1. Introduction
According to the World Health Organization, cancer is one of the leading causes of morbidity and mortality worldwide, and therefore, cancer therapy is an important objective for today’s medicinal research [
1]. Different therapeutic approaches have been taken so far, from surgical removal to radio- and chemotherapy. The use of anti-proliferative drugs in cancer therapy means that every part of the human body can be reached and, thus, not only the primary cancer can be fought, but also metastasized cells [
2]. For most compounds used in chemotherapy though, the mode of action is not exclusive to cancer cells but affects cell proliferation throughout the entire organism. This leads to adverse side effects and limits the doses that can be applied to patients [
3,
4]. Current efforts in chemotherapeutic drug development aim at increasing the selectivity for cancer cells while reducing the systemic exposure.
Many strategies have been applied to direct small molecules to the target cells. These include drug delivery systems like nanoparticles. Nanoparticles are 3-dimensional supramolecular entities, which are assumed to passively accumulate in the tumor tissue due to the enhanced permeability and retention effect [
5]. In detail, macromolecules including nanoparticles are passively enriched in neovascularized tumor tissue as the newly formed blood vessels exhibit not only enhanced perfusion, but also increased permeability. In addition, the lymphatic system in tumors is assumed to be less effective, which is leading to decreased lymphatic drainage from the tumor site. Although the enhanced permeability and retention effect (EPR) is a very interesting phenomenon, it is also assumed to be heterogeneous in humans [
6].
Hitherto, a variety of nanoparticles using many different materials has been developed, investigated and some have been approved for the use in humans [
7,
8]. Since 1995, the United States Food and Drug Administration (FDA) approved over 50 nanopharmaceuticals; the majority being liposomal, nanocrystal, or polymeric formulations. The latter are comprised of amphiphilic polymers consisting of subunits also known as block copolymers. In detail, linear block copolymers can be divided into the categories of diblock copolymers or of triblock copolymers. Diblock copolymers possess a polar and a non-polar homopolymeric subunit (A-B). Triblock copolymers consist of three homopolymeric subunits (A-B-A or A-B-C), where two subunits can be similar [
9]. Nanoparticles formed by block copolymers are known as polymersomes or polymeric nanoparticles depending on the membrane.
An entity of polymersomes, investigated for their application as a drug delivery system, are PDMS-PMOXA polymersomes where the diblock copolymer used for the formulation is poly(dimethyl siloxane)-poly-
b-(methyloxazoline). So far, these polymersomes were investigated for their toxicological and biocompatibility profile, showing no significant toxicity in in vitro models [
10,
11] and the subunits have been reported to be biocompatible [
12]. A previous study has described that the hydrophilic PMOXA-block is cleared by the kidney from the circulation [
13]. Although the PDMS-block is hydrophobic, as long as the molecular weight does not surpass 5 kDa, it undergoes renal elimination [
14]. Importantly, polymersomes have the ability to encapsulate bioactive molecules like chemotherapeutics [
7], and are therefore, suitable for drug delivery.
Most of the current approved nanoformulations are aiming at improving the pharmacokinetics and pharmacodynamics by packaging/conjugating drugs in/to nanoparticles [
15]. However, incorporating an active or triggered release strategy could lead to a dose reduction and therefore, decrease side effects [
16,
17]. A mechanism that could be utilized for such an approach is triggered release of the payload using tumor-specific elicitors [
18]. Here, the tumor-associated expression of enzymes in or close to the targeted tissue offers potential candidates. Especially, enzymes exerting proteolytic activity could be employed to cleave drug–peptide conjugates or to trigger changes in the drug carrier’s outer layer [
16]. An enzyme qualifying as an elicitor is cathepsin B.
In physiological conditions, cathepsin B is located in the lysosomes [
19], where it is involved in the degradation and, therefore, regulation of proteins. Besides proteolysis within the lysosome, cathepsin B is involved in cell death mediation [
20], and it contributes to the degradation of the extracellular matrix [
21]. Indeed, in cancer, cathepsin B is secreted by tumor cells where it contributes to the degradation and remodeling of the extracellular matrix and whereby facilitating tumor cell invasion into the surrounding tissue [
22]. In tumor tissue, cathepsin B seems to be expressed predominantly in areas bordering the extracellular matrix [
23]. By analyzing patient samples, increased protein content or activity of cathepsin B have been detected in ovarian [
19] and colorectal [
24] cancer. A similar enhancement has been observed in a B16 mouse melanoma in vivo model [
25]. Jedeszko et al. summarized the core-statements of many publications in a review showing increased expression and activity of cathepsin B in breast, colon, lung, prostate cancer, glioblastoma, and melanoma [
26].
In this study, we aimed at using cathepsin B as an elicitor for enzyme-triggered drug release from PDMS-PMOXA-based nanoparticles. The PDMS-PMOXA nanoparticles were used as a platform for surface-modifications. The separate surface-modification steps were surveyed by Fourier-transform infrared (FT-IR) spectroscopy. Because cathepsin B (CTBS) plays a pivotal role in the release mechanism, its mRNA expression was determined in patient derived tumor samples. Accordingly, a suitable in vitro cell model system was identified and subsequently used for in vitro studies on the pharmacological activity of the CTSB-degradable nanoparticles (
Supplemental Tables S1 and S2).
2. Materials and Methods
2.1. Materials
Poly(dimethylsiloxane)-b-poly(methyloxazoline) PDMS-PMOXA was purchased from Polymer Source Inc., Ottawa, Canada. Organic solvents were obtained from J.T. Baker, (Deventer, Netherlands), Carl Roth GmbH + Co. KG (Arlesheim, Switzerland), or from Sigma Aldrich (Buchs, Switzerland). p-Maleidoimino phenylisocyanate was purchased from Invitrogen (distributed by Thermo Fisher, Reinach, Switzerland). Double distilled water (ddH2O) with a resistivity of 18.2 mΩ was generated with a Barnstead Nanopure DiamondTM System (Thermo Fischer).
2.2. Quantitative PCR for Gene Expression Analysis in Cancer Tissue Samples and Representative Cell Lines
To determine CTSB mRNA expression in cancer tissue a cDNA array (CSRT103, Origene, Rockville, MD, USA) was commercially obtained. Furthermore, mRNA of cancer cell lines was isolated using peqGOLD RNA pure (Axon Lab, Baden, Switzerland) and reverse transcribed using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher). The amount of CTSB was assessed by quantitative real-time PCR (qPCR) using the pre-developed TaqManTM assays (Thermo Fischer) Hs00947439_m1, the TaqMan® gene expression mastermix, and the ViiATM 7 Real-Time PCR System. The reaction was carried out in a volume of 15 µL composed of 0.75 µL of the Hs00947439_m1-FAM TaqMan assay, 6.75 µL H2O, and 7.5 µL TaqMan® Gene Expression Mastermix (Applied Biosystems, LubioSciences, Lucerne, Switzerland). For quantification of copy numbers, a standard curve using cloned PCR amplicon was recorded.
