3.2. Rheological Properties of Spinning Solutions
The basic properties of each spinning solution, which demonstrated its quality, were rheological properties. To modify the composition of these solutions, it was necessary to determine the effect of modifications on these properties. When analyzing the rheological properties of spinning solutions, the dependence of tangential stress on shear rate (A power-law fluid—the Ostwald–de Waele relationship) is analyzed (1):
where n is the flow behavior index (-), and k is the flow consistency index (Pa·s
n). These two basic rheological parameters of each spinning solution were determined experimentally (
Table 1).
The value of the flow behavior index depended on the way in which polymer macromolecules were placed in motion, and the way they formed a system of mutually sliding layers. The linear polymer chains in the spinning solution (with a sufficiently high concentration) can be highly tangled. The determined value of n (
Table 1) indicated that all the obtained spinning solutions were shear thinned liquids. This is desirable for technological reasons (the ease of pressing the solution through the nozzles). The flow rates of solutions containing polyaniline were slightly higher. This means that polyaniline particles minimally affected the orientation of polyacrylonitrile macromolecules. This gentle effect on the
n-value may be due to the difference in the molecular structure of both polymers. The flow behavior index depended on several factors, e.g., the average molecular weight of the polymer and its degree of polydispersity, the degree of branching of macromolecules, and the content of additional components (especially fillers) and plasticizers. In the studied system, both polymers were linear, but the PAN macromolecules formed entangled structures, and the PANI macromolecules were largely rigid due to the presence of quinone rings in them. This parameter, in addition to temperature, pressure, and concentration, had a significant impact on the value of the flow consistency index k. Taking into account technological requirements, the k value of spinning solutions should be greater than 20 Pa·s
n. In comparison to changes in the n value, the presence of polyaniline molecules in the spinning solution caused a significant change in the k value. This change was mainly due to the addition of a polymer with a much lower average molecular weight relative to the spinning solution. The average molecular weight of PANI was 50 times less than PAN. Moreover, a significantly lower value of the consistency coefficient of spinning solutions containing PANI may be related to the occurrence of interactions between the PAN and PANI chains [
19,
20].
3.3. Fiber Morphology
It is well known that polyaniline obtained by aniline polymerization can occur in three oxidation states [
21]:
leucoemeraldine—(C6H4NH)n—completely reduced, white or colorless;
emeraldine—([C6H4NH]2[C6H4N]2)n—blue for the emeraldine base, green for the emeraldine salt;
(per)nigraniline—(C6H4N)n—completely oxidized, blue or violet.
Of all forms of polyaniline, only emeraldine salt (green) has an electrical conductivity at the level of semiconductors, i.e., around 1 S/cm. Emeraldine salt is obtained as a result of emeraldine base modification by HCl or HBr. Each type of polyaniline can be transformed in a reversible process of oxidation or reduction, during which changes in color and electrical conductivity occur [
22].
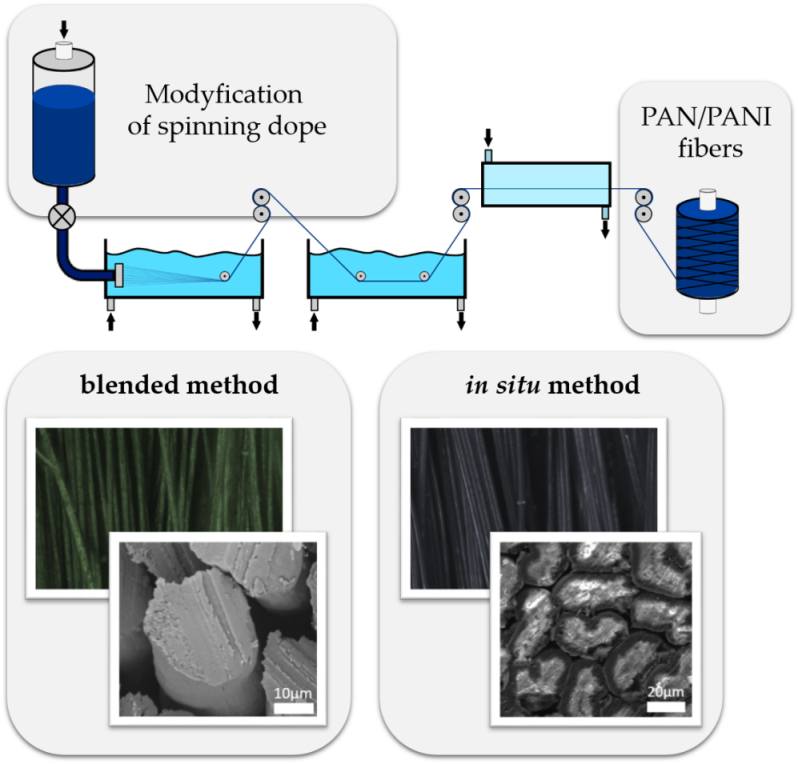
The PAN/PANI blended fibers obtained by adding polyaniline powder to the spinning solution had a dark green color (
Figure 3a), which, after immersing them in a 3% aqueous solution of hydrochloric acid, turned to dark blue (
Figure 3b). After rinsing again in water, their color returned to the previous color. The fibers obtained as a result of the synthesis of polyaniline in situ in the spinning solution were dark navy (
Figure 3c).
