Monometallic Cerium Layered Double Hydroxide Supported Pd-Ni Nanoparticles as High Performance Catalysts for Lignin Hydrogenolysis

 , , , and
, , , and
Abstract
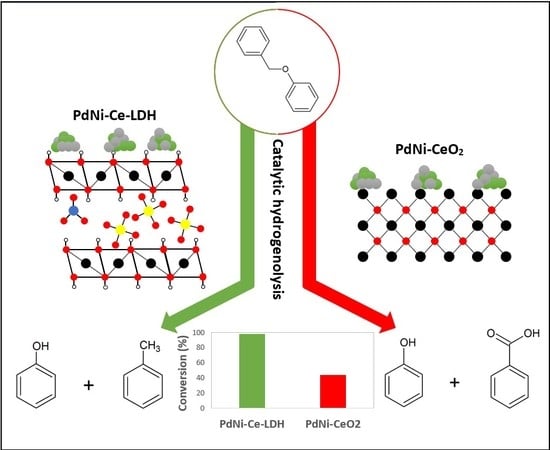
:
1. Introduction
2. Materials and Methods
2.1. Synthesis Procedures
2.1.1. Monometallic Cerium LDHs
2.1.2. Cerium Oxide Support
2.1.3. Palladium Nickel Catalysts
2.2. Chemical Analysis and Characterization
2.2.1. X-Ray Diffraction
2.2.2. Scanning Electron Microscopy and Energy-Dispersive X-Ray Spectroscopy
2.2.3. Thermogravimetric Analysis
2.2.4. Fourier-Transform Infrared Spectroscopy
2.2.5. Nitrogen Adsorption
2.2.6. X-Ray Fluorescence
2.2.7. X-Ray Photoelectron Spectroscopy
2.2.8. Iodometric Titration
2.3. Reaction Procedure
2.3.1. Reactor Set-Up
2.3.2. Catalyst Pre-Reduction.
2.3.3. Reductive Cleavage of α-O-4 Model Component
2.3.4. Reversed Phase High Performance Liquid Chromatography
3. Results and Discussion
3.1. Characterization of the Ce-LDH Support
3.1.1. X-Ray Diffraction
3.1.2. Scanning Electron Microscopy
3.1.3. X-Ray Photoelectron Spectroscopy
3.1.4. Iodometry
3.1.5. Thermogravimetric Analysis
3.1.6. Ce-LDH Structural Formula
3.2. Effect of Support Calcination Temperature
3.2.1. High Temperature X-ray Diffraction
3.2.2. Nitrogen Sorption
3.2.3. X-Ray Fluoresence
3.3. Characterization of the PdNi-Ce-LDH Catalyst
3.3.1. X-Ray Diffraction
3.3.2. Scanning Electron Microscopy and Energy-Dispersive X-Ray Spectroscopy
3.3.3. Catalytic Performance of the Cerium Supported PdNi Catalysts
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Van Geem, K.M.; Galvita, V.V.; Marin, G.B. Making chemicals with electricity. Science 2019, 364, 734–735. [Google Scholar] [CrossRef]
- Hernández, W.Y.; Lauwaert, J.; Van der Voort, P.; Verberckmoes, A. Recent advances on the utilization of layered double hydroxides (LDHs) and related heterogeneous catalysts in a lignocellulosic-feedstock biorefinery scheme. Green Chem. 2017, 19, 5269–5302. [Google Scholar] [CrossRef] [Green Version]
- Chheda, J.N.; Huber, G.W.; Dumesic, J.A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Edit. 2007, 46, 7164–7183. [Google Scholar] [CrossRef] [PubMed]
- Schutyser, W.; Renders, T.; Van den Bosch, S.; Koelewijn, S.F.; Beckham, G.T.; Sels, B.F. Chemicals from lignin: An interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. [Google Scholar] [CrossRef] [PubMed]
- Gosselink, R.J.A.; de Jong, E.; Guran, B.; Abacherli, A. Co-ordination network for lignin - standardisation, production and applications adapted to market requirements (EUROLIGNIN). Ind. Crop. Prod. 2004, 20, 121–129. [Google Scholar] [CrossRef]
- Tejado, A.; Pena, C.; Labidi, J.; Echeverria, J.M.; Mondragon, I. Physico-chemical characterization of lignins from different sources for use in phenol-formaldehyde resin synthesis. Bioresour. Technol. 2007, 98, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Zakzeski, J.; Bruijnincx, P.C.A.; Jongerius, A.L.; Weckhuysen, B.M. The Catalytic Valorization of Lignin for the Production of Renewable Chemicals. Chem. Rev. 2010, 110, 3552–3599. [Google Scholar] [CrossRef]
- Lauwaert, J.; Stals, I.; Lancefield, C.S.; Deschaumes, W.; Depuydt, D.; Vanlerberghe, B.; Devlamynck, T.; Bruijnincx, P.C.A.; Verberckmoes, A. Pilot scale recovery of lignin from black liquor and advanced characterization of the final product. Sep. Purif. Technol. 2019, 221, 226–235. [Google Scholar] [CrossRef]