2.3. Immunohistochemical Staining of CTSB in Human Ovary Cancer Tissue
For the detection of CTSB by immunohistochemical staining a commercially obtained array of paraffin embedded tissue sections was used. This array included malignant transformed and non-malignant transformed samples of various tissues (MTU951, BioCat GmbH, Heidelberg, Germany). To deparaffinize the tissue sections, two changes of xylol and rehydration in a decreasing ethanol series ranging from 96% to 0% was executed, followed by a heat induced epitope retrieval in 0.1 M citrate buffer (pH = 6.0, 20 min). The endogenous peroxidase was quenched in a 3%-H2O2-methanol-bath for 20 min. To reduce unspecific binding of the antibody, the slides were exposed for 1 h to blocking solution (5% FCS and 1% BSA in PBS). CTSB was detected with the primary anti-CTSB-antibody (sc-13985, Santa Cruz Biotechnology, Inc., Texas, TX, USA) at a dilution of 1:1000 in blocking solution. After incubation with primary antibody overnight at 4 °C, the tissue slides were washed repeatedly in PBS. The tissue slides were then exposed to the secondary HRP-coupled goat-anti-rabbit antibody (Bio-Rad Laboratories Laboratories, Cressier, Switzerland; 1:100) for two hours at room temperature (RT). After several washing steps in PBS, 1 mg/mL of diaminobenzidine (DAB) diluted in 0.05 M phosphate buffer containing 0.02% H2O2 was added for visualization of epitope-bound antibody. Nuclei were stained with hematoxylin solution (Carl Roth GmbH + Co. KG) and slides were mounted with Roti®-Histokitt II (Carl Roth GmbH + Co. KG). Finally, the stained tissue slides were imaged with a Leica DMi8 microscope equipped with a DFC 365 FX camera (Leica, Heerbrugg, Switzerland) and the LAS software Version 4.6 (Leica).
2.4. Western Blot Analysis
Protein samples were collected from cultured cells seeded at a density of 1 × 106 cells/ 10 cm dish. After reaching 80% confluence, the culture medium was replaced by the respective medium containing no FCS to avoid contamination with serum proteins. After 24 h, the culture supernatant was collected to enrich the secreted protein. In detail, the supernatant was supplemented with ice-cold 10% trichloroacetic acid, kept on ice for 15 min, and was then centrifuged for 20 min at 17,000 × g and 4 °C. The precipitate was washed twice with ice-cold acetone (5 mL), and was then air-dried for 30 min at room temperature. The enriched secreted proteins were finally solubilized in 6 M urea. To determine the protein content, the Bradford assay (Thermo Fischer) was used. Cell lysate was collected harvesting the cells in 5 mM Tris-HCL supplemented with protease inhibitor cocktail (Sigma Aldrich, Buchs, Switzerland), followed by three cycles of freezing thawing in liquid nitrogen. For Western blot analysis protein samples were supplemented with Laemmli and then separated by a 10% SDS-PAGE. Afterwards, the proteins were electrotransferred to a nitrocellulose membrane (Bio-Rad Laboratories). After blocking with 5% FCS/1% albumin in TBS-T, the membranes were exposed overnight and at 4 °C to the primary antibody sc-365558 (Santa Cruz Biotechnology, diluted 1:500) for cathepsin B or sc-47778 (Santa Cruz Biotechnology, diluted 1:1000) for actin. Thereafter, the blot was exposed to the respective HRP-conjugated secondary antibodies (Bio-Rad Laboratories) for 1 h at RT. Pierce™ ECL Western Blotting Substrate (Thermo Fisher) and the ChemiDoc™ MP Imaging System equipped with the image lab software (version 4.1) both from Bio-Rad Laboratories were used for image acquisition.
2.5. Detection of CTSB Activity by Enzymatic Assay
CTSB’s activity in OVCAR-3 and OVCAR-5 cells was detected by the liberation of the fluorescent 7-amino-4-methylcoumarin from Z-Arg-Arg 7-amido-4-methylcoumarin (Sigma-Aldrich). The assay was performed according to the instruction manual and as previously described by Barrett et al. [
27]. In short, 60 μL of 8 mM L-cysteine-HCl in 352 mM potassium phosphate buffer (including 48 mM sodium phosphate, and 4.0 mM ethylenediaminetetraacetic acid), 70 μL 0.1% Brij 35 solution in purified water, 10 μL of cell lysate or supernatant and 60 μL 0.02 mM of Nα-CBZ-Arg-Arg-7-amido-4-methylcoumarin in 0.1% Brij 35 solution. The release of 7-amino-4-methylcoumarin was measured with the microplate reader Tecan Infinite M200 Pro (Tecan, Männedorf, Switzerland; excitation = 348 nm, emission = 440 nm).
2.6. Cell Culture
The cell lines OVAR-5 (RRID:CVCL_1628), and OVCAR-3 (ATCC HTB-161) were commercially obtained from the American Tissue Culture collection, (Manassas, MA, USA). OVCAR-3 were cultured in RPMI-1640 (BioConcept) supplemented with 20% FCS, 1% non-essential amino acids (MEM-NEAA, BioConcept), and 1% GlutaMAX. OVCAR-5 were cultured in DMEM supplemented with (v/v) 10% FCS, 1% MEM-NEAA, and 1% GlutaMAX. In viability assays, 1% Penicillin/Streptomycin (BioConept) was added to the media. All cell lines were kept at 37 °C in a humidified atmosphere with 5% CO2.
2.7. Immunofluorescence of Cathepsin B in Ovarian Cancer Cells
OVCAR-5 and OVCAR-3 cells were seeded at a density of 2 × 105 cells/well on cover slips placed in 12 well plates. One day after seeding, cells were fixed with ice-cold methanol-acetone (1:1, v/v) for 5 min at −20 °C. After several washing steps with PBS, unspecific antibody binding was prevented by incubation with 5% normal goat-serum/ 0.3% Triton X-100 in PBS for 1 h at room temperature. Cells were incubated with the primary antibody anti-Cathepsin B (sc-366558, Santa Cruz Biotechnology Inc., Dallas, Texas, TX, USA) diluted in antibody dilution buffer (1% BSA/0.3% Triton X-100 in PBS, 1:10) overnight at 4 °C. After washing with PBS, cells were incubated with the secondary Alexa Fluor™ goat-anti-mouse 568 antibody (Thermo Fisher) diluted in antibody dilution buffer (1:200). After incubation for 1 h at room temperature, cells were washed with PBS and mounted using Roti®-Mount FluorCare containing DAPI for nuclei stain (Carl Roth GmbH + Co. KG). For antibody control, the primary antibody was omitted. Images were taken with the Leica DMi8 Microscope with the MC 170HD camera and LASV4.8 software (Leica Microsystems, Heerbrugg, Switzerland).