Based on the color of the obtained fibers, it can be concluded that polyaniline in the PAN/PANI blended fibers was in an emeraldine base form, and in an HCl solution it transformed into emeraldine salt (
Figure 3b). Moreover, in the PAN/PANI in situ fibers, polyaniline was in the emeraldine base form.
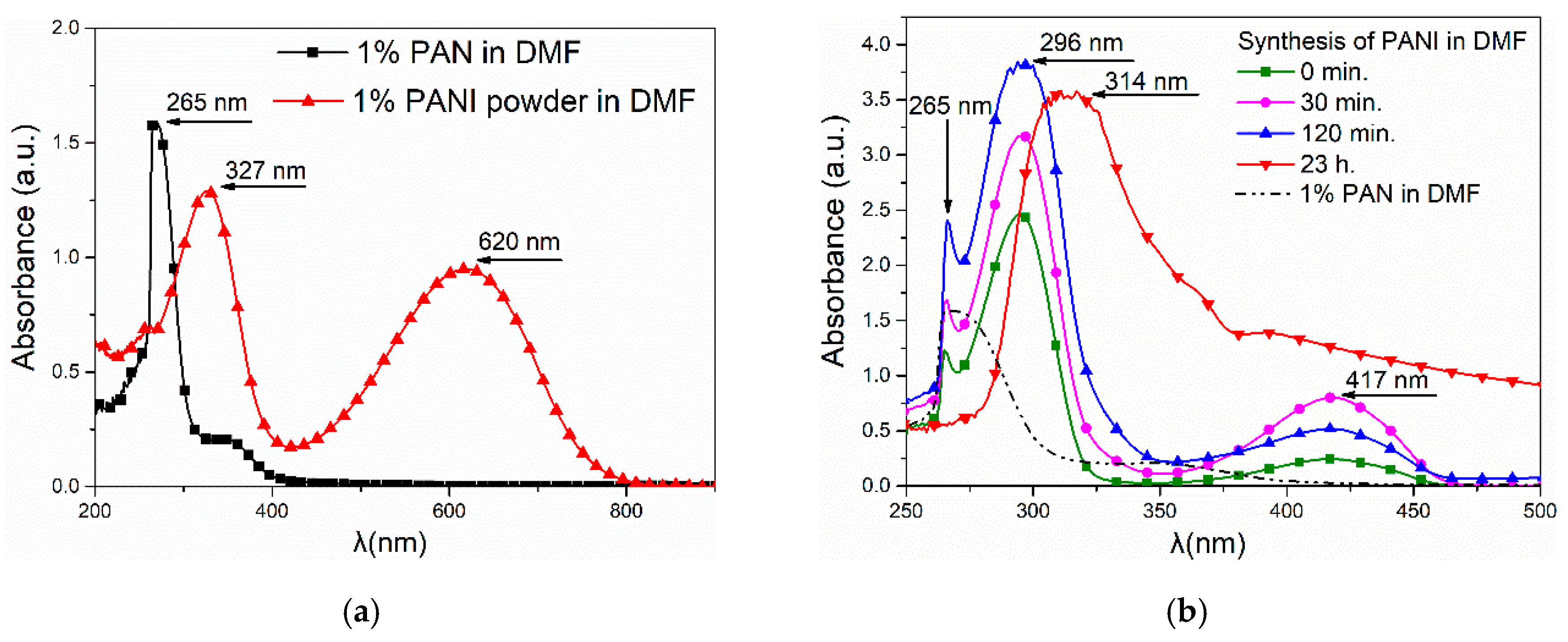
(Per)nigraniline—(C6H4N)n—a completely oxidized form of PANI—also has a blue or violet color. However, taking into account that the synthesis of polyaniline was carried out in a solvent (DMF), which is a strong reducer, it cannot be assumed that a fully oxidized form of PANI was formed. In addition, during the synthesis of PANI in DMF, colorless crystals were formed, which indicated that a part of polyaniline precipitated in the form of leucoemeraldine—a completely reduced form. A significant part of the leucoemeraldine was filtered during the synthesis, and, therefore, the fibers were mostly polyaniline fractions with shorter chains.
3.5. FTIR Spectroscopy
The chemical structure of the polyaniline, polyacrylonitrile, and obtained fibers was determined on the basis of infrared spectroscopy (
Figure 5). A description of the characteristic vibrations is shown in
Table 2 [
15,
25].
On the fibers’ FTIR spectra, the bands characteristic of polyaniline and polyacrylonitrile were visible. In the case of PAN/PANI blended fibers, all absorption bands characteristic of polyacrylonitrile were clearly visible.
In the case of PAN/PANI blended fibers, all absorption bands characteristic of polyacrylonitrile were clearly visible at 3445, 2926, 2243, 1731, 1615, 1453 and 1250 cm−1. The peaks corresponded to N–H stretching and C–H stretching in the polymer structure, stretching in C≡N, stretching in C=O, stretching in C=C, bending C–H in CH2, and stretching in C–N. The gentle absorption bands characteristic of PANI were also visible at 1588, 1160 and 804 cm−1, which were characteristic of C=C stretching in the quinoid rings, C–N bond stretching in quinoid and benzenoid rings, and δ C–H in 1,2,4 trisubstituted benzene rings. FTIR analysis indicated that these fibers were composed of polyacrylonitrile and had a low concentration of polyaniline incorporated in them, as evidenced by the low intensity of the bands characteristic of this polymer.