- Constant, S.; Wienk, H.L.J.; Frissen, A.E.; Peinder, P.D.; Boelens, R.; van Es, D.S.; Grisel, R.J.H.; Weckhuysen, B.M.; Huijgen, W.J.J.; Gosselink, R.J.A.; et al. New insights into the structure and composition of technical lignins: A comparative characterisation study. Green Chem. 2016, 18, 2651–2665. [Google Scholar] [CrossRef] [Green Version]
- Crestini, C.; Lange, H.; Sette, M.; Argyropoulos, D.S. On the structure of softwood kraft lignin. Green Chem. 2017, 19, 4104–4121. [Google Scholar] [CrossRef]
- Deuss, P.J.; Lancefield, C.S.; Narani, A.; de Vries, J.G.; Westwood, N.J.; Barta, K. Phenolic acetals from lignins of varying compositions via iron(iii) triflate catalysed depolymerisation. Green Chem. 2017, 19, 2774–2782. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Zhao, C.; Lercher, J.A. Ni-Catalyzed Cleavage of Aryl Ethers in the Aqueous Phase. J. Am. Chem. Soc. 2012, 134, 20768–20775. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Lu, J.; Zhang, X.; MacArthur, K.; Heggen, M.; Li, H.; Wang, F. Cleavage of the lignin β-O-4 ether bond via a dehydroxylation–hydrogenation strategy over a NiMo sulfide catalyst. Green Chem. 2016, 18, 6545–6555. [Google Scholar] [CrossRef]
- Zhang, J.-W.; Cai, Y.; Lu, G.-P.; Cai, C. Facile and selective hydrogenolysis of β-O-4 linkages in lignin catalyzed by Pd–Ni bimetallic nanoparticles supported on ZrO2. Green Chem. 2016, 18, 6229–6235. [Google Scholar] [CrossRef]
- Paone, E.; Espro, C.; Pietropaolo, R.; Mauriello, F. Selective arene production from transfer hydrogenolysis of benzyl phenyl ether promoted by a co-precipitated Pd/Fe3O4 catalyst. Catal. Sci. Technol. 2016, 6, 7937–7941. [Google Scholar] [CrossRef]
- Gao, X.; Zhu, S.; Li, Y. Selective hydrogenolysis of lignin and model compounds to monophenols over AuPd/CeO2. Mol. Catal. 2019, 462, 69–76. [Google Scholar] [CrossRef]
- Zhang, B.; Qi, Z.; Li, X.; Ji, J.; Zhang, L.; Wang, H.; Liu, X.; Li, C. Cleavage of lignin C–O bonds over a heterogeneous rhenium catalyst through hydrogen transfer reactions. Green Chem. 2019, 21, 5556–5564. [Google Scholar] [CrossRef]
- Deng, W.; Zhang, H.; Wu, X.; Li, R.; Zhang, Q.; Wang, Y. Oxidative conversion of lignin and lignin model compounds catalyzed by CeO2-supported Pd nanoparticles. Green Chem. 2015, 17, 5009–5018. [Google Scholar] [CrossRef]
- Sun, Z.H.; Fridrich, B.; de Santi, A.; Elangovan, S.; Barta, K. Bright Side of Lignin Depolymerization: Toward New Platform Chemicals. Chem. Rev. 2018, 118, 614–678. [Google Scholar] [CrossRef] [Green Version]
- Cheng, C.; Shen, D.; Gu, S.; Luo, K.H. State-of-the-art catalytic hydrogenolysis of lignin for the production of aromatic chemicals. Catal. Sci. Technol. 2018, 8, 6275–6296. [Google Scholar] [CrossRef]
- Margellou, A.; Triantafyllidis, K.S. Catalytic Transfer Hydrogenolysis Reactions for Lignin Valorization to Fuels and Chemicals. Catalysts 2019, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Rached, J.A.; Cesario, M.R.; Estephane, J.; Tidahy, H.L.; Gennequin, C.; Aouad, S.; Aboukais, A.; Abi-Aad, E. Effects of cerium and lanthanum on Ni-based catalysts for CO2 reforming of toluene. J. Environ. Chem. Eng. 2018, 6, 4743–4754. [Google Scholar] [CrossRef]