2.8. Assessment of Hydrodynamic Radius and Surface Charge of the Nanoparticles
To measure the hydrodynamic radius of the nanoparticles, the dynamic light scattering (DLS) technique was used (Malvern Zetasizer NanoSeries, Malvern Instruments GmbH, Herrenberg, Germany). Briefly, the samples were degassed utilizing a Thermo Vac sample degassing and thermostat system (MicroCalTM, Malvern Instruments GmbH). The samples were measured using a backscattering angle of 173° was used. Data were analyzed using the Zetasizer software 7.11. (Malvern).
2.9. Critical Aggregation Concentration of PDMS-PMOXA
Critical aggregation concentration (CAC) was determined by utilizing the different fluorescence characteristics of pyrene (Sigma-Aldrich) in a hydrophobic or hydrophilic environment [
28,
29]. Briefly, 500 μL 2.4 μM pyrene in aceton and corresponding amount of PDMS-PMOXA (2500, 250, 25, 2.5, 0.25, 0.025 μg) in acetone were added to a glass vial. The solvent was evaporated for 1.5 h at 40°C and under constant flow of nitrogen. After completely drying the pyrene and PDMS-PMOXA, 1 mL of ultrapure water was added, and particles were formed by ultrasonification. The fluorescence of pyrene was measured with the microplate reader Tecan Infinite M200 Pro (Tecan; excitation 332 nm, emission I
1 = 373 nm and I
3 = 384 nm). To determine the CAC, the pyrene’s intensity ratio of I
1/I
3 was plotted against the logarithm of the PDMS-PMOXA concentration.
2.10. Synthesis of Carboxyl Terminated Poly(Dimethylsiloxane)-b-Poly(Methyloxazoline)
Carboxyl terminated poly(dimethylsiloxane)-b-poly(methyloxazoline) (PDMS-PMOXA) was synthesized as described before [
30]. Briefly, 15.6 µmol (101.40 mg) of PDMS-PMOXA (PDMS
67-
b-PMOXA
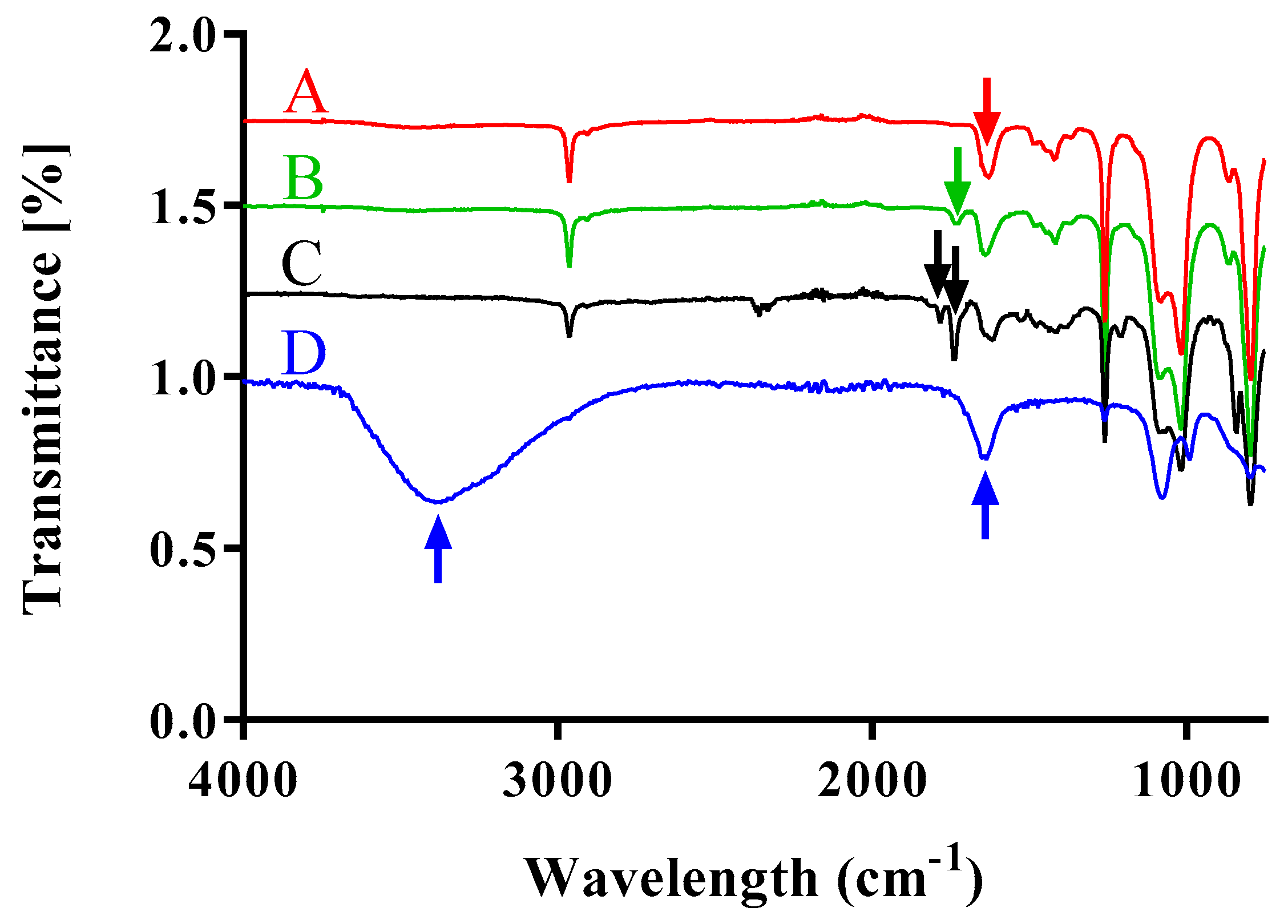
15, Mn = 5000-b-1300, polydispersity index PDI = 1.25) was solved in 5 mL of dichloromethane (DCM). 87.4 µmol (8.75 mg) of succinic anhydride, 15.6 µmol (1.91 mg) of dimethyl aminopyridine (DMAP, Sigma Aldrich), and 79.5 µmol (11 µL) of triethylamine (TEA, Sigma Aldrich) were added to the reaction. The reaction was carried out overnight slowly reaching room temperature. An ultrafiltration using a dialysis membrane (MWCO = 1000, Spectrapor, Spectrum labs, Breda, Netherlands) for 24 h was carried out to purify the reaction. Fourier transformed infrared (FT-IR) spectroscopy was performed to confirm the final compound structure.
