metal Cluster | Ttd, K | t°td, °C | Metal Cluster | Ttd, K | t°td, °C |
|---|---|---|---|---|---|
| Al2Sc2 (3-I) | 2543.0 | 2269.8 | Al2Fe2 (5-I) | 2644.7 | 2371.5 |
| Al2Ti2 (3-I) | 2463.3 | 2190.1 | Al2Co2 (5-I) | 2622.7 | 2349.5 |
| Al2V2 (3-I) | 3705.3 | 3432.1 | Al2Ni2 (1-I) | 2669.6 | 2396.4 |
| Al2Cr2 (1-I) | 1673.4 | 1400.2 | Al2Cu2 (1-I) | 2221.8 | 1948.6 |
| Al2Mn2 (1-I) | 2388.0 | 2114.8 | Al2Zn2 (1-I) | 1132.3 | 859.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mikhailov, O.V.; Chachkov, D.V. Quantum-Chemical Consideration of Al2M2 Tetranuclear Metal Clusters (M–3d-Element): Molecular/Electronic Structures and Thermodynamics. Materials 2021, 14, 6836. https://doi.org/10.3390/ma14226836
Mikhailov OV, Chachkov DV. Quantum-Chemical Consideration of Al2M2 Tetranuclear Metal Clusters (M–3d-Element): Molecular/Electronic Structures and Thermodynamics. Materials. 2021; 14(22):6836. https://doi.org/10.3390/ma14226836
Chicago/Turabian StyleMikhailov, Oleg V., and Denis V. Chachkov. 2021. "Quantum-Chemical Consideration of Al2M2 Tetranuclear Metal Clusters (M–3d-Element): Molecular/Electronic Structures and Thermodynamics" Materials 14, no. 22: 6836. https://doi.org/10.3390/ma14226836