On the other hand, in the PAN/PANI in situ fiber spectrum, the absorption bands associated with the presence of PANI-specific bonds were much more intense. A fuzzy peak above 3000 cm−1 was also visible, which was associated with the presence of water. This is not present in the PAN/PANI blended spectrum. The bands occurring at 1414, 1194 and 879 cm−1 were characteristic of stretching in C–N between benzene and quinoid rings, and bending in C–H and 1,2,4 trisubstituted benzene rings. The absorption bands characteristic for polyacrylonitrile were also visible at 2940, 2244, 1726, 1629, 1445 and 1260 cm−1, which was characteristic of C–H stretching in polymer structures, stretching in C≡N, stretching in C=O, stretching in C=C, bending C–H in CH2, and stretching in C–N.
In addition, a band corresponding to the vibrations of the deprotonated polyaniline form (1160 cm−1) was present in both fibers containing polyaniline. Moreover, pure polyaniline was in a protonated form.
From the shifts in absorption bands in the PAN/PANI in situ fiber spectra, relative to unmodified polyacrylonitrile and polyaniline, it can be concluded that electrostatic interactions arose between the chains of these polymers between the polyaniline and nitrile groups. The confirmation of the interaction between polyaniline and the nitrile group of polyacrylonitrile was in the reduction of the intensity of the absorption band around 2244 cm
−1 associated with stretching vibrations in C–N, and also the band around 3480 cm
−1 associated with stretching N–H vibrations. In both cases, this reduction was more visible in the PAN/PANI in situ fibers. Such interactions between different conductive polymers and polyacrylonitrile are known in the literature [
15]. These results confirmed observations drawn from the research of the rheological properties of the obtained solutions and UV-Vis spectra.
3.6. SEM Images
The microstructure of the fibers was observed using a scanning electron microscope (
Figure 6). Cross-sections of individual fibers were also made using a focus ion beam (FIB) microscope (
Figure 7). The surface morphology and fiber cross-sections were different for every fiber. The standard PAN fibers had a visible fibrous structure with characteristic furrows on their surface (
Figure 6a). The cross-sections of these fibers had a characteristic bean-like shape (
Figure 6b). The surface of the PAN/PANI blended fibers treated with hydrochloric acid was very smooth, and their cross-section was oval (
Figure 6c,d and
Figure 7b). Deep furrows were clearly visible on the surface of the PAN/PANI in situ fibers. The cross-sections of these fibers showed pores and cracks (
Figure 7c). The cross-section shape was also bean-like, but the surface structure was clearly different from the inside of the fibers (
Figure 6e,f). The structure of the PAN/PANI in situ fibers was discontinuous and contained many defects. It was clearly more heterogeneous than in the PAN/PANI blended fibers (
Figure 6e,f).
The reason for such differences in the microstructure of fibers can be, above all, the difference in the size of the macromolecules that have been in the solution (see
Section 3.1), and also the amount of tension during the spinning process (see
Section 2.4). As previously mentioned, during the synthesis of the PANI in the DMF and PAN solution, transparent crystals were formed (
Figure 1), which were then filtered off. Thus, only low molecular weight molecules were left in the solution, which hindered the fiber formation process. Moreover, according to FTIR results (
Figure 5) (
Table 2), during the synthesis of PANI in the DMF and PAN solution, new interactions appeared between the functional groups of both polymers. This could weaken the interaction between the polyacrylonitrile chains themselves.
3.7. Mechanical Strength
Polyaniline is a highly used material, primarily due to its high electrical conductivity, low price, and its easy method of obtaining. However, the biggest difficulty associated with its use is its low mechanical strength and processing problems. For this reason, the fibers were made from a mixture of polyaniline with fiber-forming polyacrylonitrile, which, in this system, was a polymer responsible for mechanical strength.
The fiber linear mass was determined using the following Equation (2):
where m is the average sample weight and l is the length of the sample.
The linear mass of the PAN/PANI blended fibers was slightly higher than the PAN fibers (
Table 3). However, the linear mass of the PAN/PANI in situ fibers was more than twice as high. This was because the fibers were thick as a result of them both being subjected to stretching in steam.
Based on the breaking curve, the most important parameters of the mechanical strength of the fibers were determined (
Table 3). The Young’s modulus and the specific strength of both polyaniline-containing fibers was lower than that of the pure polyacrylonitrile fibers, with the reduction being more pronounced in the case of the fibers modified in situ (
Table 3).
During the stretching of fiber-forming polymers, their crystalline structure is shaped, which increases the mechanical strength of the fibers. However, as previously mentioned, the PAN/PANI in situ fibers were not subjected to stretching in steam. While stretching, the fibers were easier to delaminate and less likely to break. This effect may occur when adding an incompatible polymer or when adding a polymer that changes the interactions between the fiber-forming polymer chains. Both the PANI/PANI blended and PAN/PANI in situ fibers contained the same polymer, and, therefore, the delamination effect was difficult to explain by the presence of an incompatible polymer.