- Liu, Z.Y.; Yao, S.Y.; Johnston-Peck, A.; Xu, W.Q.; Rodriguez, J.A.; Senanayake, S.D. Methanol steam reforming over Ni-CeO2 model and powder catalysts: Pathways to high stability and selectivity for H-2/CO2 production. Catal. Today 2018, 311, 74–80. [Google Scholar] [CrossRef]
- Van Vaerenbergh, B.; Lauwaert, J.; Vermeir, P.; De Clercq, J.; Thybaut, J.W. Chapter One - Synthesis and support interaction effects on the palladium nanoparticle catalyst characteristics. In Advances in Catalysis; Song, C., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 65, pp. 1–120. [Google Scholar]
- Cavani, F.; Trifiro, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, poroperties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Khan, A.I.; O’Hare, D. Intercalation chemistry of layered double hydroxides: Recent developments and applications. J. Mater. Chem. 2002, 12, 3191–3198. [Google Scholar] [CrossRef]
- Debecker, D.P.; Gaigneaux, E.M.; Busca, G. Exploring, tuning, and exploiting the basicity of hydrotalcites for applications in heterogeneous catalysis. Chem.-Eur. J. 2009, 15, 3920–3935. [Google Scholar] [CrossRef]
- Roelofs, J.; Lensveld, D.J.; van Dillen, A.J.; de Jong, K.P. On the structure of activated hydrotalcites as solid base catalysts for liquid-phase aldol condensation. J. Catal. 2001, 203, 184–191. [Google Scholar] [CrossRef]
- Fogg, A.M.; Rohl, A.L.; Parkinson, G.M.; O’Hare, D. Predicting guest orientations in layered double hydroxide intercalates. Chem. Mat. 1999, 11, 1194–1200. [Google Scholar] [CrossRef]
- Perez-Ramirez, J.; Abello, S.; van der Pers, N.M. Memory effect of activated Mg-Al hydrotalcite: In situ XRD studies during decomposition and gas-phase reconstruction. Chem. Eur. J. 2007, 13, 870–878. [Google Scholar] [CrossRef]
- Mishra, G.; Dash, B.; Pandey, S. Layered double hydroxides: A brief review from fundamentals to application as evolving biomaterials. Appl. Clay Sci. 2018, 153, 172–186. [Google Scholar] [CrossRef]
- Benicio, L.P.F.; Silva, R.A.; Lopes, J.A.; Eulalio, D.; dos Santos, R.M.M.; de Aquino, L.A.; Vergutz, L.; Novais, R.F.; da Costa, L.M.; Pinto, F.G.; et al. Layered double hydroxides: Nanomaterials for applications in agriculture. Rev. Bras. Cienc. Solo 2015, 39, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Chubar, N. New inorganic (an)ion exchangers based on Mg-Al hydrous oxides: (Alkoxide-free) sol-gel synthesis and characterisation. J. Colloid Interface Sci. 2011, 357, 198–209. [Google Scholar] [CrossRef]
- Muráth, S.; Somosi, Z.; Kukovecz, Á.; Kónya, Z.; Sipos, P.; Pálinkó, I. Novel route to synthesize CaAl- and MgAl-layered double hydroxides with highly regular morphology. J. Sol-Gel Sci. Technol. 2019, 89, 844–851. [Google Scholar] [CrossRef]
- Daza, C.E.; Moreno, S.; Molina, R. Ce-incorporation in mixed oxides obtained by the self-combustion method for the preparation of high performance catalysts for the CO2 reforming of methane. Catal. Commun. 2010, 12, 173–179. [Google Scholar] [CrossRef]
- Liang, J.B.; Ma, R.Z.; Sasaki, T. Layered rare earth hydroxides (LREHs): Synthesis and structure characterization towards multifunctionality. Dalton Trans. 2014, 43, 10355–10364. [Google Scholar] [CrossRef]