2.11. Synthesis of N-Hydroxisuccinimide Activated Poly(Dimethylsiloxane)-b-Poly(Methyloxazoline)
After dissolving the carbonyl terminated poly(dimethylsiloxane)-b-poly(methyloxazoline) in 5 mL of dichloromethane in a round bottom flask, the reaction mixture was cooled to 4 °C. Then, 65.4 µmol (10.2 mg) of N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDAC, Sigma Aldrich) and 100 µmol (11.5 mg) of N-hydroxylsuccinimide (NHS, Sigma Aldrich) were added to the mixture. The final compound was recovered after performing dialysis for 24 h in DCM. The N-hydroxisuccinimide activated poly(dimethylsiloxane)-b-poly(methyloxazoline) was obtained in quantitative yield and FT-IR analysis was performed to confirm the final structure.
2.12. Surface Modification of Polymer Vesicles with the NH2-Ahx-GSGFLGSC Peptide and Paclitaxel Loading
Nanoparticles holding a paclitaxel payload were formulated as described before [
30]. The nanoparticles were diluted five times (200 µL nanoparticle suspension in 800 µL PBS) leading to a 1 mg/mL of nanoparticle suspension. To 1 mL of 1 mg/mL of nanoparticle suspension, 10 µL of 4.7 mM p-maleimidophenyl isocyanate (PMPI) in dimethyl sulfoxide (DMSO) were added and stirred overnight at room temperature to allow modification of the nanoparticle surface. The peptide Fmoc-Ahx-GSGFLGSC (GSG; Ahx = aminocaproic acid; Biomatik, Cambridge, Ontario, Canada) was deprotected as described before [
30]. In brief, 76 µL of piperidine were added to 10 mg of Fmoc-protected GSG-peptide in 300 µL dimethylformamide and stirred for 1 h at room temperature. A cold ether precipitation was conducted to recover the deprotected GSG-peptide. Ten µL of a 10 mg/mL GSG-peptide solution in PBS and 10 µL EDAC (10 mg/mL in MilliQ water) were added to the 1mg/mL solution of nanoparticles to react overnight at room temperature. After coupling the peptide to the nanoparticle’s surface, an additional extrusion similar to the previously described was performed. The nanoparticles were then purified by ultrafiltration (MWCO 12-14 kDa, Spectrapor) overnight at room temperature against PBS.
2.13. Determination of the Encapsulation Efficiency.
To determine the encapsulation efficiency, high-performance liquid chromatography (HPLC) analysis was used. The GSG-modified polymer membrane was disrupted by the use of Triton-X (Merck, Zug, Switzerland) in water and acetonitrile (VWR International, Dietikon, Switzerland). In detail, 25 µL of 1 mg/mL nanoparticle (theoretically containing 0.2 mg/mL paclitaxel), 25 µL of 0.4% triton-X in water and 50 µL acetonitrile were combined, vortexed and centrifuged for 3 min at 4000 ×
g. The amount of paclitaxel encapsulated in the nanoparticles was detected with an Agilent 1100-series equipped with diode array and evaporating light scattering detector (Agilent Technologies, Basel, Switzerland). The mobile phase consisted of ddH
2O (buffer A) and acetonitrile (buffer B, VWR International, Dietikon, Switzerland). Separation was achieved with a Poroshell C18 column (3.0 × 100 mm 2.7-micron, Agilent Technologies). Starting at 50% A, 50% B changing from minute 1 to minute 11 to 0% A, 100% B, with a flow rate of 0.45 mL/min. The paclitaxel peak had a retention time of 4.8/4.9 min. To calculate the concentration of encapsulated paclitaxel a paclitaxel standard curve (Cathepsin B