However, on the basis of the FTIR results (
Figure 5), interactions between these polymers were found, which probably caused a reduction in the interactions between the PAN macromolecules, as well as a significant reduction in the consistency coefficient (
Table 1). In addition, if it was assumed that a small molecule polyaniline remained in the polyacrylonitrile solution during the fiber coagulation process, it would pass into the coagulation bath, causing pores to be visible on SEM images (
Figure 6f,c).
Typically, thermoplastic polymers such as Nylon or Kevlar [
7,
8] are used as PANI strength-enhancing materials. However, the presence of these polymers causes a decrease in the electrical conductivity of the PANI. This inversely proportional relationship is associated with the presence of micropores in the fibers, which on the one hand reduces the mechanical strength, but on the other hand facilitates the penetration of an admixture, such as HCl, into the structure of a fiber. This penetration, in turn, increases the conductivity of the fibers [
26,
27]. For this reason, the generation of electrically conductive fibers based on electronically conductive polymers has many denominations, of which, the greatest seems to be the achieving of both the desired mechanical properties and a satisfactory electrical conductivity.
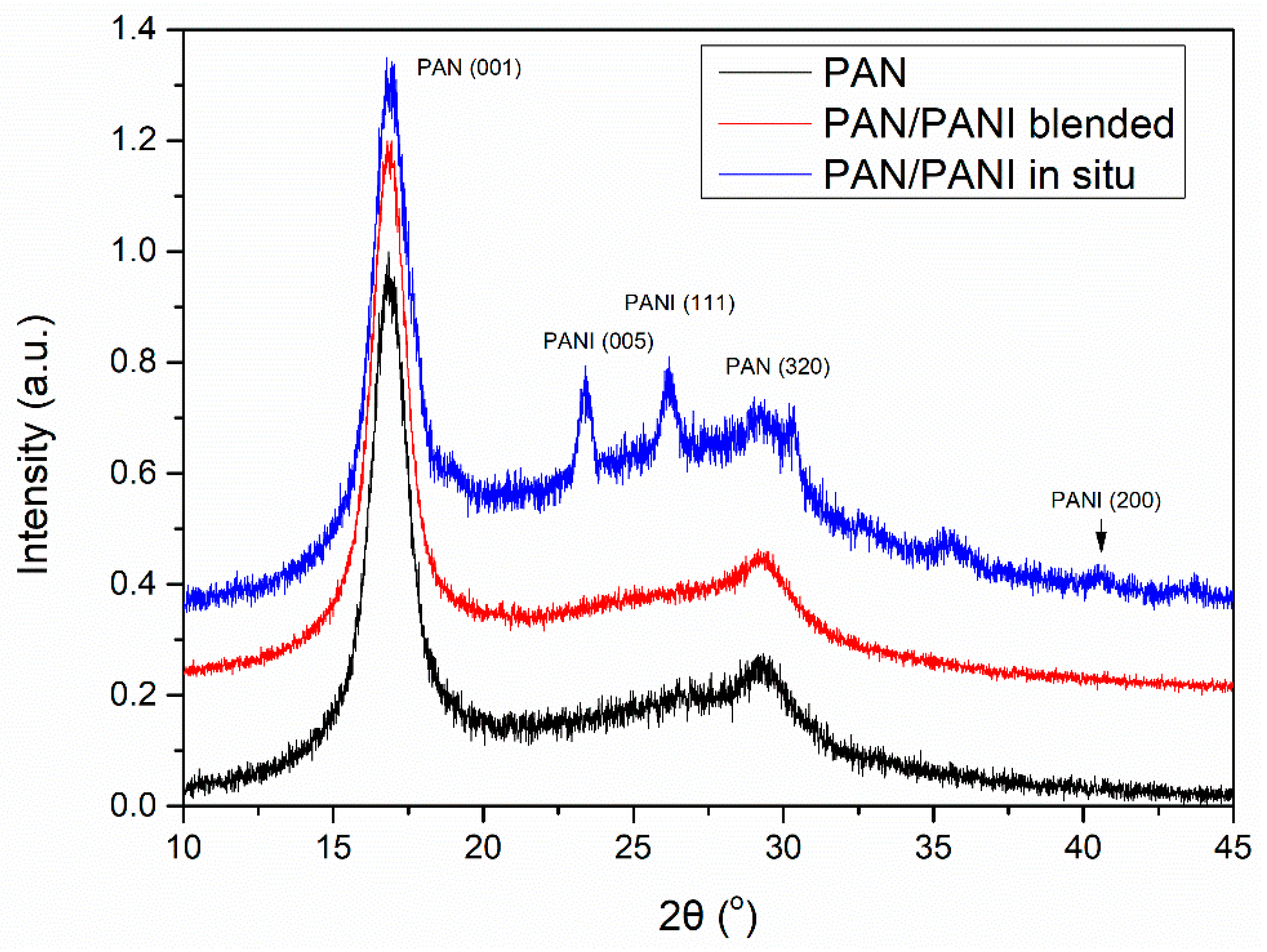
3.8. XRD
The fiber crystal structure was determined based on XRD studies (
Figure 8). On all diffractograms, peaks characteristic of polyacrylonitrile were visible. Particularly clear was the peak at 2θ~18°, which was characteristic of the hexagonal structure of PAN. In the PAN/PANI blended fiber spectrum there were no peaks characteristic of PANI, most probably due to its small amount in the fibers. However, they were clearly visible in the PAN/PANI in situ fibers, and corresponded to the pseudo-orthorhombic phase of PANI [
28].