- Ye, T.; Huang, W.M.; Zeng, L.M.; Li, M.L.; Shi, J.L. CeO2-x platelet from monometallic cerium layered double hydroxides and its photocatalytic reduction of CO2. Appl. Catal. B-Environ. 2017, 210, 141–148. [Google Scholar] [CrossRef]
- Balsamo, N.; Mendieta, S.; Oliva, M.; Eimer, G.; Crivello, M. Synthesis and Characterization of Metal Mixed Oxides from Layered Double Hydroxides. Procedia Mater. Sci. 2012, 1, 506–513. [Google Scholar] [CrossRef] [Green Version]
- Zapata, B.; Bosch, P.; Valenzuela, M.A.; Fetter, G.; Flores, S.O.; Cordova, I.R. Thermal stability of monometallic Co-hydrotalcite. Mater. Lett. 2002, 57, 679–683. [Google Scholar] [CrossRef]
- Liang, J.; Ma, R. Ln2(OH)4SO4 nH2O (Ln = Pr to Tb; n ∼ 2): A New Family of Layered Rare-Earth Hydroxides Rigidly Pillared by Sulfate Ions. Chem. Mater. 2010, 22, 6001–6007. [Google Scholar] [CrossRef]
- Vialat, P.; Rabu, P.; Mousty, C.; Leroux, F. Insight of an easy topochemical oxidative reaction in obtaining high performance electrochemical capacitor based on (CoCoIII)-Co-II monometallic cobalt Layered Double Hydroxide. J. Power Sources 2015, 293, 1–10. [Google Scholar] [CrossRef]
- Sharma, D.; Baskaran, T.; Christopher, J.; Sakthivel, A. First Report on Silicate Intercalated Monometallic Cobalt Hydrotalcite (Co-HT) Materials: Preparation and Its Applications. Nanosci. Nanotechnol. Lett. 2016, 8, 360–364. [Google Scholar] [CrossRef]
- Konnerth, H.; Zhang, J.; Ma, D.; Prechtl, M.H.G.; Yan, N. Base promoted hydrogenolysis of lignin model compounds and organosolv lignin over metal catalysts in water. Chem. Eng. Sci. 2015, 123, 155–163. [Google Scholar] [CrossRef] [Green Version]
- Dell’Agli, G.; Spiridigliozzi, L.; Marocco, A.; Accardo, G.; Ferone, C.; Cioffi, R. Effect of the Mineralizer Solution in the Hydrothermal Synthesis of Gadolinium-Doped (10% mol Gd) Ceria Nanopowders. J. Appl. Biomater. Funct. Mater. 2016, 14, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Munnik, P.; de Jongh, P.E.; de Jong, K.P. Recent Developments in the Synthesis of Supported Catalysts. Chem. Rev. 2015, 115, 6687–6718. [Google Scholar] [CrossRef]
- Ashok, A.; Kumar, A.; Bhosale, R.R.; Saleh, M.A.H.; van den Broeke, L.J.P. Cellulose assisted combustion synthesis of porous Cu–Ni nanopowders. RSC Adv. 2015, 5, 28703–28712. [Google Scholar] [CrossRef]
- Danielis, M.; Colussi, S.; de Leitenburg, C.; Trovarelli, A. The role of palladium salt precursors in Pd-PdO/CeO2 catalysts prepared by dry milling for methane oxidation. Catal. Commun. 2020, 135, 105899. [Google Scholar] [CrossRef]
- Sant, B.R. Iodometric estimation of cerium(IV) by arsenious oxide. Fresenius’ Z. Anal. Chem. 1957, 158, 252–254. [Google Scholar] [CrossRef]
- Suma, N.; Jeevananda, T.; Palanna, O.G. Determination of Cerium in Ceric Ammonium Sulphate by Novel Iodo-Potentiometric Method. Asian J. Chem. 2010, 22, 5209–5217. [Google Scholar]
- Chen, X.; Yan, N. Chapter 7 Nanoparticle Design for the Catalytic Valorization of Lignocellulosic Biomass. In Nanoparticle Design and Characterization for Catalytic Applications in Sustainable Chemistry; The Royal Society of Chemistry: London, UK, 2019; pp. 184–206. [Google Scholar] [CrossRef]