Table S3A,B; AUC versus concentration) was recorded.
2.14. Fluorescent Labeling of the Polymeric Nanoparticles
Fluorescent labeling of the polymeric nanoparticles was achieved by incorporating 3,3′-dioctadecyloxacarbocianine perchlorate (DiO, Sigma-Aldrich). In brief, 5 μL of a 25 mg/mL DiO stock solution in ethanol was added to the DCM during preparation of the thin-film layer. The dried thin-film layer was then rehydrated with PBS as described above.
2.15. Release and Uptake of Fluorescently Labeled Nanoparticles
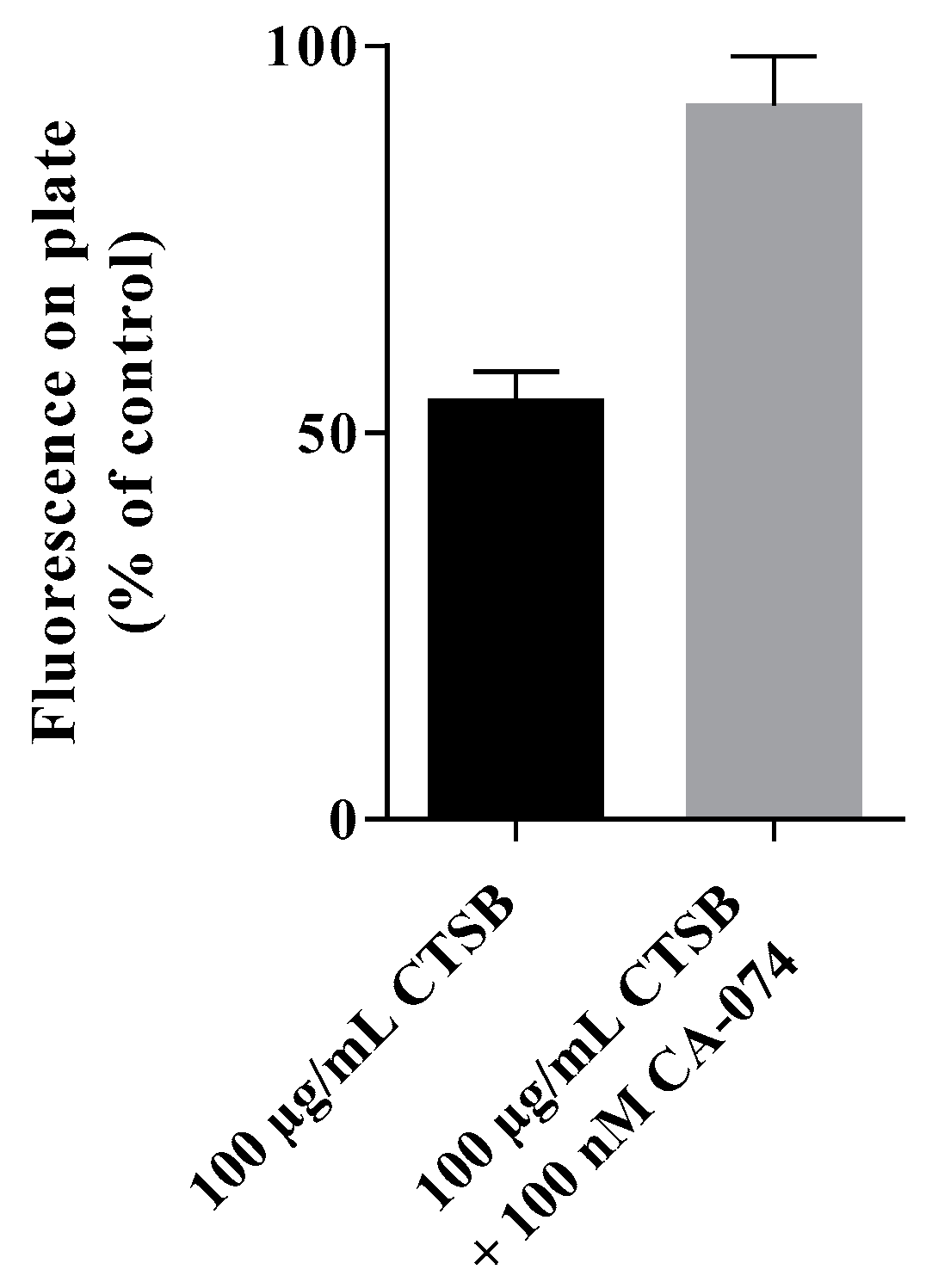
The fluorescently labeled nanoparticles were bound to a Pierce™ maleimide activated black 96-well-plate (Thermo Fisher) according to the manufacturer’s manual. In brief, after several washes with washing buffer, 100 µL of a fluorescently-labeled polymeric nanoparticle-solution (200 µg/mL in binding buffer) were added to each well and incubated over night at 4 °C to achieve nanoparticle binding to the plate’s surface. The surface binding was followed by additional washes with washing buffer. Not reacted maleimide-groups were deactivated by incubation with 100 µL 10 mg/mL L-cysteine-solution for 1 h at RT. After several washes, 100 µL of 100 µg/mL cathepsin B, 100 µg/mL cathepsin B supplemented with 100 µM CA-074, or PBS solvent control were added, respectively. After incubation for 24 h at 37 °C, the difference in fluorescent signal on the 96-well without supernatant was measures using the microplate reader Tecan Infinite M200 Pro (excitation = 490 nm, emission = 530 nm). The reduction in fluorescent signal measured on the plate’s surface was interpreted as evidence of release caused by cathepsin B.
2.16. Cell Viability Assay
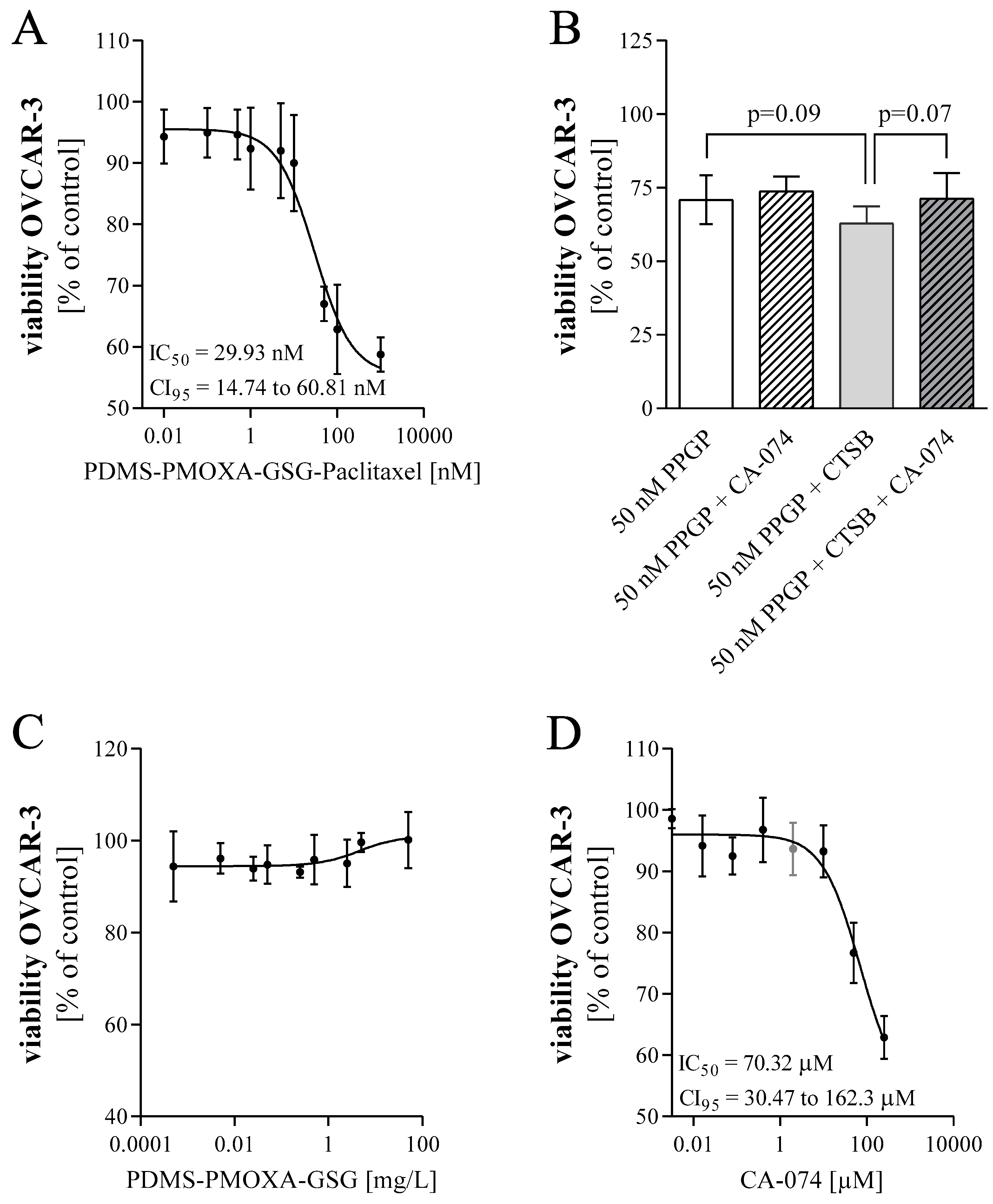
OVCAR-3 cells were seeded in 96-well plates at a density of 15,000 cells/well to assess cell viability. To determine the impact of PDMS-PMOXA-GSG-Paclitaxel particles on cell viability, cells were treated one day after seeding with increasing concentrations of paclitaxel (0.01 nM to 1 µM). The content of paclitaxel in the nanoparticles was quantified by HPLC. In order to analyze whether the release of paclitaxel is mediated by cellular cathepsin B, cells were pre-treated with 0.1 µM of the cathepsin B inhibitor CA-074 (Sigma-Aldrich) or DMSO control three hours after seeding. After 24 h, cells were exposed to PDMS-PMOXA-GSG-Paclitaxel with or without 2 μM CA-074. As positive control, cells were incubated with 25 µg/mL cathepsin B from human placenta (Sigma-Aldrich). After 48 h, cell viability was measured using the Fluorometric Cell Viability Kit I (Resazurin) from PromoKine (Vitaris AG, Baar, Switzerland). The microplate reader Tecan Infinite M200 Pro (Tecan) was used for quantification of cell viability (fluorescence, excitation = 530 nm, emission = 590 nm). Data are presented as mean ± SD as percent of control. IC50 values were estimated using a three-parameter logistic function assuming a standard slope.