The degree of fiber crystallinity (X
c) was calculated using the Hinrichen method (3):
where I
c is the integral under the peaks corresponding to the crystalline phase of the polymer, and I
a is the integral under the peaks corresponding to the amorphous phase of the polymer.
Crystal and amorphic peaks were determined based on the optimization carried out in the WAXFIT program. Curve fitting was performed using the Rosenbrock method. The degree of crystallinity for the PAN and PAN/PANI blended fibers was the same at 0.53, while for the PAN/PANI in situ fibers it was significantly lower at 0.32 (
Table 4). The significantly lower crystallinity degree of the PAN/PANI in situ fibers was caused by a lack of stretching, and was correlated with their mechanical strength.
The higher degree of crystallinity, in the case of fibers, was associated primarily with a larger ordering of polymer chains along the fiber axis [
29]. This parameter depended on the type and degree of interactions between macromolecules.
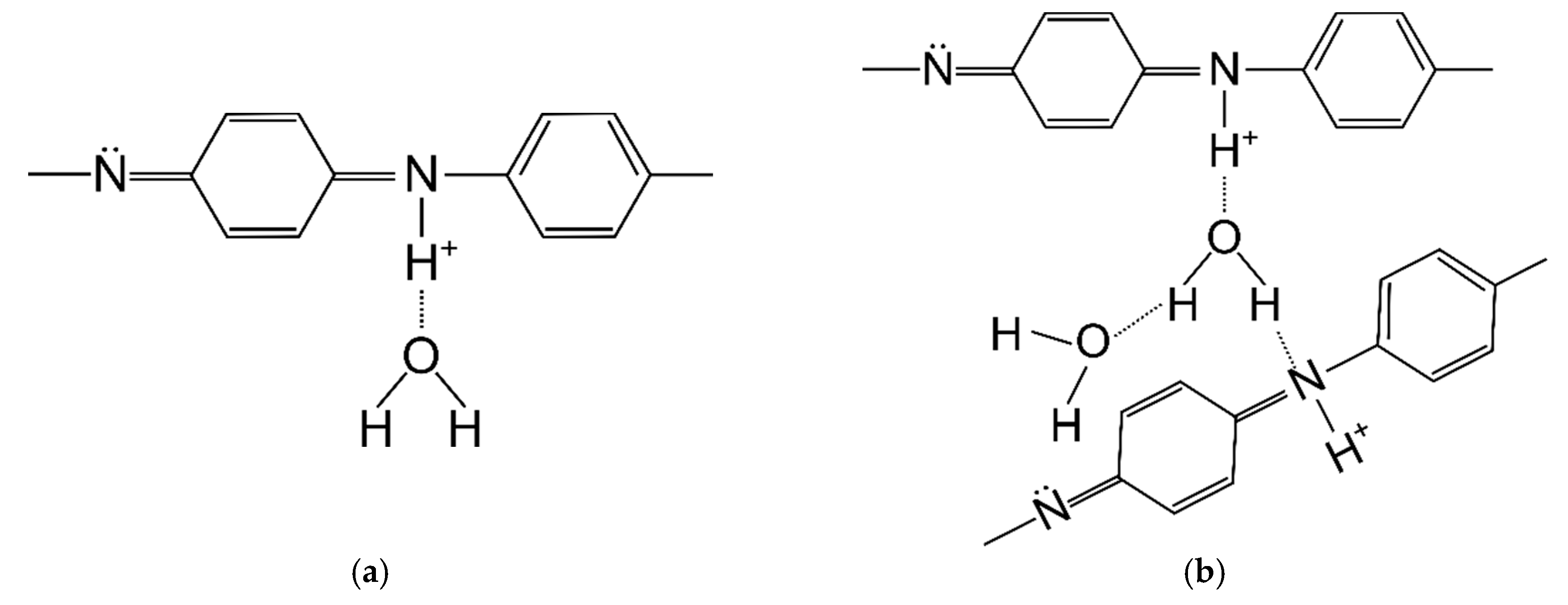
The polyacrylonitrile macromolecules contained positively charged CN groups (
Figure 9a), and caused the chains to twist into a helix, from which the crystallites are made [
30]. In the PANI, due to the presence of amine groups, hydrogen bonds and dipole-dipole interactions were formed between the macromolecules (
Figure 9b). H-bonding interactions between adjacent chains caused the stiffness of the PANI chain [
31], and an easier orientation of the chains in the material containing only polyaniline.
However, in the material containing both of these polymers, the crystallization process was difficult due to the interactions between nitrile (PAN) and amine (PANI) groups (
Figure 10). In addition, numerous cracks and voids appeared in the structure of the fiber (
Figure 6c). Analysis of the literature data [
32] indicated that the nitrile group, in some cases, may have interfered with or acted as a barrier when ordering polymer chains. This can take place in the PAN/PANI in situ fibers, where the strong interaction between the polymer chains blocks their movement, thereby limiting the crystallization process.