- Barrault, J.; Probst, S.; Alouche, A.; Percheron-Guecan, A.; Paul-Boncour, V.; Primet, M. Characterization and Catalytic Properties of Nickel Oxioe Supported on Rare Earth Oxides. Description of the Metal — Support Interaction. In Studies in Surface Science and Catalysis; Holmen, A., Jens, K.J., Kolboe, S., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; Volume 61, pp. 357–365. [Google Scholar]
- Stasinska, B.; Gac, W.; Ioannides, T.; Machocki, A. Complete Oxidation of Methane over Palladium Supported on Alumina Modified with Calcium, Lanthanum, and Cerium Ions. J. Nat. Gas Chem. 2007, 16, 342–348. [Google Scholar] [CrossRef]
- Batista, J.; Pintar, A.; Mandrino, D.; Jenko, M.; Martin, V. XPS and TPR examinations of γ-alumina-supported Pd-Cu catalysts. Appl. Catal. A: Gen. 2001, 206, 113–124. [Google Scholar] [CrossRef]
- Vialat, P.; Mousty, C.; Taviot-Gueho, C.; Renaudin, G.; Martinez, H.; Dupin, J.-C.; Elkaim, E.; Leroux, F. High-Performing Monometallic Cobalt Layered Double Hydroxide Supercapacitor with Defined Local Structure. Adv. Funct. Mater. 2014, 24, 4831–4842. [Google Scholar] [CrossRef] [Green Version]
- Mullins, D.R.; Overbury, S.H.; Huntley, D.R. Electron spectroscopy of single crystal and polycrystalline cerium oxide surfaces. Surf. Sci. 1998, 409, 307–319. [Google Scholar] [CrossRef]
- Teterin, Y.A.; Teterin, A.Y.; Lebedev, A.M.; Utkin, I.O. The XPS spectra of cerium compounds containing oxygen. J. Electron Spectrosc. Relat. Phenom. 1998, 88–91, 275–279. [Google Scholar] [CrossRef]
- Montemor, M.F.; Simões, A.M.; Ferreira, M.G.S.; Carmezim, M.J. Composition and corrosion resistance of cerium conversion films on the AZ31 magnesium alloy and its relation to the salt anion. Appl. Surf. Sci. 2008, 254, 1806–1814. [Google Scholar] [CrossRef]
- NIST Standard Reference Database 20, Version 4.1. Available online: http://dx.doi.org/10.18434/T4T88K (accessed on 3 January 2020).
- Wyon, C. X-ray metrology for advanced microelectronics. Eur. Phys. J. Appl. Phys. 2010, 49, 20101. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Peng, P.a. Surface Characterization of Aerosol Particles in Guangzhou, China: A Study by XPS. Aerosol Sci. Technol. 2009, 43, 1230–1242. [Google Scholar] [CrossRef] [Green Version]
- Dutta, P.; Pal, S.; Seehra, M.S.; Shi, Y.; Eyring, E.M.; Ernst, R.D. Concentration of Ce3+ and Oxygen Vacancies in Cerium Oxide Nanoparticles. Chem. Mat. 2006, 18, 5144–5146. [Google Scholar] [CrossRef]
- Sims, C.; Maier, R.; Johnston-Peck, A.; Gorham, J.; Hackley, V.; Nelson, B. Quantitative analysis of oxidation state in cerium oxide nanomaterials. Abstr. Pap. Am. Chem. Soc. 2017, 254, 1. [Google Scholar] [CrossRef]
- Yan, K.; Liu, Y.; Lu, Y.; Chai, J.; Sun, L. Catalytic application of layered double hydroxide-derived catalysts for the conversion of biomass-derived molecules. Catal. Sci. Technol. 2017, 7, 1622–1645. [Google Scholar] [CrossRef]
- Udupa, M.R. Thermal decomposition of cerium(IV), cerium(III), chromium(III) and titanium(IV) sulphates. Thermochim. Acta 1982, 57, 377–381. [Google Scholar] [CrossRef]
- Leskelä, M. Thermal stability of Ce2O2S. DOES Ce2O2SO4 exist? Thermochim. Acta 1985, 92, 739–742. [Google Scholar] [CrossRef]
- Poston, J.A.; Siriwardane, R.V.; Fisher, E.P.; Miltz, A.L. Thermal decomposition of the rare earth sulfates of cerium(III), cerium(IV), lanthanum(III) and samarium(III). Appl. Surf. Sci. 2003, 214, 83–102. [Google Scholar] [CrossRef]