2.17. Statistical Analysis
Statistical analysis was performed with the GraphPad prims software (version 6, GraphPad Software Inc. La Jolla, CA, USA). For statistical analysis of real time PCR a student’s t-test or one way ANOVA with multi-comparison was applied. Statistical analyses of cell viability studies were performed by column statistics with one-sample student’s t-test. In vitro data points mentioned in this publication consist of at least three independent experiments each performed with two biological replicates. A p-value ≤ 0.05 was considered statistically significant.
4. Discussion
In this study, we investigated cathepsin B digestible particles for delivery of chemotherapeutics to ovarian cancer cells. We loaded PDMS-PMOXA nanoparticles with paclitaxel and modified the particle surface with a cathepsin B cleavable peptide. A suitable in vitro model was selected and characterized before assessing the impact of the formulated nanoparticles on cellular viability.
The herein studied formulation is based on the idea to use cathepsin B as a trigger for release of a chemotherapeutic. The mode of action of this release mechanism requires increased activity of cathepsin B to ensure the triggering release close to the tumor. Comparison of expression levels of this proteolytic enzyme in healthy and malignant transformed tissue originating from various organs revealed that there is no significant difference in mRNA levels for most of the herein tested tumor entities. In this context it seems noteworthy, that the mRNA expression was assessed in a small collection of samples. Even though the sample number was low, out of the researched tumor-entities, breast cancer showed a significant reduction in cathepsin B mRNA expression compared to healthy tissue. We are not providing data on activity or protein amount. However, Berquin et al. observed decreased mRNA in RAS-transformed MCF-10A epithelial cells, while the amount and activity of the enzyme was increased compared to parenteral cells [
32]. Importantly, Krepela et al. also observed increased cathepsin B activity in malignant breast carcinoma tissue [
33].
The impact of tumor progression on cathepsin B mRNA expression levels was assessed considering the reported tumor stage. In cervical and endometrial cancer tissue, mRNA expression was not significantly different from healthy tissue. For ovarian carcinoma a trend towards increased mRNA expression levels was observed, especially in stage I tumors. Therefore, ovarian carcinoma was used as a model system for further investigation, supported by findings of a larger scale study on cathepsin B mRNA expression in malignant ovarian carcinoma compared to healthy tissue [
34]. Interestingly, their data have shown a higher distribution of mRNA expression in malignant ovarian cancer stage I/II than in stage III/IV, which is similar to our data. Benign tumors on the other hand showed no difference in cathepsin B expression compared to healthy tissue [
35]. Although mRNA expression is very interesting to discuss, it is difficult to correlate the expression level to the amount or the activity of cathepsin B due to alternative splicing variants. Concerning the cathepsin B expression, there are many regulatory steps during transcription, post-transcriptional processing, translation, post-translational processing, and trafficking [
36]. The transcriptional efficiency can be influenced by different transcriptional starting points [
37,
38], by alternative promoters [
36] and variable pre-mRNA splicing [
37,
39,
40,
41]. In tumor tissue, the mRNA splice-variant lacking exon 2 is assumed to be predominant. This splice-variant has a lower stability due to a shorter untranslated region (UTR) and it is twice as active as the mRNA including exon 2 [
37]. The mRNA lacking exon 2 seems to be prone to extracellular release, especially when the expression is increased [
42]. In short, the differences in the mRNA sequences are suggested to affect the stability of the mRNA, which influences translation, sorting, and activity of this enzyme [
37,
42].