The average size of PAN crystallites was determined based on the half-width of the peak 2θ~17°, characteristic of the plane (001) in a hexagonal structured PAN. The size of the PANI crystallites was determined based on the peaks 2θ~23° and 26°, characteristic of the planes (005) and (111). The calculations were made using the Debye–Scherrer Equation (4):
where L
(hlk) is the average size of crystallites, K is the Scherrer constants (0.89), θ
max is the angle for the maximum peak (rad), λ is the length of the radiation beam (Å), and β is the half-width of the peak (rad).
The inter-chain separation length (R) was determined based on the analysis of the most intense crystalline peak from the following Equation (5) [
31]:
The distance between planes (the d-spacing) was determined from the Bragg Equation (6):
where n is the deflection integer.
Lattice strain ε was determined from Formula (7):
Data on the crystal structure of the obtained fibers was collected in
Table 5. The crystal phase of the polyacrylonitrile in all the fibers had a similar structure. Crystallite size, d-spacing, inter-chain separation, and lattice strain all had very similar values. The size of the polyaniline crystallites was larger and amounted to 15.8 (111) and 17.9 (005) nm. Differences in the structure of both chemical and crystalline polyacrylonitrile and polyaniline present in fibers were reflected in their macroscopic structure and mechanical properties.
Inter-chain separation length is a parameter affecting the electrical conductivity of fibers. This value represents the distance between the electron jump between the chains. The smaller it is, the higher the probability of charge hopping. The distance of the order of ~4 Å corresponded to the literature concerning polyaniline, and allowed hopping to be charged between the chains [
24,
33].
3.9. Thermal Properties
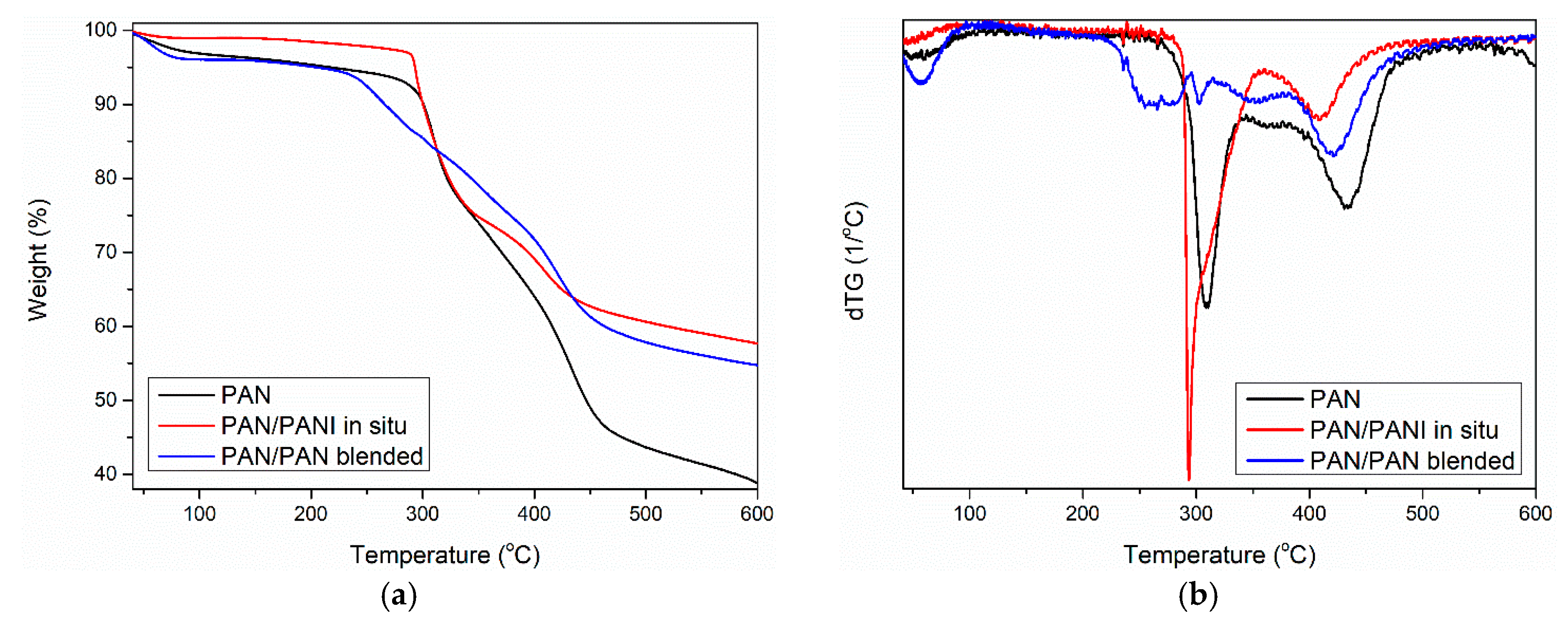
Thermogravimetric analysis of the pure PAN fiber and PANI-containing fibers showed some similarity between the samples (
Figure 11). Three mass losses were observed for all materials. The first between 25 and 100 °C (
Table 6—T
1), the second between 200 and 300 °C (
Table 6—T
2), and the third at around 420 °C (
Table 6—T
3).