- Ghanbary, F.; Jafarnejad, E. Removal of malachite green from the aqueous solutions using polyimide nanocomposite containing cerium oxide as adsorbent. Inorg. Nano-Metal Chem. 2017, 47, 1675–1681. [Google Scholar] [CrossRef]
- Trusova, E.A.; Trutnev, N.S. Cryochemical synthesis of ultrasmall, highly crystalline, nanostructured metal oxides and salts. Beilstein J. Nanotechnol. 2018, 9, 1755–1763. [Google Scholar] [CrossRef]
- Van Vaerenbergh, B.; De Vlieger, K.; Claeys, K.; Vanhoutte, G.; De Clercq, J.; Vermeir, P.; Verberckmoes, A. The effect of the hydrotalcite structure and nanoparticle size on the catalytic performance of supported palladium nanoparticle catalysts in Suzuki cross-coupling. Appl. Catal. A Gen. 2018, 550, 236–244. [Google Scholar] [CrossRef]
- Soler-Illia, G.J.D.A.A.; Jobbágy, M.; Regazzoni, A.E.; Blesa, M.A. Synthesis of Nickel Hydroxide by Homogeneous Alkalinization. Precipitation Mechanism. Chem. Mat. 1999, 11, 3140–3146. [Google Scholar] [CrossRef]
- Rocha, J.; del Arco, M.; Rives, V.; A. Ulibarri, M. Reconstruction of layered double hydroxides from calcined precursors: A powder XRD and 27Al MAS NMR study. J. Mater. Chem. 1999, 9, 2499–2503. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | a (Å) | b (Å) | c (Å) |
|---|---|---|---|
| Ce-LDH | 13.79 ± 0.02 | 7.26 ± 0.01 | 8.30 ± 0.01 |
| Ce-LDH upscaled | 13.51 ± 0.01 | 7.26± 0.01 | 8.37 ± 0.01 |
| Material | %Ce4+ (mol%) |
|---|---|
| CeO2 | 92.4% |
| Ce-LDH | 46.4% |
| Support Material | BET Specific Surface Area (m²/g) |
|---|---|
| Ce-LDH | 11.00 |
| Ce-LDH-200 | 12.44 |
| Ce-LDH-400 | 12.85 |
| Ce-LDH-600 | 16.17 |
| Ce-LDH-800 | 13.89 |
| CeO2 | 17.82 |
| Catalyst | Weight Fraction (%) | Atomic Fraction (%) | ||
|---|---|---|---|---|
| Pd | Ni | Pd | Ni | |
| PdNi-CeO2 | 48.85 | 51.15 | 34.51 | 65.49 |
| PdNi-Ce-LDH-200 | 58.75 | 41.25 | 44.01 | 55.99 |
| PdNi-Ce-LDH-400 | 65.07 | 34.93 | 50.69 | 49.31 |
| PdNi-Ce-LDH-600 | 62.77 | 37.23 | 48.19 | 51.81 |
| PdNi-Ce-LDH-800 | 48.35 | 51.65 | 34.06 | 65.94 |
| Catalyst | Conversion (%) |
|---|---|
| Ce-LDH 600 | 20 |
| PdNi/Ce-LDH 200 | 80 |
| PdNi/Ce-LDH 400 | 88 |
| PdNi/Ce-LDH 600 | 98 |
| PdNi/Ce-LDH 800 | 93 |
| PdNi/CeO2 a | 44 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Saegher, T.; Lauwaert, J.; Hanssen, J.; Bruneel, E.; Van Zele, M.; Van Geem, K.; De Buysser, K.; Verberckmoes, A. Monometallic Cerium Layered Double Hydroxide Supported Pd-Ni Nanoparticles as High Performance Catalysts for Lignin Hydrogenolysis. Materials 2020, 13, 691. https://doi.org/10.3390/ma13030691
De Saegher T, Lauwaert J, Hanssen J, Bruneel E, Van Zele M, Van Geem K, De Buysser K, Verberckmoes A. Monometallic Cerium Layered Double Hydroxide Supported Pd-Ni Nanoparticles as High Performance Catalysts for Lignin Hydrogenolysis. Materials. 2020; 13(3):691. https://doi.org/10.3390/ma13030691
Chicago/Turabian StyleDe Saegher, Tibo, Jeroen Lauwaert, Jorku Hanssen, Els Bruneel, Matthias Van Zele, Kevin Van Geem, Klaartje De Buysser, and An Verberckmoes. 2020. "Monometallic Cerium Layered Double Hydroxide Supported Pd-Ni Nanoparticles as High Performance Catalysts for Lignin Hydrogenolysis" Materials 13, no. 3: 691. https://doi.org/10.3390/ma13030691