Tumors are known for their heterogeneity in many phenotypic features including gene expression [
43]. Unsurprisingly, cathepsin B appears to be non-uniformly expressed throughout the tumor but increased expressed at the border to the extracellular matrix [
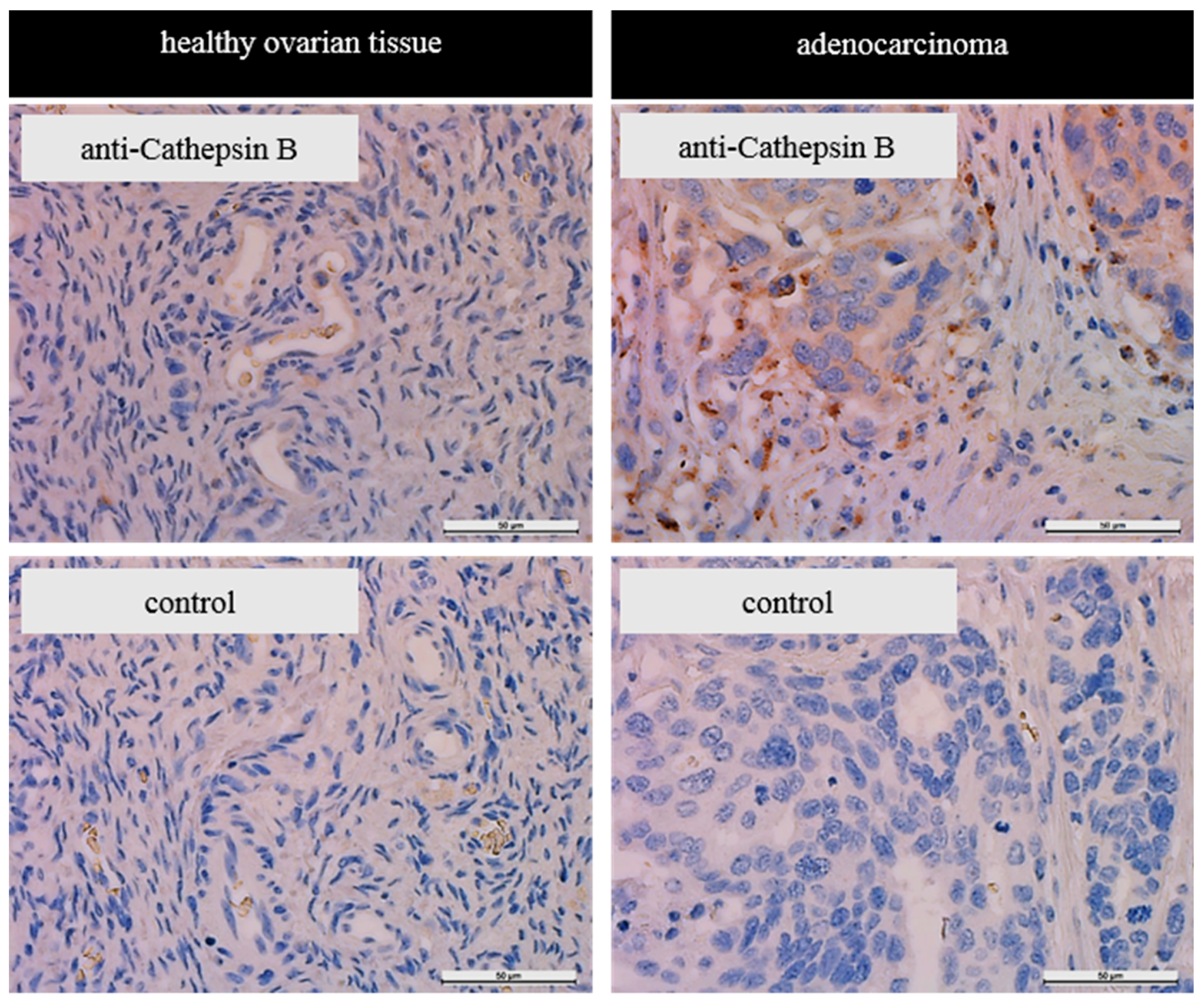
23]. Immunohistochemical staining of tissue sections supported the notion that there is an increased amount of cathepsin B in ovarian tumor tissue compared to healthy ovarian tissue. However, the herein reported immunohistochemistry data are only of limited validity as we are only reporting on one sample. However, they support findings of Scorilas et al. who reported similar results researching tumor tissue deriving ovarian cancer patients by immunohistochemistry [
44] and of Warwas et al. describing increased cathepsin B activity in the blood serum of patients with ovarian carcinoma [
35].
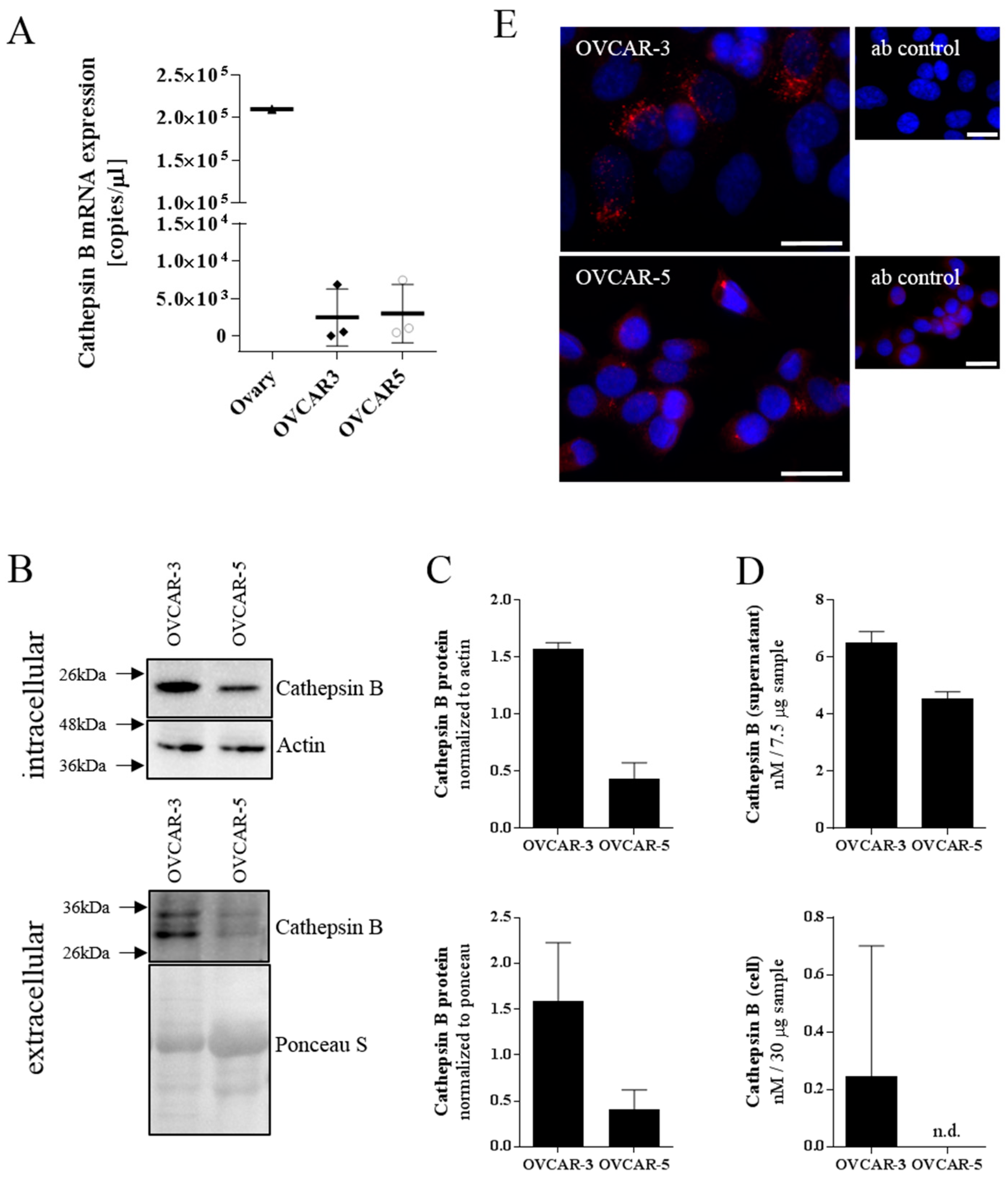
We characterized the ovarian cancer cell lines OVCAR-3 and OVCAR-5 for their cathepsin B expression. Both cell lines showed similar cathepsin B mRNA expression levels which is in accordance with the GEO dataset GDS4296/227961_at [
45]. However, Western blot analysis of intra- and extracellular proteins showed a higher amount of cathepsin B in OVCAR-3 compared to OVCAR-5 cells. This finding was confirmed by immunofluorescent staining leading to OVCAR-3 being selected as the in vitro model system of choice for our study on CTSB-peptide-modified polymersomes.