The first mass loss was caused by the loss of water bound in the material, which was due to polyaniline adsorbing large amounts of water. Water can be associated with polyaniline macromolecules in two ways: strong and weak. Poorly bound water molecules are connected to the PANI chain with one hydrogen bond (
Figure 12a). In contrast, strongly bound water is connected by two hydrogen bonds (
Figure 12b), most often between two adjacent PANI chains [
25].
Poorly adsorbed water can be removed while drying with nitrogen at room temperature, while the strongly bound one is mostly removed at a temperature in the range of 60–140 °C. Removal of this water is reversible and it can be re-absorbed. However, further removal of water from the polymer at 150–220 °C is an irreversible process and leads to polymer degradation [
34,
35].
The first derivatives of the TG curve showed that the most moisture was adsorbed in the PAN/PANI in situ fiber. Less water was present in the pure PAN fiber, and the smallest amount was found in the PAN/PANI blended fiber. As pure polyaniline can bind up to 40% by weight of water [
36], it is possible to determine its presence in fibers based on the analysis of the water-related peak. An intense peak, much larger than in the reference fiber, was associated with the presence of water in the PAN/PANI in situ fiber. This demonstrated the presence of polyaniline in fibers, which was also confirmed by the FTIR results (
Table 5) and XRD (
Table 6). A very small amount of adsorbed water in the PAN/PANI blended fiber resulted from the fact that the polyaniline, after the synthesis process, was dried before its dissolution in dimethylformamide.
Further fiber degradation was associated primarily with the phenomenon of polyacrylonitrile cyclization occurring under nitrogen [
37]. Analysis of the first derivative of TG curve (i.e., DTG curve) showed that in the case of the pure PAN and PAN/PANI blended samples, the degradation can be described as one-step and starts at around 280 °C. The PAN/PANI blended fiber was characterized by a very rapid decomposition in this temperature range, which persisted up to 350 °C.
The PAN/PANI in situ fiber was decomposed in two stages. It had a wide weight loss of about 10% in the temperature range of 200–290 °C. The initial weight loss in this range was associated with the removal of the rest of the strongly bound water. On the other hand, according to the obtained results and literature data, the weight loss was due to the presence of low-molecular polyaniline fractions in the material, which underwent gradual decomposition [
38]. This process is characteristic for PANI degradation, which is why it indicated its presence in the fiber. Another loss beginning at 290 °C and ending at 380 °C was associated with PAN cyclization. Comparing the results obtained in all the fibers showed that the in situ synthesis of polyaniline was a difficult cyclization. This was closely correlated with the FTIR conclusions (
Table 5), which indicated electrostatic interactions between the macromolecules of polyacrylonitrile and polyaniline in the PAN/PANI in situ fibers.
The third loss of fiber mass corresponded to the carbonation process of the PAN, which consisted of dehydrogenation and denitrogenation processes [
39]. As a result of these processes, a cyclic carbon structure was created. However, when combining the maximum transformation temperature, it can be seen that the lowest value of this parameter was for the PAN/PANI blended fiber. This clearly indicated that the addition of polyaniline introduced by the blended method facilitated the carbonization process.
The total weight loss (Δm
tot) after measurement was about 15% higher for the pure PAN fiber than for the modified fibers (
Table 6). Considering the high thermal resistance of PANI, this proved the effectiveness of both methods of introducing a polymer additive to the fiber.
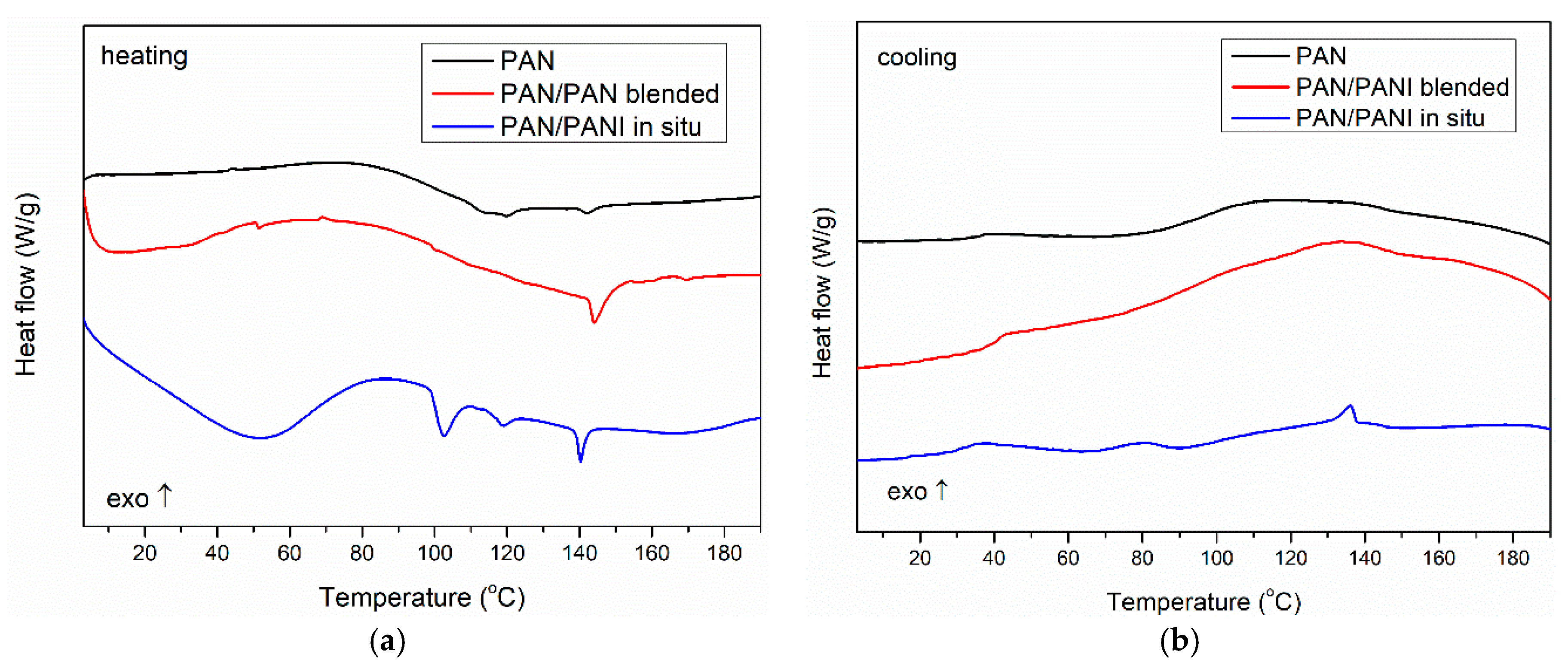
To verify the phase composition of the fibers, DSC measurements were conducted (
Figure 13). The obtained results indicated that the pure PAN fiber showed a glass transition at a temperature of about 100 °C both for heating and cooling runs, which was characteristic for polyacrylonitrile [