Following our previous research on an MMP9-sensitive drug delivery system [
30], a cathepsin B-sensitive drug delivery system was to be developed and investigated. For cathepsin B, there have been lesser approaches described, making it an interesting topic. Most of the researched formulations were polymer–drug conjugates with cathepsin B triggered release mechanisms [
46,
47,
48]. These formulations use either a cathepsin B degradable polymer [
46] or peptides like the herein used amino acid sequence Gly-Phe-Leu-Gly [
47,
48,
49,
50,
51] for the enzyme-triggered release of the payload.
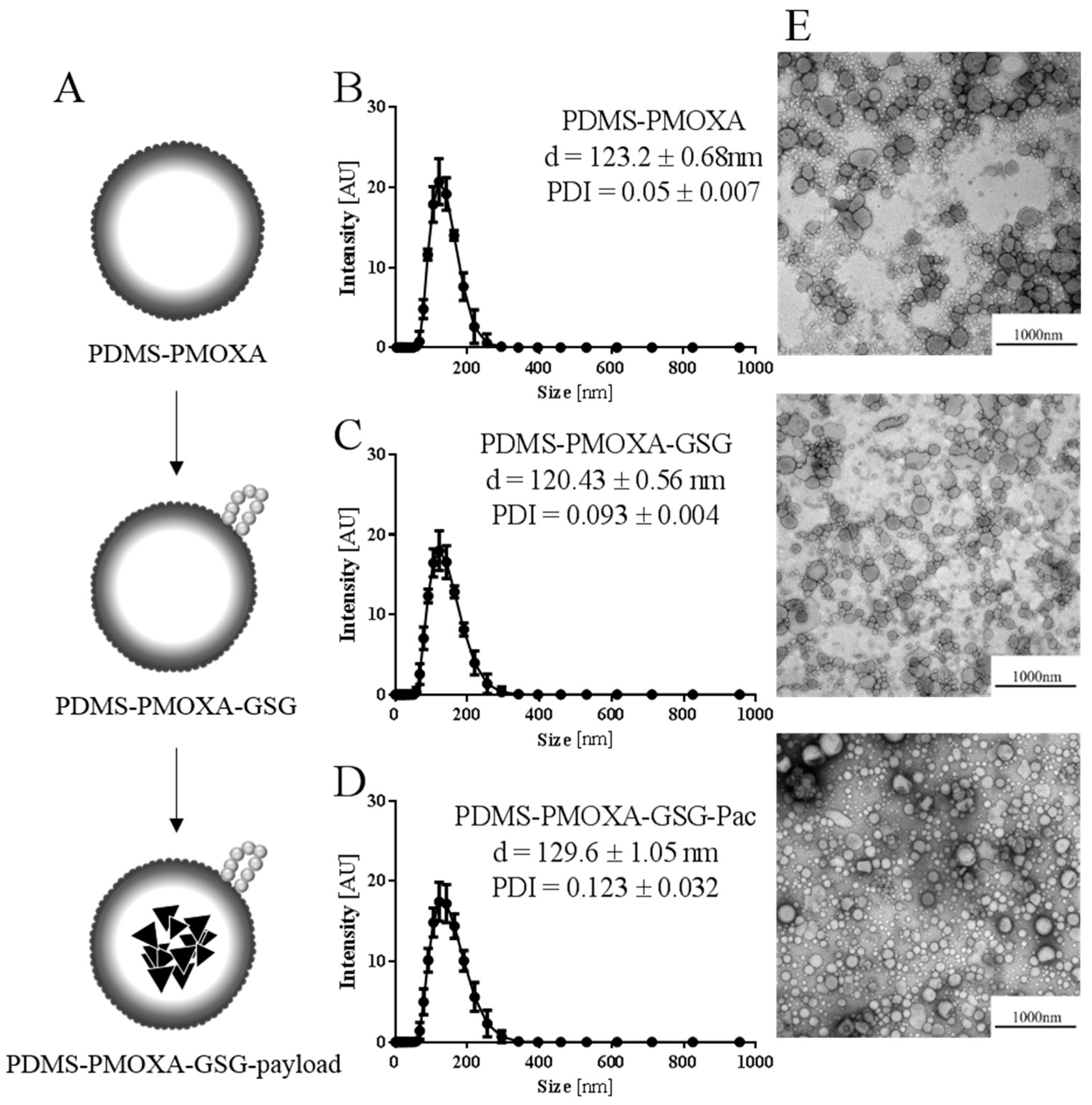
Our nanoparticles appear to be colloidally stable. However, testing the stability of the formulation over a period of 5 days revealed a significant swelling of the nanoparticles. The stability was accomplished by additional extrusion step after loading and modification. After this additional extrusion step the loading efficacy was fairly low at 9.22%, which could also depend on the used peptide.
In the herein used in vitro model system PDMS-PMOXA-GSG nanoparticles loaded with paclitaxel showed an approximately 25-fold decrease in IC50 compared to pure paclitaxel. This indicated a gradual release of paclitaxel during exposure with cells. A trend towards increasing cytotoxicity was observed when adding purified cathepsin B to the nanoparticles. This suggests that the added enzyme increases the digestion of the peptide surface layer and, therefore, accelerates release of payload. The cathepsin B inhibitor (CA-074) affects cell viability as it exhibits a cytotoxic effect itself with an IC50 of 70.32 μM. Accordingly, it is difficult to differentiate between the effect of the inhibitor and the effect of the drug paclitaxel, especially in a co-treatment. An increase in cell viability was observed after testing the nanoparticles with additional cathepsin B and inhibitor, which is in line with our expectations at a concentration of 2 μM. Treatment of cells with unloaded PDMS-PMOXA-GSG nanoparticles showed no change in cell viability.
Taken together, we report on the formulation of nanoparticles loaded with paclitaxel containing a cathepsin B digestible surface. These nanoparticles were stable with a slight tendency to swell. Furthermore, the surface-modified nanoparticles were tested in a suitable ovarian cell line for their payload release.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