40,
41].
In the case of the PAN/PANI in situ fibers’ heating curve, a wide endothermic peak was seen with a maximum at 50 °C, associated with the removal of poorly bound water. An endothermic peak was seen at 102 °C, associated with the desorption of strongly bound water to the PANI [
38]. Subsequently, the glass transition in the PAN was observed at about 110 °C, but its exact value was difficult to accurately determine because of the overlap of both processes. At 140 °C, one more endothermic peak was visible, which may be the result of the presence of low molecular weight fractions in the material remaining after the PANI polymerization process (
Figure 13a). In the cooling curve, two exothermic peaks were observed, which could be connected with crystallization of previous melted low molecular fractions during heating. Also, a glass transition of PAN at about 100 °C was present (
Figure 13b). The obtained results indicated a large heterogeneity of the system, which was also confirmed by previous SEM, mechanical strength, and TG measurements.
In the case of the PAN/PANI blended fibers, only the PAN glass transition (100 °C) was visible in the heating curve, with an additional transition at around 150 °C. Because this temperature was not identical to the glass transition of polyaniline, and was not observed in the front samples (
Figure 13a), the authors believe that it was the result of the presence of small amounts of low molecular weight PANI fractions in the material structure, which was well mixed with pure polyacrylonitrile; therefore, the obtained fibers had good mechanical parameters (
Table 3) and a smooth structure (
Figure 6b). This fraction was homogenized with the rest of the material because during the cooling run, no crystallization peaks were present. Only T
g of PAN was observed (
Figure 13b). This phenomenon also explains the problem in observing peaks from PANI to XRD, and the presence of only partial peaks in the FTIR.
3.10. Electrical Measurements
In order to determine the effect of polyaniline on the electrical properties of the fibers, resistance tests were carried out. The resistance of fibers on the 0.3 cm section was measured using the constant current method, and the results were given as mass-specific resistance Rs (Ωg/cm
2) (8). This is a parameter used to describe fiber resistance [
42]. This measure allows the electrical properties of fibers to be shown in relation to their construction, and not geometric parameters (such as diameter), which are difficult to determine in yarns composed of many fibrils.
where R is the resistance of the sample, N is the number of fibers in the yarn, m
l is the linear mass (tex) = (g/km), and l is the length of the sample (cm).
Standard polyacrylonitrile (reference) fibers are dielectrics with a resistance of 10
11 Ω (
Table 7). Both types of fibers doped with polyaniline were tested for resistivity. In the case of the PAN/PANI blended fibers, no change in the fiber resistance was observed, whereas the PAN/PANI in situ fibers were characterized by mass-specific resistances of 5.47 kΩg/cm
2. These fibers, as shown by the FTIR, XRD, and TG studies, consisted of two phases of different polymers, which were additionally visible in the SEM images. In these fibers, the polyacrylonitrile matrix was responsible for the mechanical properties and thermal stability of the fibers. Polyaniline was responsible for their electrical conductivity.
In order to compare the obtained results with literature data, it can be assumed that the conductance of the obtained PAN/PANI in situ fibers is of the order 10
−4 Scm
−1. This conductance value is lower than that of pure PANI fibers. According to literature data, polyaniline fibers have conductance on the order of 10–100 Scm
−1 [
43,
44], but mechanical properties of these fibers are weak. Better mechanical properties, but lower conductance, usually is obtained in composite fibers. Toptaş et al. obtained composite PAN/PANI fibers in a different method that was presented in this paper, but mechanical and electrical properties are comparable [
14].

 ,
, 














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}